

Abstract
In the development of heart failure, extensive remodelling of the entire myocardium takes place. In this paper, findings on structural remodelling occurring in patients with severely reduced left ventricular function due to dilated cardiomyopathy are presented. This process involves all structural proteins of the myocytes; some of them are reduced (contractile proteins and most of the sarcomeric skeleton) and others are increased (cytoskeleton and membrane-associated proteins). Likewise, the connexin43 content of gap junctions is significantly reduced. The myocyte nuclei are enlarged by 20%, but the ratio of nuclear volume to cell volume is decreased. Nuclei contain less DNA and less of the splicing factor Sc-35 than normal myocardium, which might explain the depressed transcription and translation observed in failing hearts. The connective tissue including fibronectin, laminin and the different types of collagen is augmented, whereas the number of microvessels is decreased. This results in replacement fibrosis. Cell loss is caused by either ubiquitin-related autophagic cell death (most frequent) or by acute ischemic cell death (oncosis) but to a lesser degree by apoptosis. All of these modes of cell death contribute significantly to the loss of contractile function. The morphological alterations described here are the structural correlates of the typical clinical characteristics of heart failure in humans: reduced contractile function, increased ventricular stiffness and ventricular arrhythmias.
Keywords: Heart failure, Remodelling
Recently, we were kindly invited to a symposium entitled “Between Angiogenesis and Heart Failure – 30 Years of Dedicated Research” (dedicated to Wolfgang and Jutta Schaper).
This took place in Caesarea in Israel from April 28 to May 2, 2001. The symposium was organized by Dr Gania Kessler-Icekson (Felsenstein Research Center, Petach Tikvah) and by Dr Arie Pinson (Hadassah Medical School, Jerusalem). The symposium was attended by many of our friends; it was stimulating scientifically and characterized by an extremely friendly and warm social atmosphere. Wolfgang and I were and still are very grateful to the organizers for this wonderful, very special meeting. We feel very close to our Israeli hosts and we want to express, at the beginning of this paper, our heartfelt thanks and best wishes to our friends from Israel. This paper has many co-authors because it represents long term teamwork and I am grateful to my group for an enjoyable and fruitful collaboration over many years.
In 1906, Professor Ludwig Aschoff published a monograph entitled “Die heutige Lehre von den pathologisch-anatomischen Grundlagen der Herzschwäche” (1), in which he concluded that a structural correlate of cardiac insufficiency does not exist. During the histological search for structural abnormalities in failing hearts, however, Tawara discovered the atrioventricular node of the conduction system, which was named after its discoverers ‘Aschoff-Tawara node’. The opinion that a morphological correlate of reduced cardiac function is nonexistent persisted for a long time and was changed only when failing hearts became available to the pathologist during cardiac transplantation and when improved histological techniques allowed for better tissue preparation. Furthermore, a more detailed visualization of the fine structural composition of the myocardium was possible because of the new equipment available to the morphologist, ie, the electron microscope and computer-assisted confocal microscopy, which greatly promoted the knowledge of structural changes in the diseased human heart. The use of monoclonal or polyclonal antibodies in immunofluorescence microscopy permits the identification of proteins such as those forming the cytoskeleton or the components of the intercalated disc that are not individually visible in the electron microscope. By the combined use of both techniques, however, a much more detailed analysis of all structural components of the myocytes, and of the extracellular matrix and vasculature can be carried out.
In the progression from adaptation to cardiac insufficiency, extensive remodelling of the entire myocardium takes place (2). In the present work, data are presented to substantiate the hypothesis that in the development of heart failure due to a chronic, long term disease such as dilated cardiomyopathy, all structural components of the myocardium are involved and contribute to the loss of contractile function.
REMODELLING OF CARDIOMYOCYTES
Recently, a new classification of the myocyte protein families has been proposed, which facilitates greatly the understanding of immunohistochemical results (3). This will be used throughout this text:
The contractile proteins: myosin, actin, the troponins and tropomyosin;
The sarcomeric skeleton: titin, α-actinin, and the M-band proteins including myomesin, M protein and the myosin-binding protein C;
The cytoskeletal proteins: tubulin, desmin and actin;
Membrane-associated proteins: vinculin, dystrophin, talin, spectrin and others;
Proteins of the intercalated disc: connexins, desmin, cadherins and catenins.
Remodelling of the myocardium during the progression to heart failure involves and afflicts all components of the myocytes. It is especially obvious that the contractile machinery and the sarcomeric skeleton show early and significant changes, ie, disorganization and final loss of myofilaments and their supporting proteins (Figure 1). This phenomenon affects the myocytes heterogeneously; ie, these are focal changes of different severity in the same heart. Finally, the sum of the altered cells determines the degree of loss of contractile function, which is caused basically by the loss of sarcomeres.
