

Abstract
AIM: To determine the effect of ellagic acid on apop-tosis and proliferation in pancreatic cancer cells and to determine the mechanism of the pro-survival effects of ellagic acid.
METHODS: The effect of ellagic acid on apoptosis was assessed by measuring Phosphatidylserine externalization, caspase activity, mitochondrial membrane potential and DNA fragmentation; and proliferation by measuring DNA thymidine incorporation. Mitochondrial membrane potential was measured in permeabilized cells, and in isolated mitochondria. Nuclear factor κB (NF-κB) activity was measured by electromobility shift assay (EMSA).
RESULTS: We show that ellagic acid, a polyphenolic compound in fruits and berries, at concentrations 10 to 50 mmol/L stimulates apoptosis in human pancreatic adenocarcinoma cells. Further, ellagic acid decreases proliferation by up to 20-fold at 50 mmol/L. Ellagic acid stimulates the mitochondrial pathway of apoptosis associated with mitochondrial depolarization, cytochrome C release, and the downstream caspase activation. Ellagic acid does not directly affect mitochondria. Ellagic acid dose-dependently decreased NF-κB binding activity. Furthermore, inhibition of NF-κB activity using IkB wild type plasmid prevented the effect of ellagic acid on apoptosis.
CONCLUSION: Our data indicate that ellagic acid stimulates apoptosis through inhibition of the prosu-rvival transcription factor NF-κB.
Keywords: Ellagic acid, Nuclear factor-κB, Apoptosis, Pancreatic cancer
INTRODUCTION
Pancreatic cancer is a very aggressive disease and is the fourth most common cause of death in Western countries with almost the same rate of incidence and mortality per year[1,2]. Pancreatic cancer is very resistant to radio-and chemo-therapies. One reason for that is the resistance of pancreatic cancer cells to apoptosis[3].
During the past decade, significant progress has been achieved in understanding the molecular mechanisms of apoptosis[4]. The information obtained suggests that the commitment to apoptosis occurs through activation of caspases, a unique family of cysteine proteases[4]. Caspases are synthesized as inactive precursors and are, generally, activated by proteases including caspases themselves. Thus, caspases can function in an activation cascade.
Cancer cells protect themselves from apoptosis by upregulation of prosurvival mechanisms. Activation of the transcription factor NF-κB is a key pro-survival mechanism in cancer cells[5,6]. NF-κB is constitutively active in pancreatic cancer cells, and its inhibition leads to pancreatic cancer cell death and inhibition of tumor development[7].
Ellagic acid (C14H6O8) is a polyphenolic compound present in fruits and berries such as pomegranates, strawberries, raspberries and blackberries. It has anticar-cinogenic, antioxidant and antifibrosis properties[8–11]. The anticarcinogenic effect of ellagic acid was shown in several types of cancers including skin, esophageal, and colon cancers[11,12]. However, the effects of ellagic acid on pancreatic cancer have not been studied. Furthermore, the mechanisms mediating anticancer effect of ellagic acid, in general, remain unknown.
There is a growing interest in natural compounds for enhancing cancer prevention and treatment. In this study, we show that ellagic acid induces apoptosis and decreases proliferation in pancreatic cancer cells. We demonstrate that ellagic acid stimulates apoptosis in pancreatic cancer cells through inhibiting transcription factor NF-κB activity.
MATERIALS AND METHODS
Reagents
Ac-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC) was from Peptide Institute, Inc (Osaka, Japan). Antibody against cytochrome C was from BD Biosciences (San Diego, CA). CAPE was from biomol (Plymouth meeting, PA). Ellagic acid and all other reagents were from Sigma Chemical (St. Louis, MO).
Cell culture
Human pancreatic adenocarcinoma cell lines, the poorly differentiated MIA PaCa-2 and the moderately differentiated PANC-1, were obtained from the American Type Culture Collection (Manassas, VA). MIA PaCa-2 and PANC-1 cells were grown in 1/1 D-MEM/F-12 medium (GIBCO Invitrogen Corporation, Grand Island, NY) supplemented with 15% fetal bovine serum (FBS), 4 mmol/L L-glutamine, and 1% antibiotic/antimycotic solution (Omega Scientific, Tarzana, CA). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 and were used between passages 4 and 12.
MIA PaCa-2 and PANC-1 cells were plated at a density of 2 × 106/mL on 100-mm culture dishes, cultured for up to 48 h in D-MEM/F-12 medium supplemented with FBS, glutamine and the antibiotic/antimycotic solution, collected, and processed for the specified analyses. Inhibitors or vehicle were added to the culture medium just before plating out the cells.
