

Abstract
The mechanisms responsible for collateral development have attracted considerable attention in cellular and molecular research because these vessels have the capacity to protect against myocardial infarction and reduce exercise-induced ischemia. This has led to a number of phase I trials employing angiogenic peptides, genes and cell transplantation. However, there are significant differences in the degree of collateral development among patients even with similar patterns of coronary disease. Understanding the factors responsible for this heterogeneity has important implications for the efficacy of future therapeutic strategies. Therefore, this review will examine these factors from the clinical perspective integrating the clinical evidence with recent molecular and cellular studies. The role of specific pharmacological agents and novel investigational strategies will be discussed.
Keywords: Angiogenesis, Coronary collaterals, Ischemia, Myocardial infarction
CLINICAL FACTORS INFLUENCING COLLATERAL DEVELOPMENT
The degree of collateral development in humans can be extensive, protecting large regions of myocardium (Figure 1). Collateral formation is dependant upon a combination of arteriogenesis, angiogenesis and the remodelling of pre-existing interarterial connections. Angiogenesis describes the development of new blood vessels lacking developed media from pre-existing vascular structures and results from the interplay of cell-cell, cell-matrix and cell-cytokine interactions. It is tightly regulated and transient, occuring in the female reproductive organs, during wound healing or along the border of myocardial infarction. Arteriogenesis describes the appearance of new arteries possessing fully developed tunica media. This may occur either through remodelling of pre-existing vascular structures or de novo (1). Furthermore, it provides the most clinically desirable conduits capable of producing significant increases in coronary flow reserve thereby restoring arterial inflow to the ischemic region. These processes differ fundamentally from vasculogenesis, which applies to the de novo formation of vessels by the association of angioblasts and endothelial progenitor cells (EPC). The identification of the latter cell population in the adult means that vaculogenesis may no longer be restricted to the embryo, transforming our entire perception of collateralization. Angiogenesis is characterized and modulated by the interactions between stimulators and inhibitors – many of which were first identified during studies of tumour development. Hypoxia, ischemia, mechanical stretch, shear stress and inflammation have all been shown to increase endothelial mitogen levels and expression of angiogen receptors (2–7). In fact, few patients exhibit continuous tissue level ischemia, suggesting that inflammation and shear stress may also be important factors determining arteriogenesis in individuals with multivessel coronary disease (6). Indeed, collateral growth in the canine model occurs in the epicardium where no hypoxia is present, and proceeds even when the endocardium is no longer hypoxic. This has led Schaper to postulate that a combination of shear stress and inflammatory cell recruitment is the key to arteriogenesis (recapitulated vasculogenesis) in the epicardial region. Angiogenesis is principally hypoxia driven (8,9) operating through upregulated hypoxia inducible factor (HIF)-1-α protein (10), which in turn upregulates vascular endothelial growth factor (VEGF), its receptors flt-1 and neuropilin-1, and angiopoietin-2.
Figure 1).
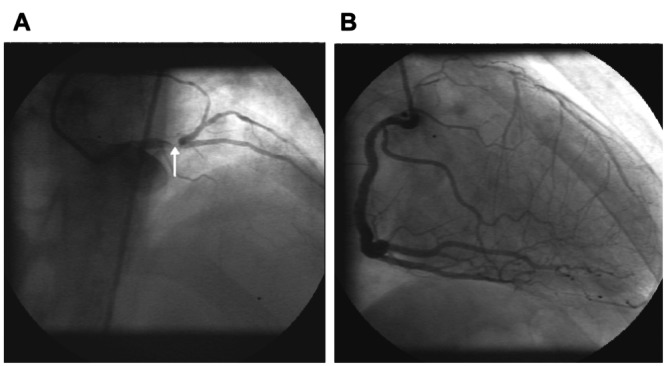
Coronary angiograms of a patient with stable angina. (A) Critical distal left main stem lesion (arrow) limiting flow to the left anterior descending and circumflex arteries. (B) Injection of the right coronary artery illustrating extensive collateralization of the anterior wall preserving normal left ventricular function and protecting against rest ischemia
A number of clinical factors are recognized to influence collateral growth. These can be divided into two principal groups – biophysical and molecular. The principal biophysical factors are percentage diameter stenosis of the feeding coronary artery, number of diseased vessels, vessel shear stress and exercise. These are the key factors modulating recapitulated vasculogenesis in the epicardium. Aging, diabetes and hypercholesterolemia all impair endothelial function and are associated with reduced collateral development. The latter influence a number of molecular processes including cellular response to hypoxia, growth factor production and receptor expression, translating into effects on nitric oxide (NO) production, chemotaxis and cellular proliferation.