Figure 1).

Degeneration of myocytes in the failing heart. (A) Slight degeneration with partial loss of myofilaments (asterisk). (B) Severe degeneration characterized by loss of myofilaments as seen in the central myocyte. At the right upper corner, a myocyte with autophagic vacuoles (arrowheads) and areas of denatured protein are evident. This cell will most probably die an autophagic cell death. (Original magnification ×3000)
In addition to the lack of sarcomeres as one of the factors responsible for the decreased compliance of failing hearts is the reduction of titin, the ‘third’ sarcomeric filament system. Reduction of titin was found in the same myocytes that lacked contractile filaments (4). Titin is the largest molecule known and it spans half a sarcomere with only one molecule. Because it contains spring-like molecular domains, it is active in passive recoil of the sarcomeres during contraction (5). Experimental studies in isolated adult myocytes in culture have shown that the expression and arrangement of titin filaments are the prerequisites for the formation of intact sarcomeres (6). We believe, therefore, that the lack of titin is operative in preventing the formation of intact sarcomeres in the remodelling process leading to heart failure.
While the ratio of unspecified cytoplasm to myofilaments increased, the cytoskeletal structures showed a significant augmentation combined with disorganization, which is most probably a compensatory mechanism for the loss of sarcomeres (Figure 2A,B). Similar changes were found for the membrane-associated proteins, even though these are the least affected by the degenerative process. In the end, however, they also show disarrangement and augmentation (Figure 2C,D). This was interpreted as a defence mechanism against the deformation of the myocyte that had lost its stability because of the loss of sarcomeres and the disturbance of the cytoskeleton. A direct correlation exists between the rate of the cytoskeletal protein increase and the left ventricular end-diastolic pressure indicating that these changes contribute to the elevated stiffness of the failing left ventricle (7).
Figure 2).
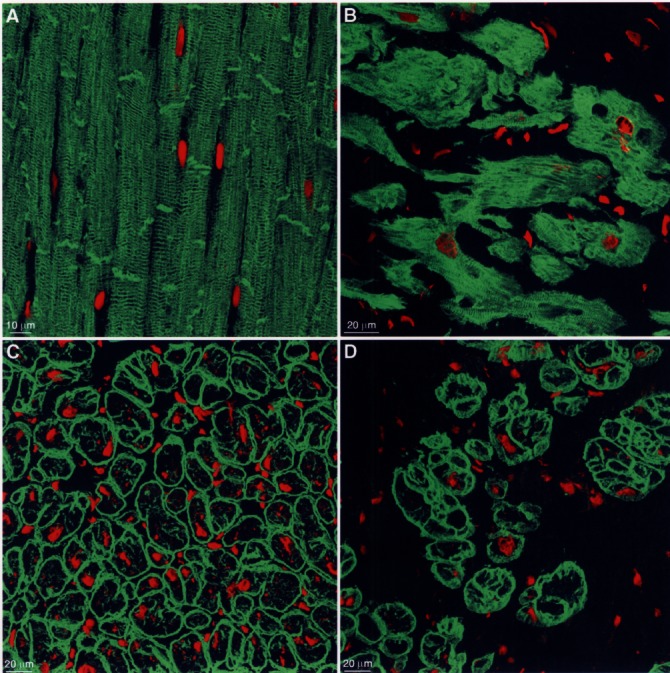
Changes of the cytoskeleton and of membrane-associated proteins (specific fluorescence green, nuclei are red). (A) Desmin, one of the major cytoskeletal proteins, shows in normal myocardium a regular cross striation and localization at the intercalated disc. (B) In failing myocardium, the cross-striation is barely recognizable because of the accumulation of desmin in myocytes of various size and shape, which are separated by a wide interstitial space. (C) Dystrophin, one of the linkage proteins, is localized in normal myocardium at the sarcolemma in a regular fashion. (D) In failing myocardium, dystrophin is increased in many cardiomyocytes
The proteins of the intercalated disc were reduced and disarranged. This applies to the cadherins as well as to the proteins of the desmosomes and the gap junctions (8). This might be clinically important because experimental studies have shown that a decrease in the major gap junctional protein connexin43 facilitates the occurrence of arrhythmias (9).