Preparation of cytosolic and membrane fractions
Cells were resuspended in a lysis buffer (250 mmol/L sucrose, 20 mmol/L HEPES, 10 mmol/L KCl, 1 mmol/L Na-EGTA, 1 mmol/L Na-EDTA, 2 mmol/L MgCl2, pH 7.0), allowed to swell for 30 min at 4°C, and then disrupted by 80 strokes in a Dounce homogenizer. Homogenates were centrifuged at 1000 g for 5 min to pellet nuclei and cell debris. Supernatants were centrifuged at 16 000 g for 30 min, and the cytosolic fractions (supernatants) were collected. Pellets (heavy membranes enriched with mitochondria) were lysed in RIPA buffer (0.15 mol/L NaCl, 50 mmol/L Tris, 1% deoxycholic acid, 1% Triton X-100, 0.1% sodium dodecyl sulfate, pH 7.2) for 1 h. To determine the quality of cytosolic and mitochondrial separation, both fractions were assessed by immunoblotting for the mitochondrial marker cytochrome C oxidase subunit IV (COX IV).
Western blot analysis
Cells were incubated in a lysis buffer (0.5 mmol/L EDTA, 150 mmol/L NaCl, 50 mmol/L Tris, 0.5% Nonidet P-40, pH 7.5) for 30 min at 4°C. The lysis buffer was supplemented with 1 mmol/L PMSF, 5 g/mL each of protease inhibitors pepstatin, leupeptin, chymostatin, antipain, and aprotinin. Cell lysates were centrifuged for 10 min at 13 000 g. Supernatants were collected and proteins were separated by SDS-PAGE (Invitrogen) and electrophoretically transferred to nitrocellulose membranes. Non-specific binding was blocked with 5% milk in Tris-buffered saline (4 mmol/L Tris base, 100 mmol/L NaCl, pH 7.5). Membranes were washed in Tris-buffered saline containing 0.05% Tween 20 (TTBS) and incubated for 2 h with the indicated primary antibodies and then for 1 h with horseradish peroxidase-conjugated secondary antibody. Blots were developed with the Supersignal Chemiluminescent Substrate (ECL) (Pierce).
Measurements of apoptosis
Apoptosis parameters were measured as previously described[13–16]: Internucleosomal DNA fragmentation was measured by using Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Manheim, Germany) according to the manufacturer’s instructions.
Phosphatidylserine (PS) externalization: PS externa-lization was analyzed with the Annexin-V (AnV)-FLUOS Staining Kit from Roche Biochemicals (Indianapolis, IN) as we described before[13]. Cells were collected and resuspended at a density of 1 × 106 cells in 500 μL of binding buffer containing 2 μL AnV and 1 μL propidium iodide (PI), incubated in the dark for 30 min at room temperature, and analyzed by flow cytometry.
Effector caspase (DEVDase) activity: DEVDase activity was measured by a fluorogenic assay in whole cell lysates using DEVD-AMC as a substrate, as we described before[13]. The lysate (50-100 μg of protein) was incubated with 10 μmol/L substrate in a reaction buffer (25 mmol/L HEPES (pH 7.5), 10% sucrose, 0.1% CHAPS, 10 mmol/L DTT) at 37°C. Caspase substrate cleavage releases AMC, which emits fluorescent signal with 380 nm excitation and 440 nm emission. Fluorescence was calibrated using a standard curve for AMC.
Measurement of mitochondrial membrane potential (ΔΨm)
Changes in ΔΨm were detected with the potential-sensitive probes 3, 3’ dihexyloxa-carbocyanine DiOC6 (3) ( Molecular Probes, Eugene, OR). Cells were incubated with 100 nmol/L DiOC6 (3) for 30 min at 37°C in the dark, washed twice with PBS, and analyzed on a FACScan using FL-1. To completely dissipate ΔΨm, cells were treated with the uncoupling agent CCCP (50 μmol/L) for 1 h before DiOC6 (3) staining.
Registration of mitochondrial respiration and membrane potential in cells permeabilized with digitonin
MIA PaCa-2 cells (5 × 106) were washed twice with PBS and resuspended in 50 μL DMEM/F12 medium without serum. The medium for mitochondrial functional assays contained 250 mmol/L sucrose, 22 mmol/L KCl, 22 mmol/L triethanolamine (pH 7.4), 3 mmol/L MgCl2, 5 mmol/L KH2PO4, 0.5% BSA. Glutamate (10 mmol/L) and malate (2 mmol/L) were used as mitochondrial respiratory substrates. Digitonin at concentration 0.001% was added to cell suspension to permeabilize plasma membrane and to allow substrates and chemicals to reach mitochondria immediately after addition into reaction medium. The measurements were performed at 25°C. Membrane potential and oxygen consumption were monitored simultaneously in a 1-mL custom-made chamber. Oxygen consumption was measured using a Clark-type electrode (Instech Lab., Plymouth Meeting, PA) connected to an oxygen meter (Yellow Springs Instruments, Yellow Springs, OH). Mitochondrial membrane potential was registered in the presence of 1 μmol/L tetraphenyl phosphonium (TPP+) using a TPP+-sensitive electrode connected to an amplifier (Vernier Software, Beaverton, OR). An increase in ΔΨm causes TPP+ uptake by mitochondria and, correspondingly, a decrease in external TPP+ measured with the electrode.
