

Although a number of genetic defects in myelodysplastic progenitor cells have been described, the intracellular signaling pathways underlying aberrant regulation of hematopoiesis remain relatively undefined. The findings of this study suggest that targeting the ID1 and C/EBPα transcriptional regulators may be of benefit in the design of novel therapies for low-risk myelodysplastic syndromes.
Keywords: myelodysplastic syndrome, myeloid, ID1, C/EBPα, hematopoiesis
Abstract
Background
In patients with myelodysplasia, a general defect in the multipotent stem-cell compartment results in disturbed proliferation and differentiation of the erythroid, megakaryocytic and myeloid lineages. Although a number of genetic defects in myelodysplastic progenitor cells have been described, the intracellular signaling pathways underlying aberrant regulation of myelopoiesis remain relatively undefined.
Design and Methods
Here, an ex vivo differentiation system was used to selectively screen for molecules improving defective hematopoiesis in myelodysplastic CD34+ progenitor cells.
Results
Bone marrow-derived CD34+ cells isolated from patients with low-risk myelodysplastic syndrome showed impaired capacity to proliferate and differentiate as well as increased levels of apoptosis. In an attempt to improve the expansion and differentiation of the myelodysplastic CD34+ progenitors, cells were treated with the p38MAPK pharmacological inhibitor SB203580, or retrovirally transduced to ectopically express active protein kinase B (PKB/c-akt), or the transcriptional regulators STAT5, C/EBPα or ID1. Whereas treatment of progenitors with SB203580, PKB or STAT5 did not enhance neutrophil development, ID1- and C/EBPα-transduced cells exhibited increased granulocyte/macrophage colony formation. Furthermore, ectopic expression of C/EBPα resulted in improved neutrophil maturation.
Conclusions
These data suggest that targeting the ID1 and C/EBPα transcriptional regulators may be of benefit in the design of novel therapies for low-risk myelodysplasia.
Introduction
Myelodysplastic syndromes (MDS) are defined as clonal stem-cell disorders characterized by ineffective hematopoiesis and with an increased risk of transformation to acute myeloid leukemia (AML).1 Leukemic transformation from normal stem cells is believed to be a multistep process during which a normal hematopoietic stem cell acquires multiple genetic and epigenetic abnormalities that ultimately lead to malignant transformation and clonal expansion.2,3 Expansion of the aberrant clone is characterized by morphological dysplasia, impaired differentiation and defective cellular functions, resulting in peripheral cytopenias that frequently involve the erythroid, myeloid, and megakaryocytic lineages.4 Programmed cell death is often up-regulated in early MDS, due to the enhanced proliferative capacity of MDS clones, and may also contribute to the peripheral cytopenias. Progression to leukemia is usually associated with abrogation of apoptosis.3 The clinical symptoms resulting from these cellular defects are transfusion-dependent anemia, an increased risk of infection or hemorrhage, and a potential progression to AML.5
The myelodysplastic syndromes can be classified into high-risk or low-risk groups according to the French American British (FAB) or the more recently established World Health Organization (WHO) classification systems.6,7 These classifications are based on the number of blast cells in the bone marrow and peripheral blood, the morphology of cells and cytogenetic abnormalities. Progression towards leukemia is rare in low-risk MDS and life expectancy is relatively long, whereas in the high-risk groups progression rates towards AML are significantly higher.8
Cytogenetic abnormalities are common and are observed in about half the cases of primary MDS and in 90% of secondary, therapy-related MDS.9 The abnormalities observed in MDS are predominantly specific chromosomal deletions, suggesting a pathogenic mechanism based on loss of tumor suppressor genes or of genes necessary for normal hematopoiesis. The most frequently observed cytogenetic abnormalities in MDS include loss of chromosome 7 or partial deletions of chromosome arms 5q, 20q, or 7q.10–12 While the majority of putative tumor suppressors in MDS remain unknown, several chromosomal translocation-mediated oncogenes and tumor suppressors have been identified. Gene inactivation is responsible for a relatively small number of MDS cases and the genes involved include p53, RB, NF1, C/EBPα, and nucleophosmin.13–19 Activating mutations in the RAS proto-oncogene, FLT3 duplications, loss-of-function point mutations in the gene encoding the AML1/RUNX1 transcription factor and p15 promoter hypermethylation have also been associated with disease progression to AML.20–23 However, none of these observed alterations is specific for MDS and the underlying molecular causes of the disease remain poorly understood.
Although a number of genetic defects in MDS progenitors have been described, the intracellular signaling pathways underlying deregulation of myelopoiesis have scarcely been investigated thus far. Through identification of the intracellular components responsible for dysfunctional hematopoiesis it will be possible to develop novel treatment strategies for MDS and AML.
To investigate defects in intracellular signaling pathways in MDS CD34+ hematopoietic progenitor cells, we developed an ex vivo hematopoiesis culture system. Furthermore, in an attempt to improve the proliferation, survival and differentiation of MDS CD34+ progenitors, cells were treated with the p38MAPK pharmacological inhibitor SB203580, or retrovirally transduced to ectopically express active protein kinase B (PKB/c-akt), or the transcriptional regulators STAT5, C/EBPα or ID1.
Design and Methods
Isolation and culture of human CD34+ cells
Mononuclear cells were isolated from bone marrow of healthy subjects and MDS patients by density centrifugation over a Ficoll-paque solution (density 1.077 g/mL). MACS immunomagnetic cell separation (Miltenyi Biotech, Auburn, CA, USA) using a hapten-conjugated antibody against CD34, which was coupled to beads, was used to isolate CD34+ cells. CD34+ cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, Paisley, UK) supplemented with 9% fetal calf serum (FCS) (Hyclone, Logan, UT, USA), 50 μM-mercaptoethanol, 10 U/mL penicillin, 10 μg/mL streptomycin, and 2 mM glutamine at a density of 0.3×106 cells/mL. Cells were differentiated towards neutrophils upon addition of stem cell factor (SCF) (50 ng/mL), FLT-3 ligand (50 ng/mL), granulocyte-macrophage colony-stimulating factor (GM-CSF) (0.1 nmol/L), and granulocyte colony-stimulating factor (G-CSF) (30 ng/mL). Every 3 days, cells were counted and fresh medium was added to a density of 0.5×106 cells/mL. After 3 days of differentiation, only G-CSF was added to the cells. Pharmacological inhibitors were freshly added to the cells every 3 or 4 days. SB203580 (Alexis Corporation, San Diego, CA, USA) at a dose of 10 μM was used to inhibit p38MAPK activity during granulopoiesis.
