

Abstract
BACKGROUND:
Various molecular mechanisms are operative in altering the sarcomeric function of the heart under increased hemodynamic workload. Expression of the atrial isoform (ALC-1) of the essential myosin light chain, a shift from α-myosin heavy chain (MHC) to β-MHC, increased phosphorylation of the regulatory myosin light chains and increased troponin I (TnI) phosphorylation have been reported to modulate cardiac contractility in rodents.
METHODS:
To assess a possible contribution of these sarcomeric proteins to cardiac performance in human myocardial hypertrophy, two different forms of cardiac hypertrophy were investigated: 19 patients with hypertropic obstructive cardiomyopathy (HOCM) and 13 patients with aortic stenosis (AS) with marked left ventricular hypertrophy and normal systolic function.
RESULTS:
There was no change in MHC gene expression, regulatory myosin light chain or TnI phosphorylation status in normal heart (NH), HOCM and AS patients. However, patients with hypertrophied myocardium expressed ALC-1 that was not detectable in NH. ALC-1 protein expression correlated positively with the left ventricular ejection fraction. In patients with hypertrophied myocardium, there was a mean ALC-1 protein expression of 12.7±3% (range 3.6% to 32%).
CONCLUSION:
In humans, ALC-1 expression is in vivo a powerful molecular mechanism of the sarcomere to maintain or improve myocardial contractility under increased hemodynamic demands.
Keywords: Myocardial hypertrophy, Myosin light chains
Cardiac hypertrophy is an adaptive response that normalizes wall stress and compensates for increased workload. It is accompanied by qualitative and quantitative changes in the expression of sarcomeric proteins. These alterations have an impact on contractility, intracellular Ca2+ homeostasis and metabolism. Changes in the myosin subunit composition improve contractility by an increase in force generation at a given Ca2+ concentration (1). Phosphorylation of the regulatory myosin light chain (MLC) can also improve cardiac performance (2). Additionally, phosphorylation of troponin I (TnI) leads to a decreased affinity of contractile proteins to Ca2+ (3). In this study, the focus is on the functional significance of myosin heavy chain (MHC) and MLC expression, and the phosphorylation of TnI and the regulatory MLC in the hypertrophied human ventricle. The myosin muscle motor consists of two heavy chains (MHC) and two pairs of light chains (MLC). The MHCs can be divided into the α- and the β-MHC isoforms (4). During development and under pathophysiological conditions, the expression of the two cardiac isoforms is regulated in a tissue-specific manner. In all mammalian species, α-MHC is expressed in the atria thoughout life. In rodents, α-MHC is also the predominant isoform in the ventricle of adult hearts. β-MHC is expressed in the embryonic ventricular tissue. In rodents, there is a switch from α-MHC to β-MHC under hemodynamic stress (5). β-MHC has reduced ATPase activity and increased economy of isometric force development (6). In humans, β-MHC is the predominant isoform in the adult heart (7). Whether there is a switch under increased workload in the human ventricle is still a matter of debate and is investigated in this study.
MLCs are classified into essential MLC (MLC-1) and regulatory (phosphorytable) MLC (MLC-2) (8,9). (Table 1) In the human heart, five MLC isoforms exist that are expressed in a tissue-specific manner: there are three regulatory MLCs, namely two ventricular specific (VLC-2 and VLC-2*) and an atrial specific (ALC-2) (10). Two essential light chains (MLC-1) have been described: ALC-1 in the atrium and VLC-1 in the ventricle (Figure 1). Human embryos express large amounts of ALC-1 in the whole heart and in skeletal muscle. For this reason the ALC-1 was designated as the embryonic MLC (MLC-1emb) (10). A positive correlation between pressure overload in adult patients with valvular heart disease and left ventricular ALC-1 expression exists. Normalization of the hemodynamic state by valve replacement significantly reduced hypertrophy and ALC-1 expression (11).
Table 1.
Nomenclature of myosin light chains and heavy chains
| MHC | Myosin heavy chain |
| MLC | Myosin light chain |
| MLC-1 | Essential myosin light chain |
| MLC-2 | Phosphorylatable (regulatory) myosin light chain |
| ALC-1 | Essential myosin light chain, atrial isoform |
| ALC-2 | Phosphorylatable (regulatory) myosin light chain, atrial isoform |
| VLC-1 | Essential myosin light chain, ventricular isoform |
| VLC-2 | Phosphorylatable (regulatory) myosin light chain, ventricular isoform |
| VLC-2* | Phosphorylatable (regulatory) myosin light chain, additional ventricular isoform |
Figure 1).