Another approach applied to measure ΔΨm is by using ΔΨm-sensitive fluorescent probe tetramethy-lrhodamine methyl ester (TMRM; Molecular Probes, Eugene, OR). Changes in the fluorescence intensity were measured in the cell suspension containing 0.5 μmol/L TMRM in the 2-mL cuvette in a RF-1501 spectrofluorophotometer (Simadzu, Japan) with 543 nm excitation, and 578 nm emission. ΔΨm-driven mito-chondrial uptake of TMRM causes TMRM quenching, which results in decreased fluorescence intensity.
Isolation of mitochondria
Mitochondria from Mia PaCa-2 cells were isolated using differential centrifugation. Cells (approximately 2 × 108) were homogenized using motor-driven tissue grinder in the isolation buffer containing 320 mmol/L sucrose, 10 mmol/L Tris-HCl (pH 7.4), 0.5 mmol/L EGTA, 0.5 mmol/L EDTA and 0.2% BSA. Unbroken cells were spun down by centrifugation at 500 g for 5 min, followed by nuclei centrifugation at 2000 g for 3 min. The resulting supernatant containing mitochondria was spun down at 12 500 g for 10 min; then mitochondria were washed in a washing buffer containing 320 mmol/L sucrose and 10 mmol/L Tris-HCL (pH 7.4); and finally the mitochondrial pellet was re-suspended in the washing buffer. The obtained mitochondrial suspension contained 8 to 12 mg protein/mL as determined by the Bradford protein assay (BioRad Laboratories). All isolation steps were performed at 4°C, and the mitochondria were kept on ice all the time. Mitochondrial membrane potential and oxygen consumption were measured using TPP+-sensitive and Clark-type electrodes correspondingly in the same incubation buffer as described above for digitonin-permeabilized cells.
Measurement of cytochrome C release from isolated mitochondria
MIA PaCa-2 cell mitochondria were incubated in the absence or presence of ellagic acid for 10 min. Aliquots of mitochondrial suspension were collected, centrifuged at 13 500 g for 10 min at 4°C, and cytochrome C levels in the mitochondria (pellet) and the medium (supern-atant) were measured by Western blot as previously described[13].
Preparation of nuclear extracts and electromobility shift assay (EMSA)
Preparation of nuclear extracts and EMSA have been described in detail[17–19]. Briefly, pancreatic cancer cells were lysed on ice in a hypotonic buffer A[17] supplemented with 1 mmol/L PMSF, 1 mmol/L DTT, and protease inhibitor cocktail containing 5 μg/mL each of pepstatin, leupeptin, chymostatin, antipain, and aprotinin. Cells were left to swell on ice for a 20 min to 25 min period: 0.3% Igepal CA-630 was then added, and the nuclei were collected by microcentrifu-gation. The nuclear pellet was resuspended in a high-salt buffer C[17] supplemented with 1 mmol/L PMSF, 1 mmol/L DTT, and the protease inhibitor cocktail described above. After incubating at 4°C, membrane debris was pelleted by microcentrifugation for 10 min, and the clear supernatant (nuclear extract) was aliquoted and stored at -80°C. Protein concentration in the extracts was determined by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
For the EMSA, aliquots of nuclear extracts with equal amounts of protein (5-10 μg) were mixed in 20-μL reactions with a buffer containing 10 mmol/L HEPES (pH 7.8), 50 mmol/L KCl, 0.1 mmol/L EDTA, 1 mmol/L DTT, 10% glycerol, and 3 μg poly (dI-dC). Binding reactions were started by the addition of 32P-labeled DNA probe and incubated at room temperature for 20 min. The oligo probe 5’-GCAGAGGGGACTTTCCGAGA-3’ containing κB binding motif (underlined) was annealed to the complementary oligonucleotide and end-labeled by using T4 polynucleotide kinase. Samples were electrophoresed on a native 4.5% polyacrylamide gel at 200 V in 0.5 TBE buffer (1 × TBE: 89 mmol/L Tris base, 89 mmol/L boric acid, 2 mmol/L EDTA). Gels were dried and densitometrically quantified in the Phosphor-Imager (Molecular Dynamics, Sunnyvale, CA). In pancreatic cancer cells, the NF-κB band has two components: the upper component corresponds to the p50/p65 heterodimer and the lower component to the p50/p50 homodimer. In the present study, we quantified the total (combined) intensity of the NF-κB band.
Cell transfection
For this a luciferase reporter gene system was used. Briefly, MIA PaCa-2 cells were simultaneously transfected with the 4KBwt-pRL-TK luciferase plasmid, which expresses the NF-κB inhibitor I-κB and pRL-TK luciferase (as a reference) using the NucleofectorTM II (Amaxa Inc, Gaithersburg, MD) according to the manufacturer protocol. The transfection efficiency and NF-κB transcriptional activity was assessed by using the Dual-Luciferase Reporter Assay System (Promega Corporation, Madison WI).