Patients
Heparinized human bone marrow cells were collected from MDS patients with a mean age of 62 years (range, 37–78 years) after informed consent had been obtained in accordance with the Declaration of Helsinki. Bone marrow specimens were obtained at diagnosis before treatment. According to the FAB classification, the patients were categorized as having refractory anemia (n=3), refractory anemia with ringed sideroblasts (n=6) or refractory anemia with an excess of blasts (n=1). The patient’s characteristics are described in Table 1. None of these patients was treated with G-CSF. Normal bone marrow was obtained from patients undergoing orthopedic surgery who gave informed consent to this collection prior to their operation. The protocols were approved by the human subject review board of the University Medical Center, Groningen.
Table 1.
Patients’ characteristics.

RA: refractory anemia; RARS: refractory anemia with ring sideroblasts;
RAEB: refractory anemia with excess blasts;Transf: transfusion-dependent;
Gran: granulocyte percentage in the peripheral blood. None of the patients had cytogenetic abnormalities.
Histochemical staining of hematopoietic cells
May-Grünwald Giemsa staining was used to analyze myeloid differentiation. Cytospins were prepared from 5×104 differentiating granulocytes and were fixed in methanol for 3 min. After fixation cytospins were stained in a 50% eosin methylene blue solution according to May-Grünwald (Sigma-Aldrich GmbH, Seelze, Germany) for 20 min, rinsed in water for 5 seconds, and the nuclei were counterstained with 10% Giemsa solution (Merck kGaA, Darmstadt, Germany) for 15 min. During neutrophil differentiation, cells could be characterized as differentiating from myeloblast, promyelocyte I, promyelocyte II, myelocyte, and metamyelocytes towards neutrophils with banded or segmented nuclei. These stages can be distinguished by the size of the cells, ratio of cytoplasm versus nucleus present, presence of azurophilic granules and the shape of the nuclei. Differentiated neutrophils were characterized as cells containing either banded or segmented nuclei.
Viral transduction of CD34+ cells
Bicistronic retroviral DNA constructs were utilized, expressing the gene of interest and an internal ribosomal entry site (IRES) followed by the gene encoding for enhanced green fluorescent protein (eGFP) (LZRS-eGFP).24–26 The retrovirus was produced by stable transfection of the retroviral packaging cell line, Phoenix-ampho by calcium-phosphate co-precipitation. Cells were plated in 6-cm dishes, 24 h before transfection. Ten micrograms of DNA were used per transfection. Medium was refreshed, 16 h after transfection. After an additional 24 h, cells were split into 75-cm2 culture flasks (Greiner, Frickenhausen, Germany), and 2 μg/mL puromycin was added to the cells. After 2 weeks of selection, cells were grown to a confluence of 90%. Subsequently, cells were grown in a minimal amount of medium for 24 h. Viral supernatants were collected and filtered through a 0.2 μm filter. CD34+ cells were transduced in 24-well dishes precoated with 1.25 μg/cm2 recombinant human fibronectin fragment CH-296 (RetroNectin; Takara, Otsu, Japan) overnight at 4°C. Transduction was performed by addition of 0.5 mL viral supernatant to 0.5 mL medium containing 0.3×106 cells. Twenty-four hours after transduction, 0.7 mL medium was removed from the cells, and 0.5 mL fresh virus supernatant was added together with 0.5 mL fresh medium.
Colony-forming unit assay
Freshly isolated CD34+ cells or retrovirally transduced cells, separated from non-transduced cells by flow cytometry, were used in colony-forming unit (CFU) assays. Cells were plated in IMDM supplemented with 35.3% FCS (Hyclone, Logan, UT, USA), 44.4% methylcellulose-based medium called Methocult (StemCell Technologies, Vancouver, Canada), 11.1 μmol/L of β-mercaptoethanol, 2.2 U/mL of penicillin, 2.2 μg/mL of streptomycin, and 0.44 mmol/L of glutamine at a density of 500 cells/well. CFU assays were done in the presence of SCF (50 ng/mL), FLT-3 ligand (50 ng/mL), GM-CSF (0.1 nmol/L), interleukin-3 (0.1 nmol/L), and G-CSF (0.2 nmol/L). Colonies were scored after 12 days of culture.
Measurement of apoptosis
Apoptotic cells were measured by staining with annexin V (Alexis, Leiden, The Netherlands) according to the manufacturer's protocol. Necrotic cells were visualized in the same assay by staining with propidium iodide.
Western blot analysis
Western blot analysis was performed using standard techniques. In brief, differentiating granulocytes were lysed in Laemmli buffer (0.12 M Tris HCl pH 6.8, 4% SDS, 20% glycerol, 0.05 μg/μL bromophenol blue, and 35 mM β-mercaptoethanol) and boiled for 5 min. Equal amounts of total lysate were analyzed by 12% SDS-polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon-P and incubated with blocking buffer (Tris buffered saline/Tween-20) containing 5% low-fat milk for 1 h before incubating with an antibody against p38MAPK (Cell Signaling Technology, Beverly, MA, USA) overnight at 4°C in the same buffer. Before incubation with antibodies against phosphorylated p38MAPK or phosphorylated MAPKAPK-2 (Cell Signaling Technology, Beverly, MA, USA) for 16 h at 4°C, blots were incubated for 1 h in blocking buffer containing 5% bovine serum albumin (BSA). Blots were subsequently incubated with peroxidase conjugated secondary antibodies for 1 h. Enhanced chemical luminescence (ECL) was used as a detection method according to the manufacturer’s protocol (Amersham Pharmacia, Amersham, UK).
Immunohistochemical staining of hematopoietic cells
Cells were first washed in phosphate-buffered saline (PBS) and resuspended in 100 μL 0.5% formaldehyde. After 15 min incubation at 37°C, 900 μL of ice-cold methanol was added to the cells. Cells were washed with PBS after 30 min of incubation on ice and resuspended in PBS/5% FCS (Hyclone, Logan, UT, USA). After 10 min incubation at room temperature, cells were washed and neutrophil progenitors were incubated with an antibody against phosphorylated p38MAPK (Cell Signaling Technology, Beverly, MA, USA) in PBS containing 5% FCS and incubated for another 30 min at 4°C. Cells were washed and subsequently incubated with a phycoerythrin (PE)-conjugated anti-rabbit antibody (Southern Biotechnology Associates, Inc., Birmingham, AL, USA) for another 30 min at 4°C. Cells were again washed cells and were analyzed by FACS (FACS Canto II, Beckton Dickinson, Alphen a/d Rijn, The Netherlands).
Statistics
An independent sample t test for was performed to compare the differences in proliferation, differentiation, and annexin-positive cells between the controls and cells transduced with STAT5, myrPKB, ID1, or C/EBPα. The same assay was performed to compare cells cultured in either the absence of presence of the pharmacological inhibitor SB203580. A p value of 0.05 or less was considered statistically significant.