Biophysical processes
Despite intensive research, the initiating event responsible for collateral growth remains controversial. In 1971, Schaper et al (11) demonstrated a relationship between myocardial ischemia, DNA synthesis, and endothelial and smooth muscle cell mitosis in canine collaterals. They proposed that arterial pressure gradients resulting from altered coronary arterial flow during ischemia are responsible for the induction of collateral growth. However, a subsequent study from this group demonstrated [3H] thymidine incorporation into adjacent arterioles, veins and capillary vessels, indicating that physical forces could not be the sole stimulus because tangential wall stress is significantly lower in veins and capillaries than in arterioles (12). Early studies implicated ischemia as a trigger for myocardial angiogenesis because the duration of repetitive coronary artery occlusions is critical to the induction of collateral growth. In several animal models, brief episodes of myocardial ischemia (15 s) were insufficient compared with longer periods of 2 to 5 min (13–15). These effects could be mediated by differences in shear stress. However, microembolization of the coronary bed is a powerful stimulus to angiogenesis without altering the pressure gradients or shear stress between large epicardial coronary arteries (16). This mechanism may operate in patients with unstable coronary plaques that release platelet microemboli into the distal coronary bed. Furthermore, pro-angiogenic cytokine expression is enhanced by hypoxic and ischemic stimuli, suggesting a strong link between myocardial ischemia and the induction of collateral growth. Shear stress is probably the most important during collateral remodelling because the ischemic signal attenuates as the vessels improve local perfusion. Increases in shear stress occur with increased flow through stenotic arteries or during periods of rapid collateral development inducing endothelia cellular activation and monocyte recruitment. Locally recruited platelet, monocyte and macrophage populations during this inflammatory response are important sources of angiogenic and mitogenic growth factors influencing vascular remodelling. Therefore ‘recapitulated vasculogenesis’ is probably more important in maintaining and remodelling newly formed collateral vessels. Schaper mphasizes this process as the key to adequate collateralization because the vessels formed are more capable of improving coronary flow reserve and it occurs in tissue outside the ischemic region independent of hypoxia. The relative contributions of angiogenesis and recapitulated vasculogenesis in collateralization of myocardium remain open to debate.
The percentage diameter stenosis of the coronary artery and symptom duration are of critical importance because these determine the depth and frequency of myocardial ischemia, as well as the shear stress stimuli crucial to collateral remodelling. A stenosis must reduce luminal diameter by at least 50% to induce tissue ischemia during exercise and thus act as a trigger to collateral growth. Because atherosclerosis is a progressively occlusive process, this allows suitable time for collateral development to occur. Sudden occlusions following abrupt plaque rupture result in significant necrosis in unsupported myocardium. This causes larger infarcts and more frequent malignant arrhythmia and mortality than in patients with progressive occlusion and thus gradual collateral growth (17–19). Therefore, one may hypothesize that repetitive ischemia on exercise may act as a stimulus to collateral formation.
Exercise
In the mid-18th century, William Heberden described a patient with angina being nearly cured of his symptoms by sawing wood for 30 min a day. This observation may be a reflection of the preconditioning phenomenon or gradual collateral development. It also emphasizes the difficulties inherent in characterizing physical factors influencing collateral development in patients, ie, selection bias. Randomized controlled trials of the influence of physical activity on collateral growth are confounded by highly motivated, physically active people declining to participate as control subjects and vice versa. Furthermore, most studies have not been able to measure collateral flow. Senti et al (20) published perhaps the most rigorous study to examine this issue. Although retrospective, this is the first study to quantify collateral flow by invasive means. In 79 patients undergoing coronary angioplasty, regional collateral flow was measured with a Doppler flow wire. A number of angiographic and clinical factors were analyzed by univariate and multivariate analyses. The key factors determining sufficient collateral growth to protect against rest ischemia during coronary occlusion were the severity of coronary artery stenosis and the duration of long term physical activity during leisure time. Todd et al (21) published a prospective study of the effects of an intensive exercise program on local myocardial perfusion assessed by scintigraphy demonstrating enhanced local myocardial perfusion in previously ischemic areas. This supports Schaper’s biophysical concept of recapitulated vasculogenesis. Repeated exercise induces a larger pressure gradient across the stenosis and hence induces greater shear stress, triggering endothelial activation, collateral channel remodelling and hypertrophy. There is evidence that repeated exercise increases endothelial NO production in humans, although Traverse et al (22) showed that coronary vasodilation on exercise can occur independently of NO.
As we discussed earlier, coronary collateral development arises as a result of a highly orchestrated interplay between molecular signals and cellular elements. Clinical factors can influence these processes at either the cellular or the molecular level. Rohan et al (23) has recently shown that the degree of neovascularization in a corneal micropocket assay can vary 20-fold between individual strains of the same mouse species. Therefore, genetic differences between patients probably also account for the variability in collateral response to a given ischemic stimulus independent of pathological processes. It is well documented that collateral development is significantly impaired in diabetic patients. Furthermore, in both clinical and animal studies, processes affecting endothelial cellular function and viability limit the degree of collateral development – principally hypercholesterolemia, aging and hypertension.
The factors responsible for these differences can be broadly classified into angiogen levels, cellular processes, and environmental influences on endothelial function.
Angiogen levels:
The current paradigm of clinical trials in angiogenesis is based on the concept that the drive to collateral formation depends upon the balance of pro- and antiangiogenic cytokines. A failure to grow collaterals is proposed to be due to impaired upregulation of proangiogenic cytokines. Therefore, variation in the ability to upregulate angiogens critical to early angiogenesis has important implications. Schieltz et al (24) investigated the responsiveness of monocytes to hypoxic stress in patients with different degrees of angiographically assessed collateral support. They found a highly significant difference in the hypoxic induction of VEGF mRNA in patients with no collaterals compared with patients with visible collaterals (mean fold induction 1.9±0.2 versus 3.2±0.3, P<0.0001). Fleisch et al (25) investigated VEGF and basic fibroblast growth factor (bFGF) levels in arterial samples obtained from patients with one to two vessel coronary artery disease (CAD) during percutaneous transluminal coronary angioplasty. This study was the first to demonstrate a significant correlation between proximal or distal coronary artery VEGF and bFGF levels and the level of collateral blood flow. Although these data are confounded by the effect of heparin on growth factor levels and inaccuracies in collateral quantification, it does support the concept that angiogen levels influence the degree of collateral support. Indeed, Porcu et al (26) recently demonstrated elevated tissue kallikrein but not VEGF levels in patients with severe peripheral vascular disease. However, a weak correlation was identified between venous VEGF and angiographic collateral score. The clinical importance of this issue remains open to debate because it only considers the proangiogenic molecular arm of the collateral growth response. We have no data describing the levels of antiangiogenic cytokines, such as angiostatin and neuropilin-1, or their effects on cell receptor modulation. The responsiveness of cellular elements recruited into the collateral growth process is also ignored.