Statistical analysis
Results are expressed as mean ± SE from at least 3 independent experiments. Statistical analysis was done using the unpaired Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
Ellagic acid stimulates apoptosis and inhibits proliferation of pancreatic cancer cells
Ellagic acid dose-dependently increased apoptosis in PaCa cells (Figure 1). To measure apoptosis we used 2 approaches. First, we showed that ellagic acid stimulates apoptotic internucleosomal DNA fragmentation in MIA PaCa-2 (Figure 1A) and PANC-1 cells (Figure 1B). Second, we used flow cytometry and AnV/PI staining to measure the percentage of dead cells as described in Experimental Procedures. We previously showed that the AnV+/PI- group includes cells at early stages of apoptosis, whereas the AnV+/PI+ group includes both necrotic cells and apoptotic cells associated with secondary necrosis[13]. Ellagic acid increased the percentage of dead MIA PaCa-2 cells (i.e. stained positively for both AnV and PI or for AnV alone) (Figure 1C).
Figure 1.

Ellagic acid stimulates apoptosis in pancreatic cancer cells. MIA PaCa-2 (A, C) and PANC-1 (B) cells were cultured for 48 h in the presence or absence of indicated doses of ellagic acid. Internucleosomal DNA fragmentation was measured using the Cell Death Detection ELISA kit (A, B); Dead cells were assessed by flow cytometry using AnV/PI staining (C). AnV+/PI- and AnV+/PI+ cells were considered dying through apoptosis and/or secondary necrosis. Values are normalized to control (A, B). Values are mean ± SE (n = 3), aP < 0.05 vs control.
Next, we measured the effect of ellagic acid on proliferation of PaCa cells. The results in Figure 2 show that ellagic acid dose-dependently inhibited proliferation of both MIA PaCa-2 and PANC-1 cell lines as measured by 3H thymidine incorporation (Figure 2A and B). The effect was most pronounced at 50 μmol/L (almost 20 fold inhibition in MIA PaCa-2 cells).
Figure 2.
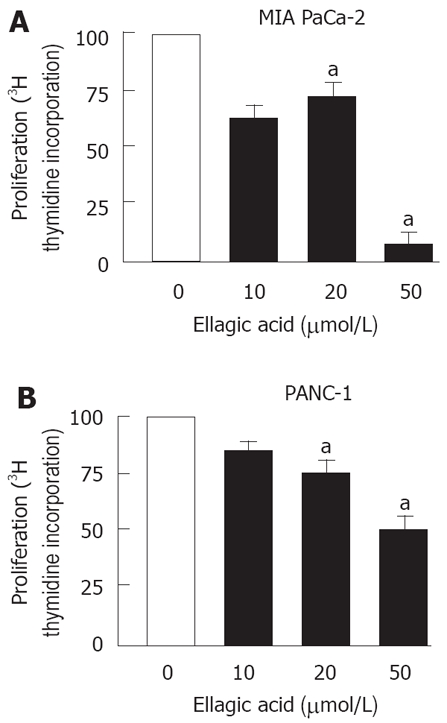
Ellagic acid inhibits proliferation in pancreatic cancer cells. MIA PaCa-2 (A, C) and PANC-1 (B) cells were cultured for 48 h in the presence or absence of indicated doses of ellagic acid. Proliferation was assessed by measuring (3H) thymidine incorporation into DNA. The results are representative of at least 3 independent experiments, aP < 0.05 vs control.
Ellagic acid induces mitochondrial depolarization, cytochrome C release, and caspase activation in pancreatic cancer cells
To determine the signaling pathway mediating the proapoptotic effect of ellagic acid, we measured the effects of ellagic acid on mitochondrial membrane potential (ΔΨm), cytochrome C release, and caspase-3 activity. Ellagic acid decreased ΔΨm as measured by flow cytometry using the potential-sensitive probe DiOC6 (3). Depolarization was already evident at 10 μmol/L, and was very pronounced at 50 μmol/L ellagic acid (Figure 3A). Ellagic acid also dose-dependently stimulated cytochrome C release, which manifests by its decrease in mitochondria-enriched membrane fractions and its increase in cytosolic fractions (Figure 3B). The increased cytochrome C release was associated with downstream activation of caspase-3 in MIA PaCa-2 (Figure 3C) and PANC-1 (Figure 3D) cells.
Figure 3.

Ellagic acid induces loss of mitochondrial membrane potential, cytochrome C release, and caspase-3 activation in pancreatic cancer cells. MIA PaCa-2 (A-C) and PANC-1 (B) cells were cultured for 48 h in the presence or absence of indicated doses of ellagic acid or broad-spectrum caspase inhibitor zVAD-fmk (100 μmol/L). A: Changes in ΔΨm were measured by flow cytometry using the potential-sensitive probe 3,3’dihexyloxa-carbocyanine DiOC6 (3); B: Cytochrome C release was assessed by measuring cytochrome C levels in both cytosolic and mitochondria-enriched membrane fractions using Western blot analysis. Blots were re-probed for cytochrome C oxidase (COX IV), a specific mitochondrial marker. Western blots of cytosolic fractions re-probed for actin to confirm equal protein loading; C and D: Caspase-3 activity was assessed by measuring the DEVDase (caspase-3 like) activities in cell lysates using a fluorometric assay with a specific substrate. The results are representative of at least 3 independent experiments. Values are normalized to control (C and D). Values are mean ± SE (n = 3), aP < 0.05 vs control.