Results
Myelodysplastic syndrome bone marrow progenitor cells show impaired progenitor expansion and increased levels of apoptosis during neutrophil differentiation
To investigate defects in intracellular signaling pathways in MDS CD34+ hematopoietic progenitor cells, an ex vivo differentiation system was utilized as described in the Design and Methods section. Human CD34+ hematopoietic progenitor cells, isolated from the bone marrow of healthy subjects and low-risk MDS patients (Table 1), were cultured in the presence of G-CSF to induce neutrophil differentiation. Cells were cultured for 17 days and differences in expansion, survival and differentiation were analyzed. Expansion of MDS CD34+ cells was dramatically decreased during neutrophil differentiation compared to the expansion of CD34+ cells from healthy controls (Figure 1A). To determine whether this impaired capacity to expand could be due to increased levels of apoptosis, the percentage of annexin-V positive cells was analyzed. The survival of MDS hematopoietic progenitor cells was significantly lower than that of control CD34+ cells during neutrophil development (Figure 1B). These results demonstrate that MDS hematopoietic progenitors have an impaired capacity to proliferate and show decreased survival during neutrophil development ex vivo.
Figure 1.
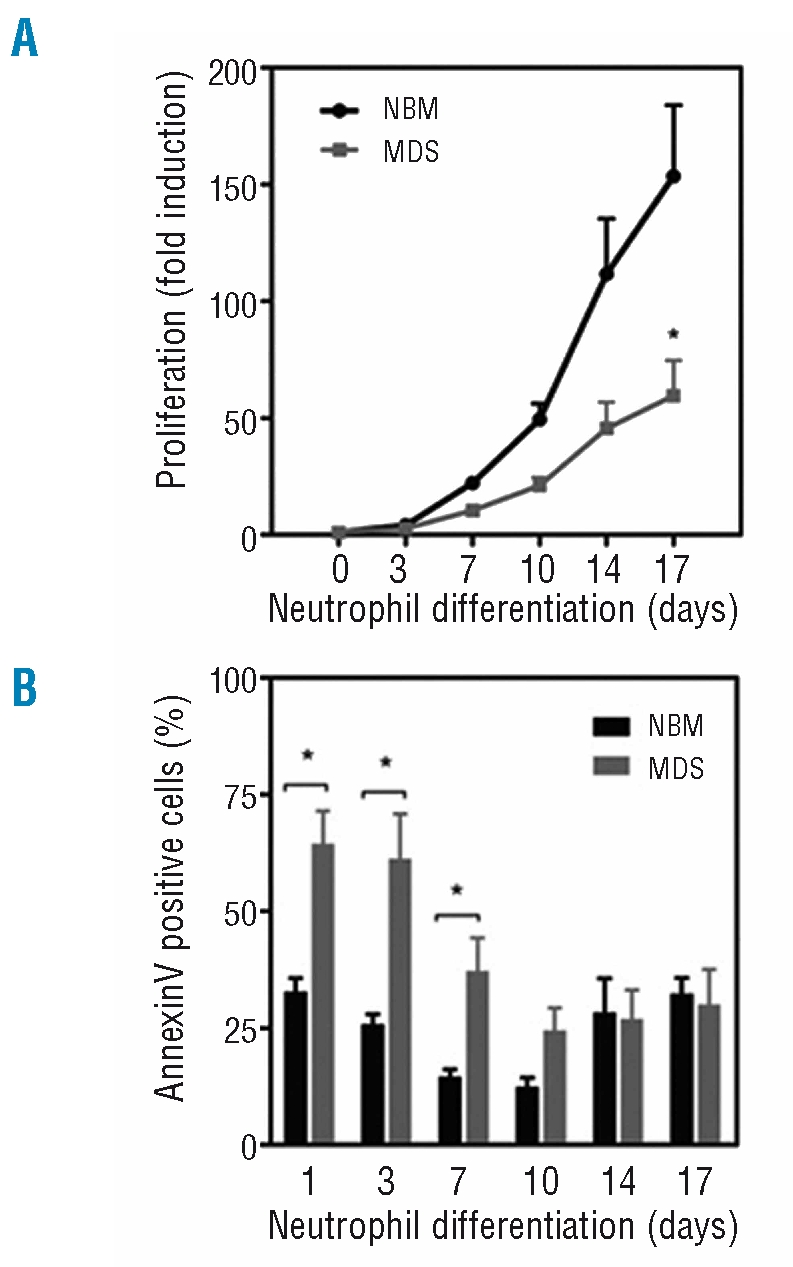
Myelodysplastic syndromes progenitor cells show impaired expansion and increased levels of apoptosis during neutrophil differentiation. CD34+ hematopoietic progenitor cells, isolated from the bone marrow of healthy subjects (NBM) and patients with low-risk myelodysplastic syndromes were cultured for 17 days in the presence of G-CSF to induce neutrophil differentiation. (A) Expansion was determined by counting the trypan blue-negative cells. (B) During the 17-day culture period the percentage apoptotic cells was determined by annexin V staining. Results are presented as means of five independent experiments. Error bars represent SEM.
Impaired differentiation and decreased colony formation capacity of myelodysplastic syndrome bone marrow progenitor cells during myelopoiesis
To determine whether MDS hematopoietic progenitor cells also had an impaired capacity to differentiate, CD34+ cells were isolated from the bone marrow of healthy subjects and low-risk MDS patients and differentiated towards neutrophils for 17 days. After 14 and 17 days of differentiation, cytospins were prepared to analyze the morphology of the differentiating granulocytes. As expected, MDS bone marrow progenitor cells showed significantly lower percentages of mature neutrophils with banded or segmented nuclei and increased numbers of undifferentiated myeloblasts after 14 and 17 days of neutrophil differentiation (Figure 2A, 2B). In addition, to assess the clonogenic potential of progenitor cells, CD34+ cells from controls and low-risk MDS patients were plated in CFU assays, and colony formation was analyzed after 12 days of culture. Granulocyte-macrophage (GM) colony-forming units (CFU-GM) of MDS CD34+ cells were reduced compared to control CD34+ cells (Figure 2C). Together these results demonstrate that CD34+ bone marrow cells from low-risk MDS patients have an impaired capacity to proliferate as well as to differentiate and show increased levels of apoptosis during neutrophil development. Importantly, this ex vivo system is representative of published observations in MDS bone marrow in vivo.
Figure 2.

Impaired differentiation and decreased GM colony formation capacity of myelodysplastic syndromes (MDS) CD34+ progenitors during myelopoiesis. CD34+ hematopoietic progenitor cells, isolated from the bone marrow of healthy subjects (NBM) and patients with low-risk MDS were cultured for 17 days in the presence of G-CSF to induce neutrophil differentiation. After 14 and 17 days of differentiation, (B) cytospins were prepared and stained with May-Grünwald Giemsa solution. (A) Data are expressed as the percentage of differentiated neutrophils. (C) CD34+ progenitor cells from controls and patients with low-risk MDS were plated in CFU assays, and colony formation was analyzed after 12 days. Results in Figures 2A and 2C are presented as means of six different MDS samples (2 with refractory anemia, 3 with refractory anemia with ringed sideroblasts, 1 with refractory anemia with excess blasts), of which four are also included in the experiments described in Figure 3 (2 with refractory anemia, 2 with refractory anemia and ringed sideroblasts). Error bars represent SEM.