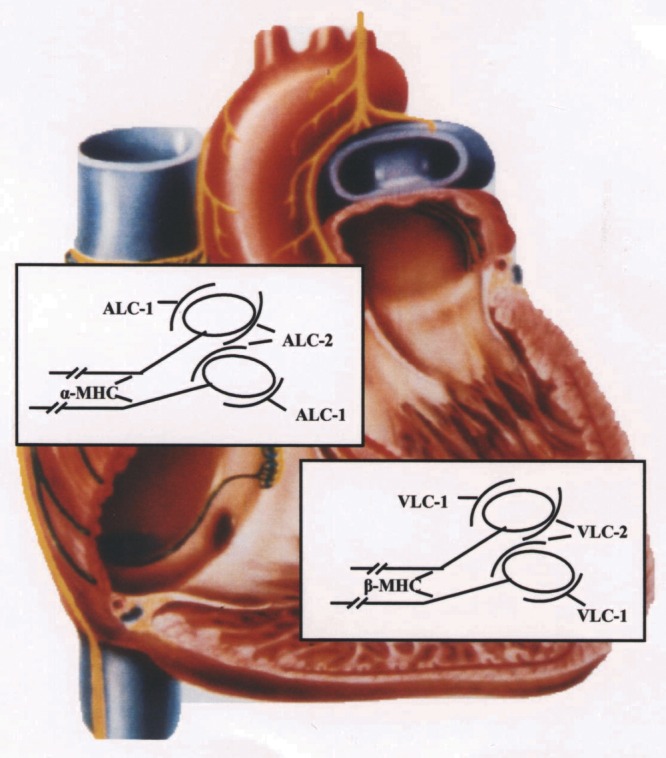
Scheme of myosin heavy chain (MHC) and myosin light chain (MLC) distribution in normal human heart. On the atrial level there is α-MHC and the artrial MLC isoforms ALC-1 and ALC-2. On the ventricular level only β-MHC and the ventricular MLC isoforms VLC-1 and VLC-2 can be detected. Each myosin molecule consists of two MHCs, and on the head of each MHC there is one molecule of MLC-1 and MLC-2
Phosphorylation of the MLC-2 has been shown to increase tension development and Ca2+ sensitivity (12). Recently, it has been demonstrated that there is a spatial gradient across the heart in MLC phosphorylation altering tension production in different wall sections (2).
The cardiac troponin complex comprises the inhibitory (TnI), the Ca2+-binding (TnC) and the tropomyosin-binding (TnT) subunits (13). Phosphorylation of TnI by Protein Kinase A (PKA), which results from β-adrenergic stimulation, leads to a decrease in the sensitivity of the contractile proteins to Ca2+, which is accompanied by an increased crossbridge cycling rate. These results indicate that PKA-induced phosphorylation of TnI increases the power output-generating capacity of cardiac myocytes (14). In terminally failing hearts, TnI phosphorylation was increased compared with normal heart (NH). This phosphorylation difference could underlie the reported decrease of myofibrillar Ca2+ sensitivity of failing myocardium (15). However, there are no data on TnI phosphorylation in hypertrophied, non-failing human myocardium. Therefore, in the present study TnI phosphorylation status of hypertrophied human myocardium was investigated.
MATERIAL AND METHODS
Patient population
Tissue from the left ventricular outflow tract was obtained from 13 patients undergoing aortic valve replacement for aortic stenosis (AS) and from 19 unrelated patients with hypertropic obstructive cardiomyopathy (HOCM) undergoing transaortal subvalvular myotomy-myectomy (16) for severe obstruction of the left ventricular outflow tract. The clinical evaluation consisted of preoperative physical examination, electrocardiography (ECG), echocardiography and cardiac catheterization. All the patients in this study had a normal ejection fractions (EF). Atrial tissue was taken from the auricle of a patient undergoing mitral valve replacement. Tissue from the interventricular septum was taken from six healthy donor hearts that could not be transplanted for technical reasons. Tissue samples were frozen in liquid nitrogen immediately after resection. Informed consent was obtained from all patients.
Tissue preparation
All tissue samples were immediately chemically skinned or frozen in liquid nitrogen after resection. All biochemical experiments were performed with demembranated multicellular heart fibres (skinned fibres) prepared as described previously (17) or with frozen tissue samples. For the preparation of the skinned fibres, ventricular fibre bundles (1 mm thick, between 2 and 5 mm long) were incubated in a solution containing 20 mM imidazole, 10 mM NaN3, 5 mM ATP, 5 mM MgCl2, 4 mM EGTA, 2 mM dithioerythritol, 50% glycerol and 1% Triton X-100, pH 7, at 4°C for 20 h. Subsequently the fibres were transferred into the same solution except Triton X-100 and stored at −20°C. Tissue specimen for analysis of phosphorylation levels were homogenized in ice-cold trichloroacetate before further experimentation. All tissue samples immediately frozen after resection were stored at −80°C.