These data together indicate that ellagic acid induces the mitochondrial pathway of apoptosis associated with mitochondrial depolarization, cytochrome C release, and downstream caspase activation.
Ellagic acid does not directly affect mitochondria function in pancreatic cancer cells
To test the effect of ellagic acid on mitochondria we isolated functional mitochondria from MIA PaCa-2 cells and measured the effect of ellagic acid on mitochondria respiration, membrane potential, and cytochrome C release (Figure 4). The respiratory control ratio of isolated mitochondria with succinate as a respiratory substrate in all the experiments was greater than 4 in all the experiments. Ellagic acid affected neither oxygen consumption measured with Clark electrode (Figure 4A) nor ΔΨm measured with TPP+ electrode (Figure 4B). The protonophore CCCP, which we used as a positive control, depolarized mitochondria (Figure 4B). In agreement with these results, ellagic acid did not increase cytochrome C release from isolated mitochondria into the incubation medium (Figure 4C).
Figure 4.
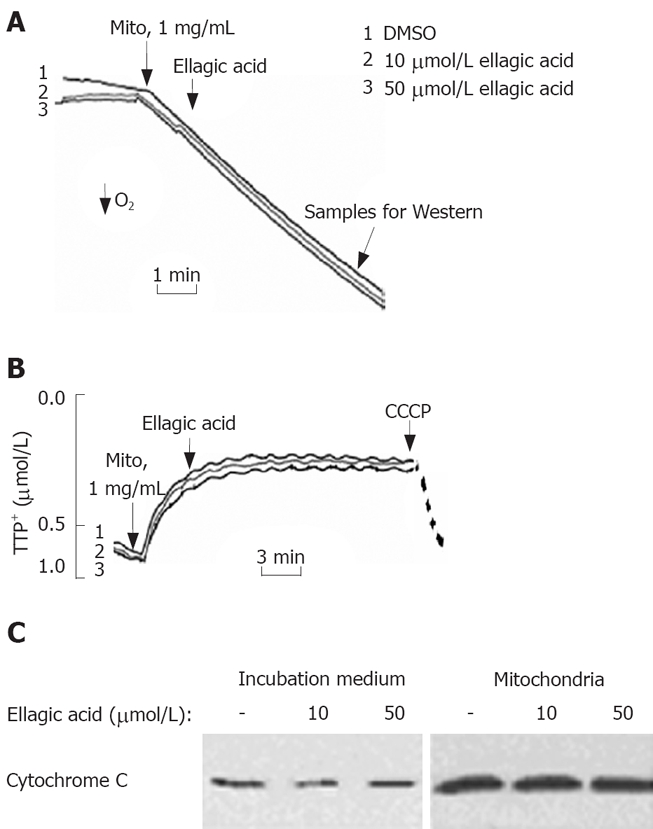
Ellagic acid does not directly affect the function of isolated mitochondria. Mitochondria were isolated from MIA PaCa-2 cells cultured in the absence of ellagic acid. A: Oxygen consumption was measured using a Clark-type electrode connected to an oxygen meter; B: Mitochondrial membrane potential (ΔΨm) was monitored in the presence of 2 μmol/L tetraphenyl phosphonium (TPP+) using a TPP+-sensitive electrode connected to an amplifier. Protonophore CCCP (10 μmol/L) was added to dissipate ΔΨm; C: Cytochrome C levels were measured in the incubation medium and the mitochondrial pellet by Western blot analysis. The results are representative of 3 independent experiments.
We next assessed the possibility of ellagic acid’s effect in permeabilized cells. For this purpose, MIA PaCa-2 cells were permeabilized with digitonin as described by Ohno et al[20]. Ellagic acid did not induce any discernable effect on the mitochondrial membrane potential in digitonin-permeabilized cells as measured with TPP+ electrode (Figure 5B) or by using TMRM fluorescent dye (Figure 5C). CCCP was applied to completely dissipate ΔΨm (Figure 5B and C). Similarly, ellagic acid did not have any effect on mitochondrial respiration in permeabilized cells (Figure 5A).
Figure 5.