Cellular processes:
A variety of cellular processes are key to collateral formation including initial recruitability to the nascent vessel (principally chemotaxis), proliferative capacity and cellular capability to release cytokines and angiogens during vessel development driving the process forward.
One of the key initiating steps in this process is the cellular response to hypoxic stress. HIF-1 is a dimeric transcription factor that binds to the promoter region of VEGF, upregulating VEGF production at low oxygen tensions. Aging exerts important influences on the availability of the HIF-1-α subunit by reducing its translocation from the cytoplasm to the nucleus, downregulating the hypoxic response (27,28). Indeed Rivard et al (29) delineated the complex effects of aging upon the cellular elements of angiogenesis in the murine ischemic hindlimb model. Older mice showed an impaired intrinsic angiogenic response to ischemia and VEGF therapy. Endothelial function was blunted in the older animals and the levels of VEGF were reduced in ischemic tissue. This was coupled with a reduction in transcriptional regulation of VEGF in the skeletal muscle and reduced T cell (CD4+) infiltrate. It is well recognized that lymphocytes are an important source of VEGF. Despite similar peripheral T cell counts in old and young mice, transendothelial migration of T cells into ischemic tissue was reduced (27). These data are reinforced by the finding in humans that migration to a VEGF stimulus is significantly impaired in monocytes from diabetic patients (30). Therefore, monocyte recruitment and the release of VEGF locally may be downregulated in both diabetes and aging in humans.
Endothelial dysfunction may be a rate-limiting factor in angiogenesis, further accounting for heterogeneous collateralization. In a canine model of collateral growth, Matsunaga et al (31) demonstrated that inhibition of NO production by Nω-nitro-l-arginine was associated with impaired collateral growth in the presence of sustained increases in local VEGF levels. This indicates that the VEGF response to ischemia requires NO to induce a proangiogenic effect and therefore in dysfunctional endothelium, reduced NO release results in suboptimal VEGF-mediated effects.
NO exerts numerous potentially competing effects on angiogenesis and collateralization. It inhibits migration and proliferation of vascular smooth muscle cells but enhances endothelial cell migration and in vitro tube formation (32). It also upregulates urokinase-type plasminogen activator expression and induces FGF-2 (33). Homozygous endothelial nitric oxide synthase (eNOS)-knockout mice show an impaired hindlimb collateral growth response to ischemia (34). Rabbits fed a diet supplemented with l-arginine improve local neovascularization in the same model. VEGF directly upregulates endothelial NO production and may be crucial for VEGF-induced angiogenesis – eNOS deficient mice are unresponsive to VEGF. Abacii et al (31) recently described impaired coronary collateral formation in diabetic patients with CAD. An impaired NO-mediated vasodilator reponse is well described in diabetes, and NO-mediated VEGF-induced vascular relaxation is reduced in coronary microvessels from patients with CAD. Therefore, factors influencing endothelial function can play a key role in limiting the collateral response through downregulation of NO and VEGF efficacy.
ENVIRONMENTAL FACTORS AND ENDOTHELIAL FUNCTION
The factors responsible for inducing endothelial dysfunction are beyond the scope of this review. They include the classic coronary artery risk factors, as well as homocysteine, free radicals and inflammatory mediators. Hypercholesterolemia in the Watanabe heritable hyperlipidemic (WHHL) rabbit hindlimb ischemia model is associated with impaired collateral formation (37). Several different mechanisms may account for the reduced capacity of WHHL endothelial cells to form new blood vessels. Both VEGF upregulation and endothelial cell migration may be potentially impaired in WHHL rabbits. Endothelial cell migratory activity, a fundamental step in angiogenesis, appears to be impaired in atherosclerosis, possibly due to the effect of oxidized low density lipoprotein (oxLDL) components on the promigratory activity of endogenous angiogenic cytokines (38). Furthermore, the response to certain agonists requiring receptor binding and signal transduction to release intracellular calcium may be impaired in dysfunctional Endothelial cells (39). Such an impairment in transmembrane signalling could compromise the responsiveness of Endothelial cells to Endothelial cell mitogens. Therefore, modulation of cholesterol levels by statin therapy may enhance collateral growth in ischemic tissue. However, hydroxymethyl glutaryl coenzyme A (HMG CoA) reductase inhibition exerts a number of effects beyond simple reductions in LDL, particularly on circulating progenitor cell populations.