Inhibition of p38 MAPK does not improve maturation or increase clonogenic capacity of myelodysplastic hematopoietic progenitor cells
A recent study showed that p38MAPK is constitutively activated in hematopoietic cells from patients with low-risk MDS.27 Inhibition of p38MAPK activity resulted in decreased apoptosis and stimulated colony formation of primary MDS progenitors, indicating a role for p38MAPK in the pathogenesis of MDS.27 To determine whether aberrant p38MAPK activation may be involved in the defective neutrophil differentiation observed in MDS primary progenitors, CD34+ hematopoietic progenitor cells isolated from controls and patients with MDS were cultured in the presence of G-CSF to induce neutrophil differentiation. After 6 days of culture, protein lysates were made and western blot analysis was performed using an antibody against phosphorylated p38MAPK or p38MAPK as a control for equal loading (Figure 3A). In contrast to the previous report, CD34+ cells from low-risk MDS patients did not show elevated levels of phosphorylated p38MAPK compared to controls. In addition, flow cytometric analysis using an antibody against phosphorylated p38MAPK was performed to further support these observations (Figure 3B).
Figure 3.

Inhibition of p38MAPK does not improve maturation or increase clonogenic capacity of myelodysplastic syndromes (MDS) progenitor cells. (A) CD34+ cells, isolated from bone marrow of healthy subjects (lanes 1 and 3) and patients with low-risk MDS (lanes 2 and 4), were cultured in the presence of G-CSF to induce neutrophil differentiation. After 6 days of culture, protein lysates were made and western blot analysis was performed using an antibody against phosphorylated p38MAPK, or p38MAPK as a control for equal loading. (B) Activation of p38MAPK was further analyzed by flow cytometry using an antibody against phosphorylated p38MAPK. (C) CD34+ cells were cultured in the presence of G-CSF to induce neutrophil differentiation. After 6 days of culture, cells were left untreated (lanes 1 and 2) or treated with SB230580 (lane 3) for 45 min before stimulation with G-CSF (lanes 2 and 3) for 15 min. Protein lysates were prepared and western blot analysis was performed with an antibody against phosphorylated MAPKAPK2 or p38MAPK as a control for equal loading. (D) After 17 days of differentiation, cytospins were prepared to analyze the morphology of the differentiating granulocytes. Data are expressed as the percentage of differentiated neutrophils. (E) CD34+ progenitor cells isolated from patients with low-risk MDS were plated in CFU assays in either the presence or absence of 10 μM SB203580 and colony formation was analyzed after 12 days. Results are presented as means of three independent experiments. Error bars represent SEM.
In order to investigate whether inhibition of p38MAPK can indeed improve final maturation of MDS progenitors during myelopoiesis, CD34+ cells isolated from patients with MDS were differentiated towards neutrophils in either the presence or absence of the specific p38MAPK inhibitor SB203580. The efficacy of SB203580 was confirmed by its ability to inhibit the phosphorylation of MAPKAPK-2 (MK2) in CD34+ cells, a direct target of p38MAPK (Figure 3C). After 17 days of differentiation, cytospins were prepared to analyze the morphology of the differentiating granulocytes. Inhibition of p38MAPK activity did not improve neutrophil development of MDS CD34+ cells (Figure 3D). In addition, to determine whether inhibition of p38MAPK can improve the clonogenic capacity of primary MDS progenitor cells, CD34+ cells from low-risk MDS patients were plated in CFU assays, and colony formation was analyzed after 12 days. Treatment of MDS hematopoietic progenitor cells with SB203580 did not result in significantly increased CFU-GM (Figure 3E). Together these results suggest that inhibition of p38MAPK is not sufficient to restore final maturation or improve the clonogenic capacity of low-risk MDS hematopoietic progenitor cells.
Increased PKB or STAT5a activity is not sufficient to restore neutrophil development in myelodysplasia
Hematopoietic cytokines can activate several signal transduction pathways, which have been shown to be involved in the regulation of myeloid differentiation, including p38MAPK, phosphatidylinositol 3 kinase (PI3K), and the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway.28 Recently we have demonstrated that during myelopoiesis PKB activity is essential for both hematopoietic progenitor survival and neutrophil development.26 We also observed that CD34+ cells from low-risk MDS patients show decreased PKB activation in response to the chemo-attractant SDF-1.29 Furthermore, neutrophils isolated from low-risk MDS patients also exhibited decreased PKB phosphorylation upon stimulation with fMLP.30 However, CD34+-derived neutrophil progenitor cells from low-risk MDS patients did not show significantly altered levels of phosphorylated PKB compared to corresponding cells from controls (Online Supplementary Figure S1). To further determine whether aberrant PKB activation may be involved in the defective neutrophil differentiation observed in MDS primary progenitors, we used of a bicistronic retroviral DNA construct co-expressing eGFP and a constitutively active form of PKBα (myrPKB). A retrovirus was generated and used to transduce low-risk MDS CD34+ cells, which were cultured in the presence of G-CSF to induce neutrophil differentiation. Three days after transduction, eGFP-positive cells were sorted by FACS from the non-transduced cells. After 14 and 17 days of differentiation, cytospins were prepared and the morphology of the cells was analyzed after May-Grünwald Giemsa staining. Activation of PKB did not improve the reduced neutrophil development from hematopoietic progenitors isolated from patients with MDS (Figure 4A), indicating that activation of PKB alone is not sufficient to rescue neutrophil development in MDS.
Figure 4.

Increased PKB or STAT5a activity is not sufficient to restore neutrophil development in myelodysplastic syndromes. CD34+ cells, isolated from patients with low-risk myelodysplastic syndromes were retrovirally transduced with (A) myrPKB, (B) STAT5a or eGFP as a control and cultured in the presence of G-CSF to induce neutrophil differentiation. After 17 days of culture, transduced cells were separated from non-transduced cells by FACS, and cytospins were prepared. Data are expressed as the percentage of differentiated neutrophils. Results are presented as means of four independent experiments. Error bars represent SEM.
We and others have also shown that activation of the transcription factor STAT5 regulates proliferation, apoptosis and differentiation during erythroid and myeloid development.24,31 To address the question of whether STAT5 signaling plays a critical role in regulating neutrophil differentiation in MDS, a bicistronic retroviral DNA construct co-expressing STAT5a and eGFP was used to transduce CD34+ cells isolated from MDS patients. After 17 days of differentiation, eGFP positive cells were sorted by FACS from the non-transduced cells and cytospins were prepared. Ectopic expression of STAT5a resulted in a modest increase in the percentage of mature neutrophils with banded or segmented nuclei compared to cells transduced with eGFP alone (Figure 4B). Together these data indicate that despite playing essential roles in granulopoiesis, ectopic expression of myrPKB or STAT5a alone is not sufficient to restore neutrophil differentiation in MDS.