Biochemical analysis
Samples for high-resolution protein gel electrophoresis for MHC analysis were prepared as follows. Briefly, samples were homogenized in low-salt buffer (20 mmol/L KCl, 2 mmol/L KH2PO4, 1 mmol/L EGTA, pH 6.8, 1 mmol/L phenylmethylsulphonylchloride and 100 μL N,N-dimethyl formamide). The samples were then centrifuged at 900 g for 10 min at 4°C. Laemmli’s buffer was added to each sample. The preparation and composition of the gel were as described earlier (10). Gel samples (0.25 to 1 μg) were loaded in a 3 μL volume onto 15-well gels. The stacking and separating gels (0.75 mm thick) consisted of 4% and 8% acrylamide, respectively; the stacking gels included 5% glycerol. The gels were run at a constant voltage of 200 V for 30 h. The gels were fixed and silver-stained A gel documentation system (Bio-Rad, Germany) was used to scan the stained gels.
MLCs were analyzed by a high-resolution two-dimensional gel electrophoresis two-dimensional polyacrylamide gel electrophoresis [2D PAGE] technique. Isoelectric focusing (first dimension) was performed in glass capillaries (12.5 cm long, 1 mm inner diameter) using the pH gradient 4.5 to 5.5 (Pharmalytes, Parmacia, Sweden). The gels were run overnight at 600 V constant load for the first dimensional separation. The second dimension was a sodium dodecylsulphate electrophoresis, using slab gels 10.5×9.5 cm, 1 mm thick. The gels were stained in Coomassie blue and the MLCs were evaluated by computer-assisted scanner densitometry (ScanPack, Biometra, Germany).
TnI phosphorylation status was analyzed by 2D PAGE technique according to the above protocol. However, the first dimension (isoelectric focusing) was a nonequilibrium gel with inversed current. TnI phosphorylation level was expressed as a percentage of total TnI, ie, TnI unphosphorylated + TnI phosphorylated = 100%.
Statistical analysis
All statistical analyses were performed using commercially available statistic programs (Enzfitter, Epistat, Germany) on Apple Macintosh or on IBM compatible PC. Values are expressed as mean ± SEM. Significance analysis was performed using the Student’s t test for unpaired values. Regression analysis was performed by the least-squares method.
RESULTS
Clinical data
In clinical terms there were homogeneous populations of patients with HOCM and with AS. All patients had left ventricular myocardial hypertrophy. The extent of hypertrophy of the interventricular septum was similar in both patient groups. There were no significant differences in hemodynamic data in the patient groups (Table 2).
Table 2.
Echocardiographic, hemodynamic and biochemical data of patients with hypertropic obstructive cardiomyopathy (HOCM) or aortic stenosis (AS)
| Diagnosis | Age (years) | Sex | IVS (mm) | EF (%) | LVEDP (mmHg) | ALC-1% |
|---|---|---|---|---|---|---|
| HOCM | 56 | f | 17 | 62 | 12 | 1 |
| HOCM | 53 | m | 14 | 63 | 21 | 3 |
| HOCM | 38 | m | 28 | 66 | 16 | 4 |
| HOCM | 34 | f | 24 | 68 | 10 | 2 |
| HOCM | 56 | f | 17 | 69 | 6 | 8 |
| HOCM | 46 | f | 20 | 73 | 13 | 5 |
| HOCM | 60 | f | 24 | 72 | 17 | 14 |
| HOCM | 80 | f | 20 | 88 | 15 | 2 |
| HOCM | 63 | f | 20 | 76 | 15 | 10 |
| HOCM | 53 | m | 31 | 77 | 19 | 6 |
| HOCM | 48 | m | 24 | 76 | 1 | 11 |
| HOCM | 43 | m | 24 | 81 | 16 | 11 |
| HOCM | 64 | f | 22 | 81 | 20 | 13 |
| HOCM | 47 | m | 13 | 79 | 11 | 16 |
| HOCM | 46 | m | 15 | 72 | 1 | 20 |
| HOCM | 42 | f | 17 | 85 | 10 | 20 |
| HOCM | 62 | f | 16 | 82 | 20 | 22 |
| HOCM | 70 | m | 22 | 86 | 6 | 25 |
| HOCM | 56 | f | 17 | 80 | 17 | 32 |
| AS | 70 | m | 22 | 65 | 6 | 1 |
| AS | 56 | f | 17 | 64 | 9 | 2 |
| AS | 46 | f | 20 | 70 | 13 | 2 |
| AS | 60 | f | 24 | 74 | 17 | 2 |
| AS | 80 | f | 20 | 78 | 15 | 5 |
| AS | 43 | m | 15 | 74 | 24 | 6 |
| AS | 42 | f | 17 | 74 | 10 | 7 |
| AS | 68 | f | 16 | 78 | 20 | 9 |
| AS | 51 | f | 17 | 82 | 16 | 6 |