One of the distinct morphological features found in myocytes of failing hearts was nuclear enlargement as recently measured by electron microscopy (10) and now reconfirmed by confocal microscopy of Feulgen-stained tissue sections. Statistical analysis showed a significant increase in the nuclear area from 64.3 μm2 in controls to 78.1 μm2 in endstage heart failure. These enlarged nuclei contained less DNA than the nuclei in control myocardium. The DNA content was reduced to 75% of control, and the DNA concentration to 60%. When calculated for the increased volume of hypertrophied cells (two-to threefold), this reduction becomes even more significant. The content and concentration of the splicing factor Sc-35, one of the major non-small nuclear ribonucleoprotein particles (11) was unchanged as compared with control, whereas a signifcant increase would have been expected for larger cell volumes (Figure 3). This quantitative nuclear alteration may be one of several explanations why transcription and translation for several protein families appear depressed during cardiac remodelling (7).
Figure 3).

Quantitative results of Feulgen and Sc-35 staining in normal and failing myocardium (DCM). (A, B) Nuclear area. In Sc-35 staining the nuclear area is somewhat smaller because Sc-35 fails to stain the entire nucleus. (C, D) DNA and Sc-35 content are unchanged. (E, F) DNA concentration is significantly reduced but Sc-35 concentration is unaltered
OCCURRENCE OF CELL DEATH
The importance of different types of cell death in the reduction of the number of myocytes in failing myocardium has been discussed extensively in recent years (12–14). In contrast to other groups, we believe that apoptosis apparently plays only a minor role in the development of heart failure but ubiquitin-related autophagic cell death (15,16) and oncosis (17) seem to be the determining factors for myocyte loss (Figure 4). Ubiquitin-related autophagic cell death is the consequence of chronic degeneration common in severely overloaded hearts. It seems that the proteasomal enzymes are inadequate for protein degradation, in amount but most probably also in activity, and that ubiquinated proteins accumulate in degenerative myocytes. In the final stage, the nucleus is fragmented and disappears from the cell. As a consequence, transcriptional activity comes to a standstill and translation ceases, leading ultimately to death of the myocyte because regeneration of proteins is impossible (unpublished data).
Figure 4).
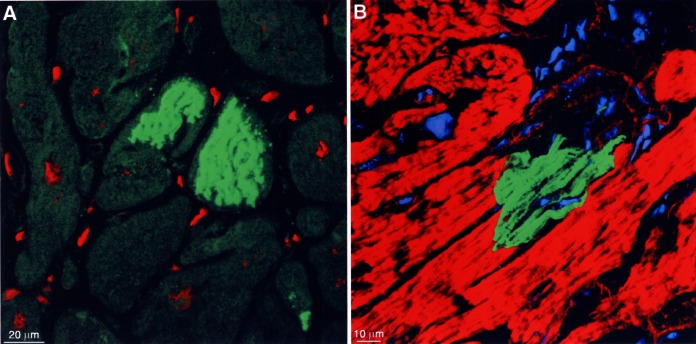
Cell death by different mechanisms. (A) Storage of ubiquitin-protein complexes in two myocytes lacking a nucleus (specific fluorescence green, nuclei red). (B) Complement 9 localization indicating acute ischemic cell death (specific fluorescence green, actin staining red, nuclei and lipofuscin granules blue). Note that only one-half of the myocyte is filled with complement 9; the other part is delineated by a fine red line (remaining actin-positive structures) and contains only unspecified cytoplasm
Oncosis, ie, acute ischemic cell death, is characterized by a positive reaction with complement 9 because the sarcolemma is leaky (18). Using this method, it is evident in the confocal microscope that oncosis occurs in single cells. This implies that microvascular defects, which are measurable as a reduction of capillary density and have been described earlier (19), may be responsible for ischemic damage of myocytes.
Necrosis is the endstage of irreversible cell injury independent of its mechanism, when the point of no return has been passed and the cellular components are degraded (17).
REMODELLING OF THE EXTRACELLULAR MATRIX
The final outcome of this process is scarring by replacement fibrosis. The connective tissue components forming the fibrosis include fibronectin as the matrix into which the fibrillar collagen fibrils and the cellular structures including fibroblasts, microvessels and macrophages are embedded (Figure 5). The increased amount of fibrotic tissue adds to the elevation of ventricular stiffness in failing hearts for which, thus, three different factors are responsible: (a) loss of contractile elements and titin; (b) increase in cytoskeletal and linkage proteins; and (c) increase in fibrosis and scar formation.
Figure 5).