Increased C/EBPα and ID1 expression results in improved neutrophil production in patients with low-risk myelodysplastic syndromes
One of the key transcriptional regulators involved in lineage choice decisions during myeloid differentiation is the CCAATT/enhancer binding protein α (C/EBPα). Genetic alterations and reduced expression of the C/EBPα gene have been found in both AML and MDS, which supports the involvement of deregulated C/EBPα expression in the inefficient granulopoiesis characteristic of MDS.18,32–34 In addition, another transcriptional regulator that has been demonstrated to play an important role in the regulation of proliferation and differentiation during myelopoiesis is inhibitor of DNA binding protein 1 (ID1).25 To investigate whether ectopic expression of ID1 or C/EBPα can indeed improve myeloid maturation of MDS progenitors, CD34+ hematopoietic progenitor cells, isolated from patients with MDS, were transduced with ID1, C/EBPα or eGFP as a control and were cultured in the presence of G-CSF to induce neutrophil differentiation. Three days after transduction, eGFP positive cells were sorted by FACS from the non-transduced cells. After 17 days of differentiation, cytospins were prepared and the morphology of the cells was subsequently analyzed after May-Grünwald Giemsa staining (Figure 5A).
Figure 5.

Ectopic expression of C/EBPα and ID1 results in improved neutrophil production in low-risk myelodysplastic syndromes (MDS). (A) CD34+ progenitor cells, isolated from the bone marrow of healthy subjects or patients with low-risk MDS were retrovirally transduced with C/EBPα, ID1 or eGFP alone and cultured in the presence of G-CSF to induce neutrophil differentiation. Subsequently, transduced cells were separated from non-transduced cells by FACS and after 17 days of culture, cytospins were prepared. (B) Data are expressed as the percentage of differentiated neutrophils. (C) CD34+ cells isolated from MDS patients were retrovirally transduced with C/EBPα, ID1 or eGFP alone. After 3 days, transduced cells were separated from non-transduced cells by FACS, plated in CFU assays and colony formation was analyzed after 12 days. Results are presented as means of three independent experiments. Error bars represent SEM.
Ectopic expression of ID1 did not improve the reduced neutrophil development of MDS CD34+ cells. However, transduction of MDS CD34+ cells with C/EBPα resulted in a dramatic increase in the percentage of neutrophils with banded or segmented nuclei (Figure 5B). These results demonstrate that ectopic expression of C/EBPα is sufficient to restore neutrophil development of MDS hematopoietic progenitors.
In addition, to determine whether ID1 or C/EBPα can improve the clonogenic capacity of MDS CD34+ cells, CFU assays were performed, and colony formation was analyzed after 12 days of culture. Interestingly, ectopic expression of both ID1 and C/EBPα resulted in increased GM colony formation (Figure 5C). Together these data suggest that targeting the ID1 and C/EBPα transcriptional regulators may be of benefit in the design of novel therapies for low-risk MDS.
Discussion
Although MDS are some of the most prevalent hematologic disorders, the defects in the intracellular signaling pathways responsible for aberrant hematopoiesis in these conditions remain largely undefined. In the present study, we investigated whether known regulators of myeloid differentiation can improve neutrophil development of MDS CD34+ hematopoietic progenitor cells utilizing a human ex vivo granulocyte differentiation system. Our data demonstrate that ID1 and C/EBPα both play a role in expansion and differentiation during myeloid development of MDS hematopoietic progenitors. Ectopic expression of ID1 and C/EBPα resulted in enhanced GM colony formation, whereas treatment of progenitors with SB203580, PKB or STAT5 did not improve colony formation. In addition, C/EBPα-transduced MDS progenitors exhibited greatly improved neutrophil maturation.
Using an ex vivo differentiation system to study the defects in intracellular signaling pathways in MDS CD34+ hematopoietic progenitor cells, we were able to demonstrate that CD34+ bone marrow cells from low-risk MDS patients have both an impaired capacity to proliferate and show increased levels of apoptosis during neutrophil development. Importantly, this ex vivo system thus mimics the published observations in MDS bone marrow in vivo.35
p38MAPK is a serine-threonine kinase, originally discovered as a stress-activated kinase, which has been demonstrated to be involved in the regulation of differentiation of various cell types, including granulocytes, with its effects being cell type- and context-specific.36 Inhibition of p38MAPK activity was shown to enhance neutrophil development, while constitutive activation of the MKK3/p38MAPK signaling module dramatically inhibited neutrophil differentiation (unpublished data). In addition, it was recently shown that p38MAPK is constitutively activated in the bone marrow of patients with low-risk MDS. Inhibition of p38MAPK activity was found to decrease apoptosis and stimulate GM colony formation in primary MDS progenitors, suggesting a role for p38MAPK in the pathogenesis of this syndrome.37,27 However, our data indicate that inhibition of p38MAPK is not sufficient to restore final maturation or improve the clonogenic capacity of low-risk MDS hematopoietic progenitors (Figure 3D, 3E). Moreover, CD34+ cells from low-risk MDS patients did not show elevated levels of phosphorylated p38MAPK compared to control cells (Figure 3A, 3B). These differences might be explained by donor variations between MDS patients’ samples as MDS comprise a heterogeneous group of stem cell disorders. In addition, although Navas et al. demonstrated that inhibition of p38MAPK stimulates GM colony formation in primary low-risk MDS progenitors,27 they did not characterize the morphology of the differentiating neutrophils.
PI3K has been demonstrated to play a critical role in the survival and proliferation of a variety of cell types and recent evidence showed that PI3K and its downstream effector PKB also play an important role in regulating hematopoiesis.26 Previously, we demonstrated that CD34+ cells from low-risk MDS patients show decreased PKB phosphorylation in response to the chemo-attractant SDF-1.29 In addition, constitutive activation of PKB in bone marrow mononuclear cells from patients with high-risk MDS was reported, while mononuclear cells from normal bone marrow and patients with low-risk MDS demonstrated low levels or no PKB activation.38 Taken together, these findings suggest that aberrant PKB activation might be one of the factors contributing to the ineffective hematopoiesis observed in MDS. However, activation of PKB in MDS CD34+ hematopoietic progenitor cells did not improve aberrant neutrophil development, indicating that activation of PKB alone is also insufficient to rescue neutrophil development in low-risk MDS (Figure 4A).
Several studies suggest that STAT5 may play a critical role in neutrophil development. Loss of STAT5 function in primary bone marrow cells, for example, leads to a reduction in CFU-G colony formation, while bone marrow cells from mice lacking STAT5 are unable to repopulate the myeloid lineage of lethally irradiated wild-type recipient mice.39–42 Furthermore, it has been demonstrated that STAT5 favors the survival of myeloid progenitors by inducing expression of the anti-apoptotic protein Bcl-xL.43 However, although STAT5 expression has been shown to be essential during myelopoiesis, our data indicate that expression of STAT5a is again not sufficient to restore neutrophil development in low-risk MDS (Figure 4B).