| AS | 72 | m | 22 | 80 | 6 | 16 |
| AS | 56 | f | 17 | 78 | 16 | 18 |
| AS | 46 | f | 20 | 76 | 13 | 24 |
| AS | 55 | m | 33 | 80 | 9 | 23 |
IVS Diameter of the interventricular septum in mm; LVEDP Left ventricular end-diastolic pressure; EF Ejection fraction; ALC-1% Expression of the atrial isofor m (ALC-1) of essential myosin light chain (MLC-1) as a percentage of total MLC-1 (ie, ALC-1 + ventricular MLC isoform = 100%); f female; m male
MLC analysis
The MLC isoforms in frozen tissue and skinned fibres were analyzed according to their isoelectric point and molecular weight by 2D PAGE. The atrial isoform of the essential light chain (ALC-1) had a higher molecular weight and was more acidic than the ventricular isoform (VLC-1). Figure 1 shows three original gels of tissue preparations of the left ventricle from patients with HOCM, AS and NH. All the fibres contained VLC-1 and the two ventricular isoforms of the regulatory light chains, VLC-2 and VLC-2*, but the preparation of normal myocardium showed no ALC-1 whereas diseased myocardium had ALC-1 (11.4% of total essential light chains [MLC-1]). The mean ALC-1 expression was 12.7±1%. Those patients who expressed ALC-1 had very different amounts ranging from 3.6% to 32.2% ALC-1 (expressed as a percentage of total MLC-1, ie, ALC-1 + VLC-1 = 100% MLC-1) (Table 2).
The ratio between essential light chains (MLC-1) and regulatory light chains (MLC-2) showed no significant difference between patients with and without ALC-1. In the NH, the MLC-1/MLC-2 ratio was 1.1±0.02%. In patients with hypertrophied myocardium, an MLC-1/MLC-2 ratio of 1.26±0.78 and a ratio of 1.21±0.48 in patients without ALC-1 (n=6), respectively was observed. The atrial isoform of the regulatory light chain (ALC-2) was never observed in our tissue. No tissue preparation-dependent differences in results were found. In all individuals investigated here, the content of ALC-1 in skinned fibres and in frozen tissue was identical, indicating total incorporation of ALC-1 protein into the sarcomere.
Phosphorylated MLC-2 promotes contractility and thus counteracts the attenuating effects of phosphorylated TnI. Therefore, MLC-2 phosphorylation in 2D PAGEs was investigated Phosphorylated proteins are more acidic, resulting in a shift of protein spots to the cathode. However, virtually no MLC-2 phosphorylation in hypertrophied or normal human myocardium was found.
MHC analysis
The relative proportions of the two forms of the MHC have been shown to be affected by a wide variety of pathological and physiological stimuli. Hearts that express the faster α-MHC produce more power than those expressing the slower β-MHC, leading to the hypothesis that MHC isoforms play a major role in the determination of cardiac contractility. In the present study, therefore, the MHC protein isoform content of our samples was determined. No α-MHC was detectable in six NH and in 32 nonfailing but hypertrophied human hearts (Figure 3).
Figure 3).
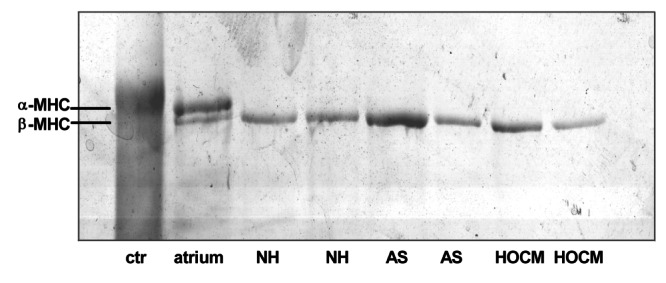
Original photographs of myosin heavy chains (MHC) in the myocardium of normal heart (NH) and in patients with aortic stenosis (AS) or with hypertropic obstructive cardiomyopathy (HOCM). Ctr Rabbit skeletal muscle myosin. Human atrial myocardium contained α-MHC and β-MHC. Human ventricular myocardium from patients with NH, AS or HOCM contained only β-MHC
TnI phosphorylation
TnI phosphorylation contributes to the impaired contractile response of the myocardium to hemodynamic stress. Using nonequilibrium 2D PAGE, TnI phosphorylation was compared in patients with NH, HOCM and AS. However, not significant change was found in the phosphorylation level of TnI in the three patient groups. In NH, phosphorylated TnI was 12±3% (n=6), in AS it was 10±4% (n=13; not significant) and in HOCM it was 13±4% (n=19; not significant) (Figure 4).