THE ROLE OF EPCs
The original paradigm of collateral development and postnatal angiogenesis involves the differentiation and growth of endothelial cells restricted to the site of vessel growth. However, this has recently been challenged by the isolation of EPCs in the peripheral circulation (40,41). The discovery of these cells has caused a complete revision of the mechanisms involved in adult vascular growth and remodelling. Both hematopoietic stem cells and EPCs share common cell surface markers including CD34, flk-1 and Tie-2. These two cell types are thought to arise from a common precursor – the hemangioblast. CD34+/flk-1+ bone marrow derived cells can be cultured in vitro demonstrating the characteristics of normal endothelial cells including uptake of LDL and in vitro angiogenesis in the presence of normal human umbilical vein endothelial cells (42–44). The CD34+/flk-1+ population actually consists of both mature endothelial cells and progenitor cells because both receptors are common to these cellular subgroups. Recently, the AC133 receptor, a specific hemopoietic stem cell surface marker, has been isolated on EPCs allowing more precise characterization of the EPC population (41). Human CD34+ cells were recently separated and cultured from both adult peripheral and umbilical cord blood samples. This EPC fraction was expanded in conditioned media and used to enhance collateral growth in an ischemic hindlimb athymic nude mouse model (45). Labelled EPCs are incorporated into the vascular beds of ischemic myocardium and skeletal muscle indicating that they play a key role in vascular growth (46). Therefore, vasculogenesis is not restricted to the embryo; ‘postnatal vasculogenesis’ also occurs in the adult through recruitment of EPCs into ischemic vascular beds.
Ischemia can promote an increase in circulating EPC numbers and incorporation into the vessels of a corneal micropocket assay in the ischemic hindlimb murine model (47). This suggests that signalling from ischemic tissue may trigger EPC recruitment. It remains controversial whether the elevated circulating EPC levels seen after myocardial infarction are due to the release of cells derived from the bone marrow directly or from a stem cell population residing in the myocardium.
Variation in the viability and proportion of this EPC population could further explain the heterogeneity in collateral development between patients. There is now emerging evidence that the circulating population of EPCs is affected by the burden of coronary risk factors. Vasa et al (48) showed has shown that there is an inverse correlation between the number of CD34/KDR positive cells and the number of risk factors (R=–0.537, P<0.001). Smoking and positive family history of coronary disease were each associated with significant reductions in EPC numbers. In addition to affecting absolute numbers of circulating cells, the migratory response of the cultured EPCs from these patients was negatively influenced by the burden of risk factors, particularly hypertension. These data provide direct evidence that EPC numbers are significantly affected by coronary risk factor burden with important implications for the capacity of patients to recruit native EPCs for revascularization of ischemic tissue.
A number of pharmacological agents exert a proangiogenic effect in vitro and in animal models. Two classes of drugs – angiotensin converting enzyme (ACE) inhibitors and statins – are routinely prescribed by cardiologists, with major mortality reduction benefits. However, it is now being realized that these agents may exert a proportion of their beneficial actions via promotion of angiogenesis in the myocardium involving mechanisms beyond their traditional pharmacological actions.
THE ROLE OF ACE INHIBITION IN ANGIOGENESIS
The effects of ACE inhibition and angiotensin II (ATII) upon angiogenesis are complex, being determined by several factors including the tissue studied, the relative degree of angiotensin (AT) 1/2 receptor expression and NO bioavailabilty. The majority of effects of ATII are anti-angiogenic. Antagonism of the ATII receptor can reverse these effects.
Even though captopril inhibits neovascularization in the rat cornea and microvascular development in hypertensive and normotensive rats (49), other ACE inhibitors have been shown to promote capillary formation in other tissues, including rat limb muscle. One agent, spirapril, demonstrates this activity in cardiac tissue.
Perhaps the most convincing study of ACE inhibition promoting angiogenesis to date was performed in the rabbit model of hindlimb ischemia (50). Quinaprilat (a non-sulfhydryl ACE inhibitor with high tissue affinity), administered subcutaneously, was compared with captopril (lower efficacy of tissue-bound ACE inhibitor) and intra-arterial recombinant human VEGF165 therapy. At day 40, both functional and morphological markers of angiogenesis were significantly elevated in the quinaprilat and recombinant VEGF-treated animals versus captopril treatment or controls. Capillary density in quinaprilat treated limbs was 50% greater than captopril or controls (214/mm2 versus 140.5/mm2). This effect was sustained at five days after cessation of quinaprilat indicating that these findings were not the result of a transient, pharmacologically mediated improvement in endothelium-dependant blood flow. The results indicated a persistent modification of the hindlimb vasculature, which was also demonstrated histologically by increased capillary density. The failure of captopril to induce an angiogenic effect was thought to be due to its lower affinity for tissue ACE. The drug doses were selected so that the same level of plasma ACE inhibition was achieved with both agents. It is possible that mobilization of endothelial cells, which typically occurs during angiogenesis, was opposed by the metalloproteinase inhibition associated with the sulfhydryl group of captopril.
The differential activation of ATII receptors could explain the degree of angiogenesis induced in any given animal model. For example, the AT2 receptor antagonist PD123319 promotes rat cremaster microvessel density to a greater extent than losartan (a specific AT1 antagonist).
The only published data describing the effect of AII receptor blockade in the heart has demonstrated that losartan promoted angiogenesis in the peri-infarct region of a rat myocardial infarction model (51). This has important implications for the prescription of AII antagonists in patients with ischemic heart disease.