ID proteins function as inhibitors of members of the basic helix-loop-helix family of transcription factors and have been demonstrated to play an important role in regulating proliferation and differentiation of a variety of cell lineages.44 It has been shown that ID1 mRNA levels are often high in proliferating cells, but are down-regulated in differentiating cells.45 We have previously shown that ID1 levels are upregulated during early granulopoiesis, then decrease during final maturation.25 Our data suggest that ectopic expression of ID1 is not sufficient to improve neutrophil differentiation in MDS; however, GM colony formation of MDS hematopoietic progenitors was significantly increased, suggesting that ID1 may exert its major effects on progenitor expansion during the early phase of granulopoiesis. Previous studies have demonstrated that aberrant activation of ID proteins can contribute to tumorigenesis by stimulating proliferation and facilitating neovascularization. In addition, analysis of various solid and leukemic human tumors have revealed that the level of expression of ID proteins is often elevated.46–49 While this suggests that targeting ID1 may be of benefit in the design of novel therapies for low-risk MDS, manipulation for therapeutic purposes will not be without risk.
C/EBPα is a leucine zipper transcription factor that plays a critical role in normal myelopoiesis. Expression of C/EBPα is detectable in early myeloid precursors and is upregulated upon commitment to granulocytes.50,51 Consistent with this expression pattern, mice deficient in C/EBPα lack mature neutrophils and accumulate immature myeloblasts in the bone marrow.52 Conversely, ectopic expression of C/EBPα in precursor cell lines triggers neutrophil differentiation.53,54 Mutations within the C/EBPα gene are found in approximately 9% of patients with AML, leading to production of C/EBPα mutants deficient in DNA binding.32,33,34 C/EBPα levels are also affected by various leukemic fusion proteins through mechanisms that involve transcriptional as well as translational repression.55–57 Although, alterations in the C/EBPα gene have been found in patients with AML, they seem to be less frequently observed in MDS patients.17 However, in patients with 5q- syndrome, a distinct clinical subgroup of MDS, the gene encoding C/EBPα was found to be extensively down-regulated in MDS progenitor cells.18 Besides mutations in the CEBPα gene itself, C/EBPα transcription may be repressed by DNA promoter hypermethylation. Methylation of DNA is a commmon epigenetic modification, which plays an important role in correct regulation of gene expression in mammalian cells. Hypermethylation of promoter residues and consequent inactivation of regulatory genes has been found to play a pathogenetic role in the development of MDS.58 Recent data have shown that in a specific subgroup of AML, which phenotypically resembles AML with mutations in C/EBPα, the CEBPα gene was silenced due to promoter hypermethylation.59 Our results demonstrate that ectopic expression of C/EBPα is sufficient to restore neutrophil development of MDS hematopoietic progenitors, supporting the hypothesis that abrogation of granulopoiesis in MDS patients is, at least in part, due to aberrant C/EBPα expression or functionality in the bone marrow. Interestingly, the major effect of C/EBPα was observed on neutrophil differentiation rather than on CFU-GM growth, arguing for a key role in granulocytic differentiation. Consistently, previous studies showing that expression of C/EBPα is detectable in early myeloid precursors and is upregulated upon commitment to granulocytes, indicate that C/EBPα may indeed exert its major effects on progenitor maturation during the late phase of granulopoiesis.50,51
G-CSF is an essential cytokine for both the proliferation of myeloid precursors and their differentiation into mature neutrophils. It is tempting to speculate that the number of G-CSF receptors expressed on the membrane of progenitors may play a critical role in the maturation defect in myelodysplastic patients. Previous studies demonstrated that CEBPα plays an important role in transcriptionally regulating G-CSF receptor expression, by direct interaction with the G-CSSFR promoter.60,61 Moreover, decreased G-CSF receptor expression on CD34+ cells was found in a significant proportion of patients with both low-risk and high-risk MDS.62 Interestingly, MDS patients with low receptor expression had a strong predisposition to develop neutropenia and a poor or absent response to G-CSF administration. It could, therefore, be hypothesized that increased expression of C/EBPα in low-risk MDS CD34+ cells may result in enhanced G-CSF receptor expression, leading to more efficient signaling in response to G-CSF, ultimately resulting in improved neutrophil development.
In conclusion, while a variety of genetic alterations have been reported to be involved in the pathogenesis of MDS, our data suggest that targeting C/EBPα may be sufficient in the design of novel therapies for low-risk MDS.
Acknowledgments
we would like to thank Dr G. Nolan (Stanford University School of Medicine, Stanford, CA, USA) for kindly providing us with the LZRS construct and the phoenix-ampho packaging cell line. We would also like thank Prof. Dr. H. Spits (AMC, Amsterdam, The Netherlands) for providing us with the LZRS construct expressing myrPKB and ID1. Finally, we thank Dr. O.A. MacDougald (University of Michigan Medical School Ann Arbor, MI, USA) for providing us with the C/EBPα construct.
Footnotes
The online version of this article contains a supplementary appendix.
Authorship and Disclosures
CRG designed the research, performed experiments, made the figures, analyzed results, and wrote the paper; MB designed the research, performed experiments, made the figures, analyzed results, and wrote the paper; EV designed the research and analyzed results; PJC designed the research, analyzed results and wrote the paper. The authors reported no potential conflicts of interest.
Funding: CG and MB were supported by a grant from the Dutch Cancer Society (RUG 2003-2929 and UU 2005-3659).