Figure 4).
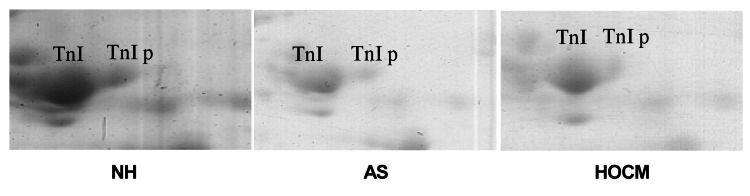
Original photographs of troponin I (TnI) in the myocardium of normal heart (NH) and in patients with aortic stenosis (AS) or hypertropic obstructive cardiomyopathy (HOCM). TnI p indicates phosphorylated TnI. There are no differences in the amount of phosphorylated TnI in the three patient groups
Correlation between ALC-1 protein expression and EF
EF showed a mean value of 73±7%. Notably, there was a statistically significant positive correlation between EF and ALC-1 content of the myocardium. This is compatible with a role of the ALC-1 in contractility in these disease states (Figure 5). The high correlation coefficient of 0.76 (P<0.01, F-test) further supports this hypothesis.
Figure 5).
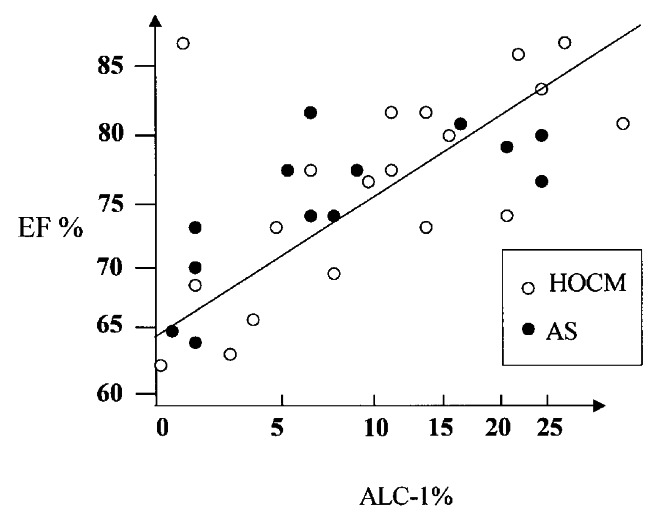
Relation between the expression of atrial isoform of the essential myosin light chain (ALC-1) and ejection fraction (EF). Each circle indicates one patient. The ALC-1 content is shown as a percentage of total myosin light chain-1 (ALC-1 + VLC-1). AS Aortic stenosis; HOCM Hypertrophic obstructive cardiomyopathy
DISCUSSION
Gene expression in the overloaded rodent heart has been studied extensively to highlight the molecular response of the sarcomere. It is generally accepted that, in terms of contractile proteins, a shift of MHC isoforms is part of the response (18). Work from several groups has shown that in the rodent heart, the MHC phenotype switches from the β-MHC isoform to the α-MHC isoform during maturation and vice versa under increased hemodynamic demands (19–24). This conversion of the phenotype is associated with alterations in the contractile and energetic behaviour of the contractile apparatus. Expression of α-MHC leads to a higher maximal shortening velocity, paralleled by a decreased economy in tension development (25). In turn, high amounts of β-MHC decrease maximal shortening velocity and increase economy of contraction (26), which is believed to improve the function of the overloaded cardiomyocyte. These findings raised the question of whether this mechanism might also be operative in the human ventricle. Immunohistochemical studies (27–29) and peptide maps (28) revealed a tissue-specific expression of both MHC isoforms, with a predominant expression of the α-MHC in the atrium and the β-MHC in the ventricle. However, quantification of the relative distribution of both isoforms in the ventricle has proved difficult.
In two previous studies, α-MHC mRNA was detected in the normal human heart. A downregulation of α-MHC mRNA and an upregulation of β-MHC mRNA was reported in the failing and hypertrophied human heart in analogy to the rodent heart (29,30). However, these studies did not address the problem of whether this switch at the RNA level is reflected in MHC protein content.
Therefore, we here quantified the expression of α-MHC protein in the normal and hypertrophied human ventricle using high-resolution gel electrophoresis. We found no traces of α-MHC protein expression in the normal or in the hypertrophied ventricular human tissue. The MHC switch for regulation of power output is not available in humans. What might be the human correlate? We recently reported that the human ventricular cardiomyocyte regulates its power output by changing essential MLC gene expression rather than MHC gene expression (31,32). These observations imposed the question of whether an increase of ALC-1 expression is also beneficial for the performance of the human ventricle in vivo.