The mechanisms responsible for these effects of tissue ACE inhibition remain to be fully elucidated. Upregulation of angiogenic cytokines is unlikely to be responsible. ATII promotes VEGF expression by endothelial and smooth muscle cells; therefore, ACE inhibitor should be antiangiogenic. The angiogenic effects of ACE inhibitor may occur by increasing NO bioavailability. At least two possible mechanisms have been proposed: first, increased bradykinin levels, which can activate the l-arginine-NO pathway or, second, prevention of accumulated AII from stimulating NADH oxidase to produce superoxide anions, which can degrade NO. The proangiogenic role of bradykinin has recently been emphasized by gene transfection experiments in a mouse model of hindlimb ischemia. Intramuscular delivery of adenovirus containing the human tissue kallikrein gene enhanced capillary density caused by ischemia, and this effect was blocked by kinin B1 or B2 receptor antagonists.
There is evidence that ACE inhibition can significantly improve myocardial blood flow acutely at rest (52), during hypoperfusion (53), during postmyocardial infarction (54) and during reperfusion in experimental animals (55). These studies of the acute effects of ACE inhibitor focused on coronary flow reserve and the vasodilation of epicardial coronary vessels, induced by the upregulation of NO production and bradykinin. The acute coronary vasodilator effects in humans were recently quantitatively assessed by Schneider et al (56) with dynamic positron emission tomography.
These findings have important implications for the promotion of myocardial perfusion and could explain some of the positive effects of ACE inhibitor on cardiovascular mortality, left ventricular remodelling after myocardial infarction and coronary event rates. Clearly, important effects on acute coronary vasomotor tone are exerted by ACE inhibitors modulating NO and bradykinin levels, as well as more permanent effects on collateral vessel formation through their proangiogenic actions.
STATINS
The proangiogenic effects of statins have begun to be recognized over the past five years. Although statins lower give cholesterol through inhibition of HMG CoA reductase and hence reduce oxLDL levels, which impair endothelial function, metabolites of the HMG CoA reductase pathway also exert angiogenic effects (57). These effects are principally achieved through modulation of eNOS and Akt activity (58,59). Akt is inhibited by mevalonate and acts as a fundamental signalling gateway at the crossroads of angiogenesis, endothelial stem cell recruitment and differentiation (60). Activation of Akt by statins occurs by reducing mevalonate accumulation downstream of HMG CoA reductase. Llevadot et al (61) recently showed that Akt in EPCs is rapidly activated by simvastatin, leading to enhanced cell migration and survival. Indeed, dominant negative Akt overexpression leads to functional blockade of EPC bioactivity. As well as preventing apoptosis and promoting EPC mobilization, Akt also upregulates endothelial NO production in response to shear stress and VEGF via phosphorylation of eNOS. Kureishi et al (59) showed that simvastatin activates Akt in ordinary endothelial cells promoting eNOS phosphorylation, inhibiting apoptosis and accelerating the formation of vascular structures in vitro as well as in a rabbit hindlimb ischemia model. Dimmeler has reinforced these observations by showing that statins increased both EPC and hemopoietic stem cell numbers in mice as well as the differentiation of CD34 positive cells into EPCs in vitro. These effects are reproducible in humans (62) and can be blocked by the addition of mevalonate occuring independently of the rhokinase/eNOS pathway. Statins were equipotent to VEGF in upregulating EPC numbers in vitro and did not upregulate either angiogenic peptide or receptor levels. These findings point to a novel mechanism of modulating EPC differentiation and hence altering neovascularization independent of angiognic peptides. Perhaps endogenous modulation of Akt activity is partially responsible for the heterogeneity in collateral formation between patients. Indeed, Akt is a novel therapeutic target for future angiogenic strategies.
EFFECT OF ANGIOGENIC GROWTH FACTORS ON MYOCARDIAL PERFUSION IN HUMANS
The promotion of collateral growth in ischemic myocardium is now entering the era of angiogenic peptide and gene transfection techniques. Phase I trials performed to assess safety and efficacy are not placebo controlled and so the 30% improvement in exercise tolerance seen in placebo patients cannot be excluded. To date the principal concerns of tumour development, retinal neovascularization and hemangioma formation have not been overcome. Plasmid transfection is extremely inefficient and so viral vectors are preferred –most commonly adenoviruses. These vectors induce local inflammation and, although transiently expressed, may exert a degree of their proangiogenic effects in animal models as a result of monocyte recruitment rather than local transfected target cell angiogen expression. The only placebo controlled trial of VEGF peptide therapy failed to improve exercise tolerance compared with an identical placebo group.
VEGF and FGF-4 have been selected for trials of angiogenic gene therapy because these peptides have a native or ligated signal sequence that facilitates active peptide secretion.
There have been four principal phase I trials of angiogenic gene therapy. Losordo et al (63) performed intramyocardial injections of plasmid VEGF165 (phVEGF) in 30 patients. Improvements in exercise tolerance, angina frequency and left ventricular ejection fraction were documented. Three trials have employed adenoviral vectors. Rosengart et al (64) delivered 4×108 to 4×1010 particle-forming units of VEGF121 to 15 patients undergoing coronary bypass surgery and to six via a mini thoracotomy alone. No serious adverse effects were reported. Although the number of subjects was too small for statistically significant data to be obtained, most patients reported an improvement in both angina class and treadmill exercise time. There was also a trend toward an increased Rentrop grade of collateral flow in both groups. The preliminary results of a multicentre double- blind trial of adenovirus encoding FGF-4 were recently reported at the 2001 meeting of the American College of Cardiology. Grines et al administered incremental doses of adenovirus encoding FGF-4 to 67 patients with mild to moderate angina. Although there was no overall difference in exercise time between treatment and placebo arms, subgroup analyses demonstrated improvements in those patients with low neutralizing antibody titres to the adenovirus vector. Finally, an adenoviral vector transcribing the HIF-1-α transcription factor has been evaluated by intramyocardial injection to regions that cannot be revascularized surgically in patients undergoing coronary artery bypass grafting. The results of this study are awaited. In animal models of hindlimb and myocardial ischemia, HIF-1-α gene transfection augments collateral development (65).