References
- 1.Lowenthal RM, Marsden KA. Myelodysplastic syndromes. Int J Hematol. 1997;65:319–38. doi: 10.1016/s0925-5710(96)00566-x. [DOI] [PubMed] [Google Scholar]
- 2.Fenaux P. Myelodysplastic syndromes: from pathogenesis and prognosis to treatment. Semin Hematol. 2004;41:6–12. doi: 10.1053/j.seminhematol.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld C, List A. A hypothesis for the pathogenesis of myelodysplastic syndromes: implications for new therapies. Leukemia. 2000;14:2–8. doi: 10.1038/sj.leu.2401618. [DOI] [PubMed] [Google Scholar]
- 4.Kouides P, Bennett J. Understanding the myelodysplastic syndromes. Oncologist. 1997;2:389–401. [PubMed] [Google Scholar]
- 5.Heaney ML, Golde DW. Myelodysplasia. N Engl J Med. 1999;340:1649–60. doi: 10.1056/NEJM199905273402107. [DOI] [PubMed] [Google Scholar]
- 6.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99. [PubMed] [Google Scholar]
- 7.Vardiman JW. The new World Health Organization classification of myeloid neoplasms: Q&A with James W. Vardiman, MD. Clin Adv Hematol Oncol. 2003;1:18–21. [PubMed] [Google Scholar]
- 8.Maschek H, Gutzmer R, Choritz H, Georgii A. Life expectancy in primary myelodysplastic syndromes: a prognostic score based upon histopathology from bone marrow biopsies of 569 patients. Eur J Haematol. 1994;53:280–7. doi: 10.1111/j.1600-0609.1994.tb01320.x. [DOI] [PubMed] [Google Scholar]
- 9.Mauritzson N, Albin M, Rylander L, Billström R, Ahlgren T, Mikoczy Z, et al. Pooled analysis of clinical and cytogenetic features in treatment-related and de novo adult acute myeloid leukemia and myelodysplastic syndromes based on a consecutive series of 761 patients analyzed 1976–1993 and on 5098 unselected cases reported in the literature 1974–2001. Leukemia. 2002;16:2366–78. doi: 10.1038/sj.leu.2402713. [DOI] [PubMed] [Google Scholar]
- 10.Van den Berghe H, Vermaelen K, Mecucci C, Barbieri D, Tricot G. The 5q-anomaly. Cancer Genet Cytogenet. 1985;17:189–255. doi: 10.1016/0165-4608(85)90016-0. [DOI] [PubMed] [Google Scholar]
- 11.Asimakopoulos FA, White NJ, Nacheva E, Green AR. Molecular analysis of chromosome 20q deletions associated with myeloproliferative disorders and myelodysplastic syndromes. Blood. 1994;84:3086–94. [PubMed] [Google Scholar]
- 12.Le Beau MM, Espinosa R, Davis EM, Eisenbart JD, Larson RA, Green ED. Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood. 1996;88:1930–5. [PubMed] [Google Scholar]
- 13.Preudhomme C, Vanrumbeke M, Lai JL, Lepelley P, Wattel E, Fenaux P. Inactivation of the p53 gene in leukemias and myelodysplastic syndrome (MDS) with 17p monosomy. Leukemia. 1994;8:2241–2. [PubMed] [Google Scholar]
- 14.Preudhomme C, Vachee A, Lepelley P, Vanrumbeke M, Zandecki M, Quesnel B, et al. Inactivation of the retinoblastoma gene appears to be very uncommon in myelodysplastic syndromes. Br J Haematol. 1994;87:61–7. doi: 10.1111/j.1365-2141.1994.tb04871.x. [DOI] [PubMed] [Google Scholar]
- 15.Cilloni D, Gottardi E, Messa F, Fava M, Scaravaglio P, Bertini M, et al. Significant correlation between the degree of WT1 expression and the International Prognostic Scoring System score in patients with myelodysplastic syndromes. J Clin Oncol. 2003;21:1988–95. doi: 10.1200/JCO.2003.10.503. [DOI] [PubMed] [Google Scholar]
- 16.Preudhomme C, Vachee A, Quesnel B, Wattel E, Cosson A, Fenaux P. Rare occurrence of mutations of the FLR exon of the neurofibromatosis 1 (NF1) gene in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) Leukemia. 1993;7:1071. [PubMed] [Google Scholar]
- 17.Fuchs O, Provaznikova D, Kocova M, Kostecka A, Cvekova P, Neuwirtova R, et al. CEBPA polymorphisms and mutations in patients with acute myeloid leukemia, myelodysplastic syndrome, multiple myeloma and non-Hodgkin’s lymphoma. Blood Cells Mol Dis. 2008;40:401–5. doi: 10.1016/j.bcmd.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson L, Edén P, Olsson E, Månsson R, Astrand-Grundström I, Strömbeck B, et al. The molecular signature of MDS stem cells supports a stem-cell origin of 5q myelodysplastic syndromes. Blood. 2007;110:3005–14. doi: 10.1182/blood-2007-03-079368. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhang M, Yang L, Xiao Z. NPM1 mutations in myelodysplastic syndromes and acute myeloid leukemia with normal karyotype. Leuk Res. 2007;31:109–11. doi: 10.1016/j.leukres.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Horiike S, Yokota S, Nakao M, Iwai T, Sasai Y, Kaneko H, et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997;11:1442–6. doi: 10.1038/sj.leu.2400770. [DOI] [PubMed] [Google Scholar]
- 21.Padua RA, Guinn BA, Al-Sabah AI, Smith M, Taylor C, Pettersson T, et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia. 1998;12:887–92. doi: 10.1038/sj.leu.2401044. [DOI] [PubMed] [Google Scholar]
- 22.Neubauer A, Greenberg P, Negrin R, Ginzton N, Liu E. Mutations in the RAS proto-oncogenes in patients with myelodysplastic syndromes. Leukemia. 1994;8:638–41. [PubMed] [Google Scholar]
- 23.Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood. 2004;103:2316–24. doi: 10.1182/blood-2003-09-3074. [DOI] [PubMed] [Google Scholar]
- 24.Buitenhuis M, Baltus B, Lammers JJ, Coffer PJ, Koenderman L. Signal transducer and activator of transcription 5a (STAT5A) is required for eosinophil differentiation of human cord blood-derived CD34+ cells. Blood. 2003;101:134–42. doi: 10.1182/blood-2002-03-0740. [DOI] [PubMed] [Google Scholar]
- 25.Buitenhuis M, van Deutekom HWM, Verhagen LP, Castor A, Jacobsen SEW, Lammers JJ, et al. Differential regulation of granulopoiesis by the basic helix-loop-helix transcriptional inhibitors ID1 and ID2. Blood. 2005;105:4272–81. doi: 10.1182/blood-2004-12-4883. [DOI] [PubMed] [Google Scholar]
- 26.Buitenhuis M, Verhagen LP, van Deutekom HWM, Castor A, Verploegen S, Koenderman L, et al. Protein kinase B (c-akt) regulates hematopoietic lineage choice decisions during myelopoiesis. Blood. 2008;111:112–21. doi: 10.1182/blood-2006-07-037572. [DOI] [PubMed] [Google Scholar]
- 27.Navas TA, Mohindru M, Estes M, Ma JY, Sokol L, Pahanish P, et al. Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood. 2006;108:4170–7. doi: 10.1182/blood-2006-05-023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–37. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- 29.Fuhler GM, Drayer AL, Olthof SGM, Schuringa JJ, Coffer PJ, Vellenga E. Reduced activation of protein kinase B, RAC, and F-actin polymerization contributes to an impairment of stromal cell derived factor-1 induced migration of CD34+ cells from patients with myelodysplasia. Blood. 2008;111:359–68. doi: 10.1182/blood-2006-11-060632. [DOI] [PubMed] [Google Scholar]