Here, we evaluated the effects of ALC-1 on cardiac performance of the human heart in vivo in a clinical homogeneous population of patients with HOCM and AS without systolic dysfunction. We observed a close and statistically significant correlation between essential MLCs and contractility of the human ventricle in vivo. Increased ALC-1 protein expression augmented cardiac contractility in the human ventricle.
Myocardial performance could be influenced not only by isoform shifting of MHCs and MLCs but also by phosphorylation of sarcomeric proteins. To determine the influence of phosphorylation of sarcomeric proteins, we investigated the phosphorylation levels of two key proteins of myocardial contractility: VLC-2 and TnI in normal and hypertrophied human heart. In our patients, we observed no phosphorylation of VLC-2 in the human ventricle and no significant changes in TnI phosphorylation levels.
Expression of ALC-1 in diseased hearts raises the question of whether there is a compensatory downregulation of the ventricular isoform. According to the generally accepted myosin model, the ratio between MLC-1 and MLC-2 is 1 (ie, each MHC monomer is associated with one essential and one regulatory light chain) (33). In our patients we did not see a difference in the MLC-1/MLC-2 ratio in any of the groups, the ratio being about 1 in each individual. Downregulation of the ventricular form of the MLC-2 is to be expected in order to maintain the normal MLC-1/MLC-2 ratio. This observation is in line with previous studies on ALC-1 expression in the diseased human myocardium. The hypertrophied right ventricle of children with congenital heart diseases has been demonstrated to express large amounts of ALC-1 up to adulthood (34). Similarly, the dilated left ventricle of patients with ischemic or dilative cardiomyopathy re-expressed ALC-1 (35). In adult patients with valvular heart disease, a positive correlation between systolic wall stress and left ventricular ALC-1 expression has been reported. Normalization of the hemodynamic state by valve replacement significantly reduced hypertrophy and ALC-1 expression (11). Recently, some studies investigated the functional consequences of re-expression of essential MLCs on the molecular level. Myosin crossbridges with high amounts of ALC-1 had a higher detachment rate and rate of force generation than myosin with VLC-1 (31). These results showed that ALC-1 increased cycling kinetics of cardiac crossbridges in demembranated multicellular heart fibres, thus regulating the contractile state of the heart. It was also shown that there is a poitive correlation between the Ca2+ sensitivity of isometric force development of skinned human heart fibres and ALC-1 content (32). The inotropic effects of ALC-1 expression could be confirmed in vivo by a recent mouse model overexpressing ALC-1 (36).
The molecular mechanism explaining the effects of essential MLC on crossbridges in the heart may reside in the mode of interaction of the essential MLC and actin. It has been demonstrated that the amino terminal domain of MLC-1 isoforms interacts with the carboxyl terminus of actin. This interaction could be of functional importance because the inhibition of this interaction increased force production and shortening velocity of human heart fibres (37). Interestingly, the primary structure of the most amino terminal end of ALC-1 and VLC-1 is different: ALC-1 contains two charges less compared with VLC-1, suggesting a different binding affinity between ALC-1 or VLC-1 and actin. In fact, binding of ALC-1 to actin was found to be weaker than binding of VLC-1 to actin (38). Weakening of the VLC-1-actin interaction by peptide competition accelerated crossbridge cycling kinetics, increased tension production and increased Ca2+ sensitivity (39). These results are analogous to studies on muscle fibres with an increased content of ALC-1 (31,32,39).
To our knowledge, this is the first study investigating MHC and MLC composition in the following subsets of patients with cardiac hypertrophy and normal systolic function: HOCM and severe AS. In view of the relative rarity of these patients (particularly HOCM with indication for operation) the numbers are large. It is noteworthy that large pieces of tissue were used for measurements as opposed to percutaneous biopsies with the inherent problem of tissue heterogeneity (fibrosis). These two factors may also explain the relatively low variability of our results compared with other studies in human tissue. We demonstrate improved intrinsic contractile function in the human ventricle in vivo, which significantly correlated with enhanced ALC-1 protein expression, excluding an MHC type of regulation. Expression of ALC-1 in the hypertrophied human ventricle therefore may be an adaptational molecular mechanism to improve the inotropic state of the human heart. ALC-1 expression turned out to be a powerful compensatory mechanism to enhance power output of the human heart.
Figure 2).