The confounding issue of placebo effects was recently addressed by Symes et al (64), who performed a percutaneous catheter based introduction of plasmid rhVEGF-2 in six patients randomized to gene transfer or a sham procedure. Both groups showed a reduction in angina symptoms over the first 30 days suggesting an initial placebo effect. However, actively treated patients improved their symptom status at 90 days and did not regress like the placebo group. This was accompanied by improvements in both myocardial perfusion and electromechanical parameters.
We await data from formal single- and double-blinded placebo controlled trials of angiogenic gene therapy – a phase II trial of VEGF-2 plasmid vector is underway. However, besides gene transfection, cellular transplantation strategies are on the verge of novel revascularization therapies. The therapeutic potential of EPCs is being evaluated and there is now evidence that stem cell therapy in murine models of myocardial infarction may allow myocyte replacement of damaged myocardium (67). Progenitor cells could be isolated from cord blood or from the recipient’s own peripheral blood, expanded ex vivo and then reintroduced. These cells could be modified by ex vivo gene transfection techniques before their reintroduction. Asahara et al (68) have already demon-strated that ex vivo tranfection with VEGF164 improves the proliferative capacity of murine EPCs. These techniques could be employed to optimize EPC engraftment into vascular beds as well as modulate local vessel development.
Therefore, in the future, clinical strategies to improve collateral perfusion may involve the introduction of endothelial progenitors or hematological stem cells programmed to migrate to target areas of ischemia and integrate into the developing vascular bed. These cells may also serve as vehicles to secrete cytokines locally, promoting further cellular recruitment to drive vascular development and even myoblast differentiation. This may form part of an integrated tissue engineering strategy to revascularize ischemic tissue or regenerate infarcted myocardium.
REFFERENCES
- 1.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 2.Patrick CW, Jr, McIntire LV. Shear stress and cyclic strain modulation of gene expression in vascular endothelial cells. Blood Purif. 1995;13:112–4. doi: 10.1159/000170194. [DOI] [PubMed] [Google Scholar]
- 3.Shyy YJ, Hsieh HJ, Usami S, Chien S. Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc Natl Acad Sci USA. 1994;91:4678–82. doi: 10.1073/pnas.91.11.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wung BS, Cheng JJ, Chao YJ, Lin J, Shyy YJ, Wang DL. Cyclical strain increases monocyte chemotactic protein-1 secretion in human endothelial cells. Am J Physiol. 1996;270:H1462–8. doi: 10.1152/ajpheart.1996.270.4.H1462. [DOI] [PubMed] [Google Scholar]
- 5.Schaper W. Tangential wall stress as a molding force in the development of collateral vessels in the canine heart. Experientia. 1967;23:595–6. doi: 10.1007/BF02137994. [DOI] [PubMed] [Google Scholar]
- 6.Sasayama S, Fujita M. Recent insights into coronary collateral circulation. Circulation. 1992;85:1197–204. doi: 10.1161/01.cir.85.3.1197. [DOI] [PubMed] [Google Scholar]
- 7.Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brogi E, Schatteman G, Wu T, et al. Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest. 1996;97:469–76. doi: 10.1172/JCI118437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Namiki A, Brogi E, Kearney M, et al. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem. 1995;270:31189–95. doi: 10.1074/jbc.270.52.31189. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–94. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 11.Schaper W, De Brabander M, Lewi P. DNA synthesis and mitoses in coronary collateral vessels of the dog. Circ Res. 1971;28:671–9. doi: 10.1161/01.res.28.6.671. [DOI] [PubMed] [Google Scholar]
- 12.Pasyk S, Schaper W, Schaper J, Pasyk K, Miskiewicz G, Steinseifer B. DNA synthesis in coronary collaterals after coronary artery occlusion in conscious dog. Am J Physiol. 1982;242:H1031–7. doi: 10.1152/ajpheart.1982.242.6.H1031. [DOI] [PubMed] [Google Scholar]
- 13.Fujita M, Sasayama S, Ohno A, Yamanishi K, Araie E, Franklin D. Importance of myocardial ischemia for coronary collateral development in conscious dogs. Int J Cardiol. 1990;27:179–86. doi: 10.1016/0167-5273(90)90157-z. [DOI] [PubMed] [Google Scholar]
- 14.Mohri M, Tomoike H, Noma M, Inoue T, Hisano K, Nakamura M. Duration of ischemia is vital for collateral development: repeated brief coronary artery occlusions in conscious dogs. Circ Res. 1989;64:287–96. doi: 10.1161/01.res.64.2.287. [DOI] [PubMed] [Google Scholar]
- 15.Yamanishi K, Fujita M, Ohno A, Sasayama S. Importance of myocardial ischaemia for recruitment of coronary collateral circulation in dogs. Cardiovasc Res. 1990;24:271–7. doi: 10.1093/cvr/24.4.271. [DOI] [PubMed] [Google Scholar]