- 30.Fuhler GM, Drayer AL, Vellenga E. Decreased phosphorylation of protein kinase B and extracellular signal-regulated kinase in neutrophils from patients with myelodysplasia. Blood. 2003;101:1172–80. doi: 10.1182/blood.V101.3.1172. [DOI] [PubMed] [Google Scholar]
- 31.Schuringa JJ, Chung KY, Morrone G, Moore MAS. Constitutive activation of STAT5A promotes human hematopoietic stem cell self-renewal and erythroid differentiation. J Exp Med. 2004;200:623–35. doi: 10.1084/jem.20041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPα), in acute myeloid leukemia. Nat Genet. 2001;27:263–70. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 33.Gombart AF, Hofmann W, Kawano S, Takeuchi S, Krug U, Kwok SH, et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99:1332–40. doi: 10.1182/blood.v99.4.1332. [DOI] [PubMed] [Google Scholar]
- 34.Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P, Preudhomme C. CEBPA point mutations in hematological malignancies. Leukemia. 2005;19:329–34. doi: 10.1038/sj.leu.2403614. [DOI] [PubMed] [Google Scholar]
- 35.Raza A, Alvi S, Borok RZ, Span L, Parcharidou A, Alston D, et al. Excessive proliferation matched by excessive apoptosis in myelodysplastic syndromes: the cause-effect relationship. Leuk Lymphoma. 1997;27:111–8. doi: 10.3109/10428199709068277. [DOI] [PubMed] [Google Scholar]
- 36.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 37.Katsoulidis E, Li Y, Yoon P, Sassano A, Altman J, Kannan-Thulasiraman P, et al. Role of the p38 mitogen-activated protein kinase pathway in cytokine-mediated hematopoietic suppression in myelodysplastic syndromes. Cancer Res. 2005;65:9029–37. doi: 10.1158/0008-5472.CAN-04-4555. [DOI] [PubMed] [Google Scholar]
- 38.Follo MY, Mongiorgi S, Bosi C, Cappellini A, Finelli C, Chiarini F, et al. The Akt/mammalian target of rapamycin signal transduction pathway is activated in high-risk myelodysplastic syndromes and influences cell survival and proliferation. Cancer Res. 2007;67:4287–94. doi: 10.1158/0008-5472.CAN-06-4409. [DOI] [PubMed] [Google Scholar]
- 39.Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood. 2002;99:479–87. doi: 10.1182/blood.v99.2.479. [DOI] [PubMed] [Google Scholar]
- 40.Snow JW, Abraham N, Ma MC, Abbey NW, Herndier B, Goldsmith MA. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood. 2002;99:95–101. doi: 10.1182/blood.v99.1.95. [DOI] [PubMed] [Google Scholar]
- 41.Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, et al. STAT5a and STAT5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–50. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 42.Ilaria RLJ, Hawley RG, Van Etten RA. Dominant negative mutants implicate STAT5 in myeloid cell proliferation and neutrophil differentiation. Blood. 1999;93:4154–66. [PubMed] [Google Scholar]
- 43.Kieslinger M, Woldman I, Moriggl R, Hofmann J, Marine JC, Ihle JN, et al. Antiapoptotic activity of STAT5 required during terminal stages of myeloid differentiation. Genes Dev. 2000;14:232–44. [PMC free article] [PubMed] [Google Scholar]
- 44.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–14. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 45.Cooper CL, Newburger PE. Differential expression of Id genes in multipotent myeloid progenitor cells: Id-1 is induced by early- and late-acting cytokines while Id-2 is selectively induced by cytokines that drive terminal granulocytic differentiation. J Cell Biochem. 1998;71:277–85. doi: 10.1002/(sici)1097-4644(19981101)71:2<277::aid-jcb12>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 46.Han S, Gou C, Hong L, Liu J, ZheyiHan, Liu C, et al. Expression and significances of Id1 helix-loop-helix protein overexpression in gastric cancer. Cancer Lett. 2004;216:63–71. doi: 10.1016/j.canlet.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q, Tsao SW, Fu S, Xue W, Meng X, Feng H, et al. Over-expression of Id-1 in gastric adenocarcinoma: implication for a novel diagnostic marker. Anticancer Res. 2004;24:881–6. [PubMed] [Google Scholar]
- 48.Ishiguro A, Spirin K, Shiohara M, Tobler A, Norton JD, Rigolet M, et al. Expression of Id2 and Id3 mRNA in human lymphocytes. Leuk Res. 1995;19:989–96. doi: 10.1016/0145-2126(95)00084-4. [DOI] [PubMed] [Google Scholar]
- 49.Suh HC, Leeanansaksiri W, Ji M, Klarmann KD, Renn K, Gooya J, et al. Id1 immortalizes hematopoietic progenitors in vitro and promotes a myeloproliferative disease in vivo. Oncogene. 2008 Jun 9; doi: 10.1038/onc.2008.175. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 51.Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Scott E, Sawyers CL, Friedman AD. C/EBPalpha bypasses granulocyte colony-stimulating factor signals to rapidly induce PU.1 gene expression, stimulate granulocytic differentiation, and limit proliferation in 32D cl3 myeloblasts. Blood. 1999;94:560–71. [PubMed] [Google Scholar]
- 54.Zhang P, Nelson E, Radomska HS, Iwasaki-Arai J, Akashi K, Friedman AD, et al. Induction of granulocytic differentiation by 2 pathways. Blood. 2002;99:4406–12. doi: 10.1182/blood.v99.12.4406. [DOI] [PubMed] [Google Scholar]
- 55.Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, et al. AML1/ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 2001;7:444–51. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- 56.Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 57.Helbling D, Mueller BU, Timchenko NA, Hagemeijer A, Jotterand M, Meyer-Monard S, et al. The leukemic fusion gene AML1-MDS1-EVI1 suppresses CEBPA in acute myeloid leukemia by activation of calreticulin. Proc Natl Acad Sci USA. 2004;101:13312–7. doi: 10.1073/pnas.0404731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Silverman LR. DNA methyltransferase inhibitors in myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17:585–94. doi: 10.1016/j.beha.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Wouters BJ, Jordà MA, Keeshan K, Louwers I, Erpelinck-Verschueren CAJ, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood. 2007;110:3706–14. doi: 10.1182/blood-2007-02-073486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Upregulation of interleukin 6 and granulocyte colony-stimulating factor receptors by transcription factor CCAAT enhancer binding protein alpha (C/EBP alpha) is critical for granulopoiesis. J Exp Med. 1998;188:1173–84. doi: 10.1084/jem.188.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood. 1996;88:1234–47. [PubMed] [Google Scholar]
- 62.Sultana TA, Harada H, Ito K, Tanaka H, Kyo T, Kimura A. Expression and functional analysis of granulocyte colony-stimulating factor receptors on CD34++ cells in patients with myelodysplastic syndrome (MDS) and MDS-acute myeloid leukaemia. Br J Haematol. 2003;121:63–75. doi: 10.1046/j.1365-2141.2003.04261.x. [DOI] [PubMed] [Google Scholar]