Original photographs of myosin light chains (MHC) in the myocardium of normal heart (NH) and in patients with aortic stenosis (AS) or hypertropic obstructive cardiomyopathy (HOCM). All fibres contained VLC-1, VLC-2 and VLC-2*, but patients with hypertrophied myocardium contained ALC-1 additionally. ALC-1 Atrial essential light chain; TM Tropomyosin; VLC-1 Ventricular essential light chain; VLC-2 and VLC-2* Isoforms of the ventricular regulatory light chain. No phosphorylation of the regulatory light chains was detectable
REFERENCES
- 1.Schaub MC, Hirzel HO, Tuchschmid CR, et al. Atrial and ventricular isomyosin compositions in patients with different forms of cardiac hypertrophy. Basic Res Cardiol. 1987;2(Suppl):357–67. doi: 10.1007/978-3-662-11289-2_35. [DOI] [PubMed] [Google Scholar]
- 2.Davis JS, Hassanzadeh H, Winitsky S, et al. The overall pattern of cardiac contraction depends on a gradient of MLC-2 phosphorylation. Cell. 2001;107:631–41. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 3.Solaro RJ, Rarick HM. Troponin and tropomyosin. Circ Res. 1998;83:471–80. doi: 10.1161/01.res.83.5.471. [DOI] [PubMed] [Google Scholar]
- 4.Weeds AG, Pope B. Chemical studies on light chains from cardiac and skeletal muscle myosins. Nature. 1971;234:85–8. doi: 10.1038/234085a0. [DOI] [PubMed] [Google Scholar]
- 5.Cummins P. Contractile proteins in muscle disease. J Muscle Res Cell Motility. 1983;4:5–24. doi: 10.1007/BF00711956. [DOI] [PubMed] [Google Scholar]
- 6.Schier JJ, Adelstein RS. Structural and enzymatic comparison of human cardiac muscle myosin isolated from infants, adults and patients with hypertrophic cardiomyopathy. J Clin Invest. 1982;69:816–25. doi: 10.1172/JCI110521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price KM, Littler WA, Cummins P. Human atrial and venticular myosin light chains subunits in the adult and during development. Biochem J. 1980;191:571–80. doi: 10.1042/bj1910571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurabayashi M, Komuro I, Tsuchimochi I, Takaku F, Yazaki Y. Molecular cloning and characterization of human atrial and ventricular myosin alkali light chain cDNA clones. J Biol Chem. 1988;263:13930–6. [PubMed] [Google Scholar]
- 9.Barton P, Buckingham ME. The myosin alkali light chain proteins and their genes. Biochem J. 1992;231:249–61. doi: 10.1042/bj2310249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cummins P, Lambert SJ. Myosin transitions in the bovine and human heart. A developmental and anatomical study of heavy and light subunits in the atrium and ventricle. Circ Res. 1986;58:846–58. doi: 10.1161/01.res.58.6.846. [DOI] [PubMed] [Google Scholar]
- 11.Sütsch G, Brunner UT, Von Schulthess C, et al. Hemodynamic performance and myosin light chain-1 expression in the hypertrophied left ventricle in aortic valve disease before and after valve replacement. Circ Res. 1992;70:1035–43. doi: 10.1161/01.res.70.5.1035. [DOI] [PubMed] [Google Scholar]
- 12.Sweeney HL, Stull JT. MLC phosphorylation in striated muscle: regulation and function. Am J Physiol. 1993;264:1085–95. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 13.Brisson JR, Golosinska K, Smillie LB, Sykes BD. Interaction of tropomyosin and troponin T: a proton nuclear magnetic resonance study. Biochemistry. 1986;25:4548–55. doi: 10.1021/bi00364a014. [DOI] [PubMed] [Google Scholar]
- 14.Herron TJ, Korte FS. Power output is increased after phosphorylation of myofibrillar proteins. Circ Res. 2001;89:1184–90. doi: 10.1161/hh2401.101908. [DOI] [PubMed] [Google Scholar]
- 15.Boder GS, Anderson PA. TnI phosphorylation. Circ. 1997;96:1495–500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 16.Schulte HD, Bircks WH, Loesse B, Godehardt EAJ, Schwartzkopff B. Prognosis of patients with hypertrophic obstructive cardiomyopathy after transaortic myectomy. J Thorac Cardiovasc Surg. 1993;106:709–17. [PubMed] [Google Scholar]
- 17.Ritter O, Haase H, Schulte HD, Lange PE, Morano I. Remodeling of the hypertrophied human myocardium by bHLH factors. J Cell Biochem. 1999;74:551–61. doi: 10.1002/(sici)1097-4644(19990915)74:4<551::aid-jcb5>3.3.co;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. J Clin Invest. 1989;84:1693–700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swynghedauw B. Developmental and functional adapation of contractile proteins in cardiac and skeletal muscles. Physiol Rev. 1986;66:710–71. doi: 10.1152/physrev.1986.66.3.710. [DOI] [PubMed] [Google Scholar]