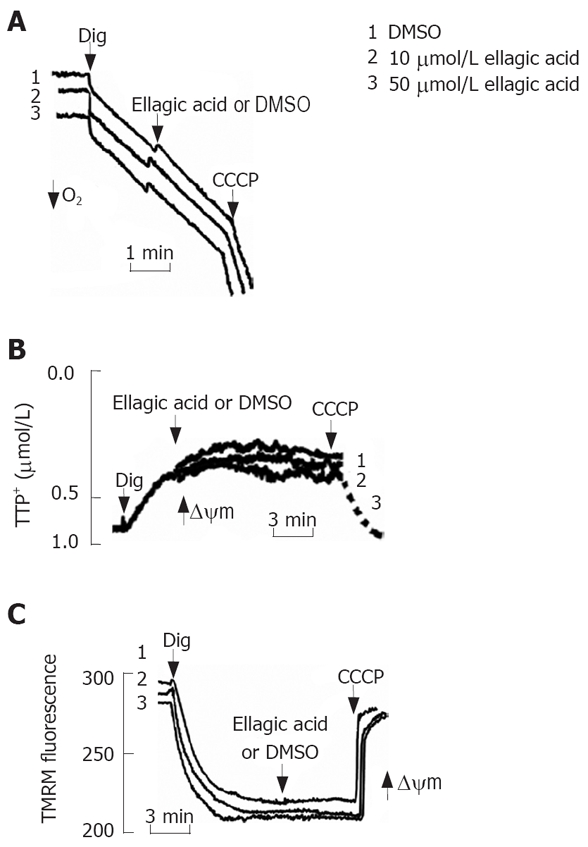
Ellagic acid does not directly affect mitochondria function inpermeabilized MIA PaCa-2 cells. MIA PaCa-2 cells were permeabilized with 0.001% digitonin. A: Oxygen consumption was measured using a Clark-type electrode connected to an oxygen meter; B: Changes in ΔΨm were monitored in the presence of 2 μmol/L tetraphenyl phosphonium (TPP+) using a TPP+-sensitive electrode connected to an amplifier; C: Changes in ΔΨm were monitored using the ΔΨm-sensitive fluorescent probe tetramethylrhodamine methyl ester (TMRM); changes in the fluorescence intensities were measured using the excitation at 543 nm and the emission at 578 nm. Protonophore CCCP (10 μmol/L) was added to dissipate ΔΨm.
The results in Figures 4 and 5 indicate that ellagic acid does not directly affect mitochondria functions in pancreatic cancer cells.
Ellagic acid and NF-κB inhibition act through the same mechanism to stimulate apoptosis
We hypothesized that ellagic acid induced the mitochondrial pathway of apoptosis through blocking a key upstream prosurvival mechanisms, namely NF-κB. NF-κB is a key transcription factor, which is usually activated and has anti-apoptotic role in cancer cells including pancreatic cancer[4]. We found that ellagic acid dose-dependently decreased NF-κB binding activity in both MIA PaCa-2 and PANC-1 cell lines (Figure 6A).
Figure 6.

Ellagic acid decreases the activity of the transcription factor NF-κB. The effects of ellagic acid and pharmacological or molecular inhibition of NF-κB on apoptosis are not additive. MIA PaCa-2 (A and B) cells were cultured for 48 h in the presence or absence of indicated doses of ellagic acid. MIA PaCa-2 cells were transfected with 4KBwt-pRL-TK luciferase plasmid and pRL-TK luciferase as a control using the NucleofectorTM II electroporation system(C and D). A: NF-κB binding activity was measured as described in Experimental procedures; B and D: Internucleosomal DNA fragmentation was measured using the Cell Death Detection ELISA kit; C: NF-κB transcriptional activity was assessed using the dual-Luciferase Reporter Assay System assay. Results are representative of at least 3 independent experiments. Values are normalized to control (B and D). Values are mean ± SE (n = 3), aP < 0.05 vs control.
We further showed that a pharmacologic inhibitor of NF-κB caffeic acid phenethyl ester (CAPE) stimulated apoptosis in PaCa cells, and that in the presence of CAPE there was no additional stimulation of apoptosis by ellagic acid (50 μmol/L) (Figure 6B). Further, transfection of MIA PaCa-2 cells with 4KBwt-pRL-TK plasmid completely blocked NF-κB transcriptional activity (Figure 6C), and at the same time stimulated apoptosis as measured by DNA fragmentation by > 5-fold (Figure 6D). The addition of ellagic acid to the transfected cells did not further increase DNA fragmentation (Figure 6D), confirming that ellagic acid causes apoptosis through inhibition of NF-κB.
DISCUSSION
Our study aimed to investigate the effect of ellagic acid on cell death and proliferation of pancreatic cancer cells and to determine the mechanism through which ellagic acid affects cell survival. We used the poorly differentiated MIA PaCa-2 and moderately differentiated PANC-1 human pancreatic carcinoma cell lines, which both display K-ras and p53 mutations characteristic of pancreatic cancer.
We found that ellagic acid: (1) stimulated apoptosis and inhibited proliferation of pancreatic cancer cells; (2) activated the mitochondrial death pathway associated with loss of ΔΨm, cytochrome C release and caspase-3 activation without directly affecting the mitochondria; and (3) inhibited NF-κB activity in pancreatic cancer cells.
Mechanisms through which ellagic acid inhibits NF-κB remain to be investigated.
One mechanism through which NF-κB inhibits apoptosis is up-regulation of the anti-apoptotic Bcl-xL protein. Bcl-xL, in turn, blocks mitochondrial permea-bilization resulting in inhibition of cytochrome C release as well as preventing mitochondrial depolarization[21–23]. Our data indicate that ellagic acid decreases NF-κB acivity, leading to activation of the mitochondrial pro-apoptotic pathway and resulting in cytochrome C release and caspase activation. This scheme is depicted in Figure 7.
Figure 7.