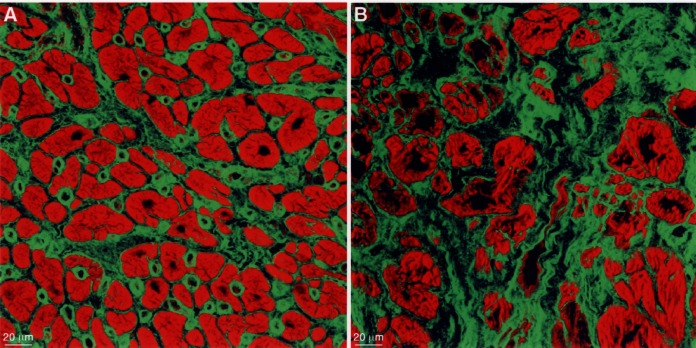
The extracellular matrix stained for fibronectin (specific fluorescence green, actin staining red). (A) In normal myocardium, the amount of fibronectin is moderate and capillaries are distinctly labelled. (B) In failing myocardium, the amount of fibronectin is greatly increased and the myocytes are of different size and shape and are isolated in the fibrotic tissue
Abnormal mechanical stresses exerted by the fibrotic tissue on the surrounding myocytes result in an increase in cardiomyocyte linkage proteins thereby stabilizing the sarcolemma and preserving myocyte integrity. However, degeneration of myocytes will continue and a vicious circle is thus established. This will then further contribute to the reduction of cardiac function.
REFERENCES
- 1.Aschoff L, Tawara S. Die heutige Lehre von den pathologisch-anatomischen Grundlagen der Herzschwäche. Jena: Fischer; 1906. [Google Scholar]
- 2.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 3.Kostin S, Heling A, Hein S, Scholz D, Klövekorn W-P, Schaper J. The protein composition of the normal and diseased cardiac myocyte. Heart Fail Rev. 1998;2:245–60. [Google Scholar]
- 4.Hein S, Scholz D, Fujitani N, et al. Altered expression of titin and contractile proteins in failing human myocardium. J Mol Cell Cardiol. 1994;26:1291–306. doi: 10.1006/jmcc.1994.1148. [DOI] [PubMed] [Google Scholar]
- 5.Labeit S, Kolmerer B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–6. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 6.Person V, Kostin S, Suzuki K, Labeit S, Schaper J. Antisense oligonucleotide experiments elucidate the essential role of titin in sarcomerogenesis in adult rat cardiomyocytes in long-term culture. J Cell Sci. 2000;113:3851–9. doi: 10.1242/jcs.113.21.3851. [DOI] [PubMed] [Google Scholar]
- 7.Heling A, Zimmermann R, Kostin S, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–53. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 8.Rieger M, Richter M, Hein S, Schaper J, Kostin S. Is downregulation of connexin 43 a universal feature of different chronic cardiac diseases in human patients. Circulation. 2000;102(Suppl):653. (Abstract) [Google Scholar]
- 9.Thomas SA, Schuessler RB, Berul CI, et al. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: Evidence for chamber-specific molecular determinants of conduction. Circulation. 1998;97:686–91. doi: 10.1161/01.cir.97.7.686. [DOI] [PubMed] [Google Scholar]
- 10.Scholz D, Diener W, Schaper J. Altered nucleus/cytoplasm relationship and degenerative structural changes in human dilated cardiomyopathy. Cardioscience. 1994;5:127–38. [PubMed] [Google Scholar]
- 11.Fu XD, Maniatis T. The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3’splice site. Proc Natl Acad Sci USA. 1992;89:1725–9. doi: 10.1073/pnas.89.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–9. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 13.Anversa PL, Kajstura J, Guerra S, Beltrami CA. Myocyte death and growth in the failing heart. Lab Invest. 1998;78:767–86. [PubMed] [Google Scholar]
- 14.Elsässer A, Suzuki K, Schaper J. Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. J Mol Cell Cardiol. 2000;32:711–24. doi: 10.1006/jmcc.2000.1125. [DOI] [PubMed] [Google Scholar]
- 15.Blommaart EFC, Luiken JJFP, Meijer AJ. Autophagic proteolysis: control and specificity. Histochem J. 1997;29:365–85. doi: 10.1023/a:1026486801018. [DOI] [PubMed] [Google Scholar]
- 16.Bursch W, Hochegger K, Török L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of skeletal filaments. J Cell Sci. 2000;113:1189–98. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 17.Majno G, Joris I. Apoptosis, oncosis and necrosis. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 18.Doran JP, Howie AJ, Townend JN, Bonser RS. Detection of myocardial infarction by immunohistological staining for C9 on formalin fixed, paraffin wax embedded sections. J Clin Pathol. 1996;49:34–7. doi: 10.1136/jcp.49.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devaux B, Scholz D, Hirche A, Klövekorn WP, Schaper J. Upregulation of cell adhesion molecules and the presence of low grade inflammation in human chronic heart failure. Eur Heart J. 1997;18:470–9. doi: 10.1093/oxfordjournals.eurheartj.a015268. [DOI] [PubMed] [Google Scholar]