- 20.Mercardier JJ, Schwartz K. Myosin isoenzymic changes in several models of rat cardiac hypertrophy. Circ Res. 1981;49:525–32. doi: 10.1161/01.res.49.2.525. [DOI] [PubMed] [Google Scholar]
- 21.Izumo S. Myosin heavy chain mRNA and protein isoform transitions during cardiac hypertrophy. J Clin Invest. 1987;79:970–7. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lompré AM, Schwartz K, d’Albis A, Lacombe G, Van Thiem N, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature. 1979;282:105–17. doi: 10.1038/282105a0. [DOI] [PubMed] [Google Scholar]
- 23.Mahdavi V, Izumo S, Nadal-Ginard B. Developmental and hormonal regulation of sarcomeric myosin heavy chain gene familiy. Circ Res. 1987;60:804–14. doi: 10.1161/01.res.60.6.804. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz K, Lecarpentier Y, Martin JL, Lompré AM, Mercardier JJ, Swynghedauw B. Myosin isoenzyme distribution correlates with speed of myocardial contraction. J Mol Cell Cardiol. 1981;13:1071–5. doi: 10.1016/0022-2828(81)90297-2. [DOI] [PubMed] [Google Scholar]
- 25.Alpert NR, Mulieri LA. Increased myothermal economy of isometric force generation in compensated cardiac hypertrophy induced by pulmonary artery constriction in rabbit. Circ Res. 1982;50:491–500. doi: 10.1161/01.res.50.4.491. [DOI] [PubMed] [Google Scholar]
- 26.Tsuchimochi H, Sugi M, Kuro M, et al. Isozymic change in myosin of human atrial myocardium induced by overload. J Clin Invest. 1984;74:662–5. doi: 10.1172/JCI111466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorza L, Mercardier JJ, Schwartz K, Thornell LE, Sartore S, Schiaffino S. Myosin types in the human heart. Circ Res. 1984;54:694–702. doi: 10.1161/01.res.54.6.694. [DOI] [PubMed] [Google Scholar]
- 28.Bouvagnet P, Mairhofer H, Leger JO, Puech P, Leger JJ. Distribution pattern of (αand βmyosin in normal and diseased human ventricular myocardium. Basic Res Cardiol. 1989;84:91–102. doi: 10.1007/BF01907006. [DOI] [PubMed] [Google Scholar]
- 29.Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–70. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowes BD. Changes in gene expression in the intact human heart. J Clin Invest. 1997;100:2315–24. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morano M, Zacharzowsky U, Maier M, et al. Regulation of human contractility by essential myosin light chain isoforms. J Clin Invest. 1996;98:467–73. doi: 10.1172/JCI118813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morano I, Hädicke K, Haase H, et al. Changes in essential myosin light chain isoform expression provide a molecular basis for isometric force regulation in the failing human heart. J Mol Cell Cardiol. 1997;29:1177–87. doi: 10.1006/jmcc.1996.0353. [DOI] [PubMed] [Google Scholar]
- 33.Hirzel HO, Tuchschmid CR, Schneider J, Krayenbuehl HP, Schaub MC. Relationship between composition, hemodynamics and myocardial structure in various forms of human cardiac hypertrophy. Circ Res. 1985;57:729–40. doi: 10.1161/01.res.57.5.729. [DOI] [PubMed] [Google Scholar]
- 34.Auckland LM, Lambert SJ, Cummins P. Cardiac myosin light and heavy chain isotypes in tetralogy of Fallot. Cardiovasc Res. 1986;20:828–63. doi: 10.1093/cvr/20.11.828. [DOI] [PubMed] [Google Scholar]
- 35.Trahair T. Myosin light chain gene expression associated with disease states of the human heart. J Mol Cell Cardiol. 1993;26:577–85. doi: 10.1006/jmcc.1993.1067. [DOI] [PubMed] [Google Scholar]
- 36.Fewell JG, Hewett HE, Sambe A, et al. Functional significance of cardiac myosin light chain isoform switching in transgenic mice. J Clin Invest. 1998;101:2630–9. doi: 10.1172/JCI2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trayer IP, Trayer AP, Levine BA. Evidence that the N-terminal region of the A1-light chain of myosin interacts directly with the C-terminal region of actin. Eur J Biochem. 1987;164:259–66. doi: 10.1111/j.1432-1033.1987.tb11019.x. [DOI] [PubMed] [Google Scholar]
- 38.Morano I, Haase H. Different actin affinities of human cardiac essential myosin light chain isoforms. FEBS Lett. 1997;408:71–4. doi: 10.1016/s0014-5793(97)00390-6. [DOI] [PubMed] [Google Scholar]
- 39.Morano I, Ritter O, Bonz A, et al. Myosin light chain interaction regulates cardiac contractility. Circ Res. 1995;76:720–5. doi: 10.1161/01.res.76.5.720. [DOI] [PubMed] [Google Scholar]