- 16.Chilian WM, Mass HJ, Williams SE, Layne SM, Smith EE, Scheel KW. Microvascular occlusions promote coronary collateral growth. Am J Physiol. 1990;258:H1103–11. doi: 10.1152/ajpheart.1990.258.4.H1103. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Castellano N, Garcia EJ, Abeytua M, et al. Influence of collateral circulation on in-hospital death from anterior acute myocardial infarction. J Am Coll Cardiol. 1998;31:512–8. doi: 10.1016/s0735-1097(97)00521-4. [DOI] [PubMed] [Google Scholar]
- 18.Habib GB, Heibig J, Forman SA, et al. Influence of coronary collateral vessels on myocardial infarct size in humans. Results of phase I thrombolysis in myocardial infarction (TIMI) trial. The TIMI Investigators. Circulation. 1991;83:739–46. doi: 10.1161/01.cir.83.3.739. [DOI] [PubMed] [Google Scholar]
- 19.Hirai T, Fujita M, Nakajima H, et al. Importance of collateral circulation for prevention of left ventricular aneurysm formation in acute myocardial infarction. Circulation. 1989;79:791–6. doi: 10.1161/01.cir.79.4.791. [DOI] [PubMed] [Google Scholar]
- 20.Senti S, Fleisch M, Billinger M, Meier B, Seiler C. Long-term physical exercise and quantitatively assessed human coronary collateral circulation. J Am Coll Cardiol. 1998;32:49–56. doi: 10.1016/s0735-1097(98)00181-8. [DOI] [PubMed] [Google Scholar]
- 21.Todd IC, Bradnam MS, Cooke MB, Ballantyne D. Effects of daily high-intensity exercise on myocardial perfusion in angina pectoris. Am J Cardiol. 1991;68:1593–9. doi: 10.1016/0002-9149(91)90315-c. [DOI] [PubMed] [Google Scholar]
- 22.Traverse JH, Wang YL, Du R, et al. Coronary nitric oxide production in response to exercise and endothelium-dependent agonists. Circulation. 2000;101:2526–31. doi: 10.1161/01.cir.101.21.2526. [DOI] [PubMed] [Google Scholar]
- 23.Rohan RM, Fernandez A, Udagawa T, Yuan J, D’Amato RJ. Genetic heterogeneity of angiogenesis in mice. FASEB J. 2000;14:871–6. doi: 10.1096/fasebj.14.7.871. [DOI] [PubMed] [Google Scholar]
- 24.Schultz A, Lavie L, Hochberg I, et al. Interindividual heterogeneity in the hypoxic regulation of VEGF: significance for the development of the coronary artery collateral circulation. Circulation. 1999;100:547–52. doi: 10.1161/01.cir.100.5.547. [DOI] [PubMed] [Google Scholar]
- 25.Fleisch M, Billinger M, Eberli FR, Garachemani AR, Meier B, Seiler C. Physiologically assessed coronary collateral flow and intracoronary growth factor concentrations in patients with 1- to 3-vessel coronary artery disease. Circulation. 1999;100:1945–50. doi: 10.1161/01.cir.100.19.1945. [DOI] [PubMed] [Google Scholar]
- 26.Porcu P, Emmanueli C, Kapatsoris M, Chao J, Chao L, Mededdu P. Reversal of angiogenic growth factor upregulation by revascularisation of lower limb ischemia. Circulation. 2002;105:67–72. doi: 10.1161/hc0102.101360. [DOI] [PubMed] [Google Scholar]
- 27.Rivard A, Berthou-Soulie L, Principe N, et al. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem. 2000;275:29643–7. doi: 10.1074/jbc.M001029200. [DOI] [PubMed] [Google Scholar]
- 28.Waltenberger J, Mayr U, Pentz S, Hombach V. Functional upregulation of the vascular endothelial growth factor receptor KDR by hypoxia. Circulation. 1996;94:1647–54. doi: 10.1161/01.cir.94.7.1647. [DOI] [PubMed] [Google Scholar]
- 29.Rivard A, Fabre JE, Silver M, et al. Age-dependent impairment of angiogenesis. Circulation. 1999;99:111–20. doi: 10.1161/01.cir.99.1.111. [DOI] [PubMed] [Google Scholar]
- 30.Waltenberger J, Lange J, Kranz A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation. 2000;102:185–90. doi: 10.1161/01.cir.102.2.185. [DOI] [PubMed] [Google Scholar]
- 31.Matsunaga T, Warltier DC, Weihrauch DW, Moniz M, Tessmer J, Chilian WM. Ischemia-induced coronary collateral growth is dependent on vascular endothelial growth factor and nitric oxide. Circulation. 2000;102:3098–103. doi: 10.1161/01.cir.102.25.3098. [DOI] [PubMed] [Google Scholar]
- 32.Murohara T, Horowitz JR, Silver M, et al. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- 33.Ziche M, Parenti A, Ledda F, et al. Nitric oxide promotes proliferation and plasminogen activator production by coronary venular endothelium through endogenous bFGF. Circ Res. 1997;80:845–52. doi: 10.1161/01.res.80.6.845. [DOI] [PubMed] [Google Scholar]
- 34.Murohara T, Asahara T, Silver M, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–78. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abaci A, Oguzhan A, Kahraman S, et al. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–42. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 36.Metais C, Li J, Li J, Simons M, Sellke FW. Effects of coronary artery disease on expression and microvascular response to VEGF. Am J Physiol. 1998;275:H1411–8. doi: 10.1152/ajpheart.1998.275.4.H1411. [DOI] [PubMed] [Google Scholar]
- 37.Van Belle E, Rivard A, Chen D, et al. Hypercholesterolemia attenuates angiogenesis but does not preclude augmentation by angiogenic cytokines. Circulation. 1997;96:2667–74. doi: 10.1161/01.cir.96.8.2667. [DOI] [PubMed] [Google Scholar]