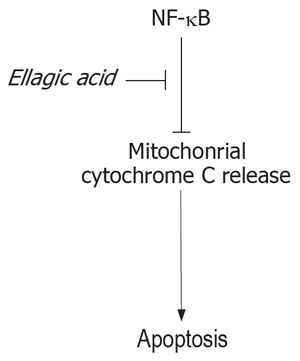
Representative scheme of the proposed mechanism of induction of apoptosis by ellagic acid.
Our data indicate that ellagic acid inhibits proliferation of pancreatic cancer cells, similar to that in published data of other cells[21,24–28]. Ellagic acid completely abolished proliferation at high concentration, whereas it only increased apoptosis by 2.5-fold. The contribution of necrosis might account for the decreased proliferation.
Our results as well as the published data on the potent inhibitory effect of ellagic acid on the vascular endothelial growth factor receptor and platelet-derived growth factor receptor leading to the inhibition of their signaling[29], indicate that ellagic acid is a powerful phenolic compound with pro-apoptotic and anti-proliferation effects in cancer cells. Understanding the mechanism of action of ellagic acid will allow us to test the effect of the compound alone or in combination with other compounds on cancer growth and survival in an orthotopic model of pancreatic cancer[30].
In summary, the present study shows that ellagic acid induces apoptosis and decreases proliferation in pancreatic cancer cells. This phenolic compound stimulates mitochondrial depolarization, cytochrome C release and caspase activation. Ellagic acid has no direct effect on mitochondria. One mechanism by which it stimulates the mitochondrial death pathway is through inhibiting the transcription factor NF-κB, a major prosurvival factor in pancreatic cancer cells. There is an increasing interest in the use of natural products for cancer treatments. Our results suggest a potential therapeutic role for ellagic acid in the treatment of pancreatic cancer.
COMMENTS
Background
Pancreatic cancer is very resistant to radio- and chemo- therapies. Recently, there is a growing interest in natural compounds for enhancing cancer prevention and treatment. Ellagic acid is a phenolic compound present in fruits and berries such as pomegranates, strawberries, raspberries and blackberries. It has anticarcinogenic, antioxidant and antifibrosis properties. In the present study the authors investigate the effect of ellagic acid on pancreatic cancer cell proliferation and resistance to death.
Research frontiers
The article focuses on the regulation of pancreatic cancer cell death by a phenolic compound, ellagic acid.
Innovations and breakthroughs
The present study shows that ellagic acid induces apoptosis and decreases proliferation in pancreatic cancer cells. It was shown for the first time that ellagic acid stimulates mitochondrial depolarization, cytochrome C release and caspase activation. Ellagic acid has no direct effect on mitochondria. One mechanism by which it stimulates the mitochondrial death pathway is through inhibiting transcription factor NF-κB, a major prosurvival factor in pancreatic cancer cells.
Applications
The data of this article demonstrate the anti-cancer properties of ellagic acid as well as its mechanism of action. By knowing the mechanism of action of ellagic acid, it can be used in combination with other drugs that target other pro-survival proteins to increase apoptosis in pancreatic cancer cells.
Peer review
This is a carefully performed study with novel findings that have the potential for therapeutic application in pancreatic cancer. It examines the effect of a naturally occurring polyphenolic compound, ellagic acid, on pancreatic cancer cell function, in particular, apoptosis and proliferation. The authors report that ellagic acid stimulates apoptosis of two pancreatic cancer cell lines and the decreased proliferation of ellagic acid-treated cells.
Supported by the Department of Veterans Affairs Merit Review (to A.S.G), the Hirshberg foundation and the NIH/NCCAM (1P01AT003960-01)
Peer reviewer: Minoti Vivek Apte, Pancreatic Research Group, South Western Sydney Clinical School, the University of New South Wales, Level 2, Thomas and Rachel Moore Education Centre, Liverpool Hospital, New South Wales 2170, Liverpool, Australia
S- Editor Li DL L- Editor Roberts SE E- Editor Lin YP
References
- 1.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 2.Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics, 1997. CA Cancer J Clin. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Westphal S, Kalthoff H. Apoptosis: targets in pancreatic cancer. Mol Cancer. 2003;2:6. doi: 10.1186/1476-4598-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;4:567–586. doi: 10.1159/000082182. [DOI] [PubMed] [Google Scholar]
- 5.Ayala GE, Dai H, Ittmann M, Li R, Powell M, Frolov A, Wheeler TM, Thompson TC, Rowley D. Growth and survival mechanisms associated with perineural invasion in prostate cancer. Cancer Res. 2004;64:6082–6090. doi: 10.1158/0008-5472.CAN-04-0838. [DOI] [PubMed] [Google Scholar]
- 6.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004;101:2351–2362. doi: 10.1002/cncr.20605. [DOI] [PubMed] [Google Scholar]