- 38.Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest. 1996;97:2736–44. doi: 10.1172/JCI118728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bossaller C, Habib GB, Yamamoto H, Williams C, Wells S, Henry PD. Impaired muscarinic endothelium-dependent relaxation and cyclic guanosine 5′-monophosphate formation in atherosclerotic human coronary artery and rabbit aorta. J Clin Invest. 1987;79:170–4. doi: 10.1172/JCI112779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 41.Peichev M, Naiyer AJ, Pereira D, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–8. [PubMed] [Google Scholar]
- 42.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 43.Kalka C, Masuda H, Takahashi T, et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ Res. 2000;86:1198–202. doi: 10.1161/01.res.86.12.1198. [DOI] [PubMed] [Google Scholar]
- 44.Kalka C, Masuda H, Takahashi T, et al. Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ Res. 2000;86:1198–202. doi: 10.1161/01.res.86.12.1198. [DOI] [PubMed] [Google Scholar]
- 45.Kalka C, Masuda H, Takahashi T, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci USA. 2000;97:3422–7. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isner JM, Asahara T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J Clin Invest. 1999;103:1231–6. doi: 10.1172/JCI6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 48.Vasa M, Fichtlscherer S, Aicher A, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 49.Volpert OV, Ward WF, Lingen MW, et al. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest. 1996;98:671–9. doi: 10.1172/JCI118838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fabre JE, Rivard A, Magner M, Silver M, Isner JM. Tissue inhibition of angiotensin-converting enzyme activity stimulates angiogenesis in vivo. Circulation. 1999;99:3043–9. doi: 10.1161/01.cir.99.23.3043. [DOI] [PubMed] [Google Scholar]
- 51.Sladek T, Sladkova J, Kolar F, et al. The effect of AT1 receptor antagonist on chronic cardiac response to coronary artery ligation in rats. Cardiovasc Res. 1996;31:568–76. [PubMed] [Google Scholar]
- 52.Sudhir K, Chou TM, Hutchison SJ, Chatterjee K. Coronary vasodilation induced by angiotensin-converting enzyme inhibition in vivo: differential contribution of nitric oxide and bradykinin in conductance and resistance arteries. Circulation. 1996;93:1734–9. doi: 10.1161/01.cir.93.9.1734. [DOI] [PubMed] [Google Scholar]
- 53.Kitakaze M, Minamino T, Node K, et al. Beneficial effects of inhibition of angiotensin-converting enzyme on ischemic myocardium during coronary hypoperfusion in dogs. Circulation. 1995;92:950–61. doi: 10.1161/01.cir.92.4.950. [DOI] [PubMed] [Google Scholar]
- 54.Ertl G, Kloner RA, Alexander RW, Braunwald E. Limitation of experimental infarct size by an angiotensin-converting enzyme inhibitor. Circulation. 1982;65:40–8. doi: 10.1161/01.cir.65.1.40. [DOI] [PubMed] [Google Scholar]
- 55.Piana RN, Wang SY, Friedman M, Sellke FW. Angiotensin-converting enzyme inhibition preserves endothelium-dependent coronary microvascular responses during short-term ischemia-reperfusion. Circulation. 1996;93:544–51. doi: 10.1161/01.cir.93.3.544. [DOI] [PubMed] [Google Scholar]
- 56.Schneider CA, Voth E, Moka D, et al. Improvement of myocardial blood flow to ischemic regions by angiotensin-converting enzyme inhibition with quinaprilat IV: a study using [15O] water dobutamine stress positron emission tomography. J Am Coll Cardiol. 1999;34:1005–11. doi: 10.1016/s0735-1097(99)00316-2. [DOI] [PubMed] [Google Scholar]
- 57.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101:207–13. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 58.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 59.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- 61.Llevadot J, Murasawa S, Kureishi Y, et al. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–90. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 63.Losordo DW, Vale PR, Symes JF, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–4. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 64.Rosengart TK, Lee LY, Patel SR, et al. Angiogenesis gene therapy: phase I assessment of direct intramyocardial administration of an adenovirus vector expressing VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation. 1999;100:468–74. doi: 10.1161/01.cir.100.5.468. [DOI] [PubMed] [Google Scholar]
- 65.Vincent KA, Shyu KG, Luo Y, et al. Angiogenesis is induced in a rabbit model of hindlimb ischemia by naked DNA encoding an HIF-1alpha/VP16 hybrid transcription factor. Circulation. 2000;102:2255–61. doi: 10.1161/01.cir.102.18.2255. [DOI] [PubMed] [Google Scholar]
- 66.Symes JF, Losordo DW, Vale PR, et al. Gene therapy with vascular endothelial growth factor for inoperable coronary artery disease. Ann Thorac Surg. 1999;68:830–6. doi: 10.1016/s0003-4975(99)00807-3. [DOI] [PubMed] [Google Scholar]
- 67.Orlic D, Kajstura J, Chimenti S, Bodine DM, Leri A, Anversa P. Transplanted adult bone marrow cells repair myocardial infarcts in mice. Ann NY Acad Sci. 2001;938:221–9. doi: 10.1111/j.1749-6632.2001.tb03592.x. [DOI] [PubMed] [Google Scholar]
- 68.Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–72. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]