- 8.Mukhtar H, Das M, Khan WA, Wang ZY, Bik DP, Bickers DR. Exceptional activity of tannic acid among naturally occurring plant phenols in protecting against 7,12-dimethylbenz(a)anthracene-, benzo(a)pyrene-, 3-methylcholanthrene-, and N-methyl-N-nitrosourea-induced skin tumorigenesis in mice. Cancer Res. 1988;48:2361–2365. [PubMed] [Google Scholar]
- 9.Thresiamma KC, Kuttan R. Inhibition of liver fibrosis by ellagic acid. Indian J Physiol Pharmacol. 1996;40:363–366. [PubMed] [Google Scholar]
- 10.Osawa T, Ide A, Su JD, Namiki M. Inhibition of lipid peroxidation by ellagic acid. J. Agric. Food Chem. 1987;35:808–812. [Google Scholar]
- 11.Stoner GD, Gupta A. Etiology and chemoprevention of esophageal squamous cell carcinoma. Carcinogenesis. 2001;22:1737–1746. doi: 10.1093/carcin/22.11.1737. [DOI] [PubMed] [Google Scholar]
- 12.Larrosa M, Tomas-Barberan FA, Espin JC. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J Nutr Biochem. 2006;17:611–625. doi: 10.1016/j.jnutbio.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Vaquero EC, Edderkaoui M, Nam KJ, Gukovsky I, Pandol SJ, Gukovskaya AS. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology. 2003;125:1188–1202. doi: 10.1016/s0016-5085(03)01203-4. [DOI] [PubMed] [Google Scholar]
- 14.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 15.Edderkaoui M, Hong P, Vaquero EC, Lee JK, Fischer L, Friess H, Buchler MW, Lerch MM, Pandol SJ, Gukovskaya AS. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1137–G1147. doi: 10.1152/ajpgi.00197.2005. [DOI] [PubMed] [Google Scholar]
- 16.Edderkaoui M, Hong P, Lee JK, Pandol SJ, Gukovskaya AS. Insulin-like growth factor-I receptor mediates the prosurvival effect of fibronectin. J Biol Chem. 2007;282:26646–26655. doi: 10.1074/jbc.M702836200. [DOI] [PubMed] [Google Scholar]
- 17.Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem. 2006;281:3370–3381. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 18.Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275:G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 19.Labrecque L, Lamy S, Chapus A, Mihoubi S, Durocher Y, Cass B, Bojanowski MW, Gingras D, Beliveau R. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis. 2005;26:821–826. doi: 10.1093/carcin/bgi024. [DOI] [PubMed] [Google Scholar]
- 20.Chauvin C, De Oliveira F, Ronot X, Mousseau M, Leverve X, Fontaine E. Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J Biol Chem. 2001;276:41394–41398. doi: 10.1074/jbc.M106417200. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 22.Dobbeling U. Transcription factor profiling shows new ways towards new treatment options of cutaneous T cell lymphomas. Curr Drug Discov Technol. 2007;4:24–30. doi: 10.2174/157016307781115467. [DOI] [PubMed] [Google Scholar]
- 23.Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554–561. doi: 10.1078/0171-9335-00182. [DOI] [PubMed] [Google Scholar]
- 24.Mertens-Talcott SU, Talcott ST, Percival SS. Low concentrations of quercetin and ellagic acid synergisti-cally influence proliferation, cytotoxicity and apoptosis in MOLT-4 human leukemia cells. J Nutr. 2003;133:2669–2674. doi: 10.1093/jn/133.8.2669. [DOI] [PubMed] [Google Scholar]
- 25.Mertens-Talcott SU, Bomser JA, Romero C, Talcott ST, Percival SS. Ellagic acid potentiates the effect of quercetin on p21waf1/cip1, p53, and MAP-kinases without affecting intracellular generation of reactive oxygen species in vitro. J Nutr. 2005;135:609–614. doi: 10.1093/jn/135.3.609. [DOI] [PubMed] [Google Scholar]
- 26.Chang WC, Yu YM, Chiang SY, Tseng CY. Ellagic acid suppresses oxidised low-density lipoprotein-induced aortic smooth muscle cell proliferation: studies on the activation of extracellular signal-regulated kinase 1/2 and proliferating cell nuclear antigen expression. Br J Nutr. 2008;99:709–714. doi: 10.1017/S0007114507831734. [DOI] [PubMed] [Google Scholar]
- 27.Ross HA, McDougall GJ, Stewart D. Antiproliferative activity is predominantly associated with ellagitannins in raspberry extracts. Phytochemistry. 2007;68:218–228. doi: 10.1016/j.phytochem.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 28.Losso JN, Bansode RR, Trappey A 2nd, Bawadi HA, Truax R. In vitro anti-proliferative activities of ellagic acid. J Nutr Biochem. 2004;15:672–678. doi: 10.1016/j.jnutbio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Labrecque L, Lamy S, Chapus A, Mihoubi S, Durocher Y, Cass B, Bojanowski MW, Gingras D, Beliveau R. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis. 2005;26:821–826. doi: 10.1093/carcin/bgi024. [DOI] [PubMed] [Google Scholar]
- 30.Eibl G, Reber HA, Wente MN, Hines OJ. The selective cyclooxygenase-2 inhibitor nimesulide induces apoptosis in pancreatic cancer cells independent of COX-2. Pancreas. 2003;26:33–41. doi: 10.1097/00006676-200301000-00007. [DOI] [PubMed] [Google Scholar]