

Abstract
A highly efficient drug vector for photodynamic therapy (PDT) drug delivery was developed by synthesizing PEGylated gold nanoparticle conjugates, which act as a water-soluble and biocompatible “cage” that allows delivery of a hydrophobic drug to its site of PDT action. The dynamics of drug release in vitro in a two-phase solution system and in vivo in cancer-bearing mice indicates that the process of drug delivery is highly efficient, and passive targeting prefers the tumor site. With the Au NP–Pc 4 conjugates, the drug delivery time required for PDT has been greatly reduced to less than 2 h, compared to 2 days for the free drug.
Introduction
Photodynamic therapy (PDT) is a promising technique for treating various cancers that involves light, photosensitizers, and tissue oxygen.1 Most photosensitizing agents, such as porphyrins and phthalocyanines (Pc's), are hydrophobic and locate preferentially in apolar sites such as the lipid bilayer membranes of cells.2 After intravenous injection and accumulation in the target tissue, the photosensitizers, which can be excited with light of an appropriate wavelength, can transfer energy to surrounding tissue oxygen, generate highly reactive oxygen species (e.g., singlet oxygen, 1Δg), and induce apoptosis or necrosis directly.3 On one hand, such PDT drugs need to be lipophilic to get through the lipophilic membranes in order to incorporate into the relevant sites (e.g., mitochondria, endoplasmic reticulum, and the Golgi apparatus), so that the initial oxidative damage can occur on proteins that reside within the membranes of those organelles.4 On the other hand, lack of solubility under physiological conditions constitutes a significant problem for intravenous PDT drug delivery in vivo.5 The administration of such photosensitizers usually takes 24 h or more to reach the maximum accumulation in the tumor sites.1 This poses an additional risk for toxicity and side effects. Therefore, one needs a delivery vector that can confer hydrophilicity during the drug delivery without destroying the hydrophobic characteristics of the drug itself. There are several delivery strategies known to stabilize PDT drugs in aqueous systems such as liposomes, polymeric micelles, conjugated polymer nanoparticles, and colloidal silica-based nanoparticles.6 Liposomes, which contain phospholipids and cholesterol, are able to deliver hydrophobic PDT drugs through the lipid bilayers. However, the circulation half-lives are as short as several minutes due to the disintegration through lipoprotein exchange and fast uptake by the mononuclear phagocyte system.6a
Among the various delivery systems, PEGylated gold nanoparticles (Au NPs) hold the promise to be a highly efficient PDT drug delivery platform.7 Au NPs are well known for their chemical inertness and have minimum toxicity.8 The size of the NPs can be tuned from 2 to 100 nm, with correspondingly large surface-to-volume ratios.9 By using water-soluble polyethylene glycol (PEG), which has been approved for human intravenous application, NPs can be stabilized by steric repulsion to inhibit colloid aggregation in physiological conditions.10,11 Due to the high degree of hydration and randomly coiled PEG molecules, it is reported that PEGylated Au NPs show remarkable resistance in protein adsorption.12,13 Drugs on the NPs could be shielded from being uptaken by the reticuloendothelial system (RES).14 Therefore, the circulation time of NPs in the blood can be extended. Furthermore, in vivo distribution of drugs could be modulated as well. Such drug vectors can preferentially accumulate in tumor sites through the leaky tumor neovasculature and do not return to the circulation, the so-called “enhanced permeability and retention” (EPR) effect.14
Here, we report the synthesis of PEGylated Au NP–Pc 4 conjugates and the dramatically improved delivery of the drug, which can, as a Au NP conjugate, be well dispersed in aqueous solutions (Figure 1). Silicon phthalocyanine 4 (Pc 4) is a hydrophobic PDT drug currently under phase I clinical trials.15,16 When Pc 4 is injected for in vivo PDT, it usually takes 1 or 2 days until sufficient Pc 4 reaches the tumor site, which then can be irradiated with 672 nm light for therapy.17 With the NP–Pc 4 conjugates, the time for the maximum drug accumulation to the target tumor has been greatly reduced to only <2 h, compared to 2 days for the free Pc 4.
Figure 1.

Design of the water-soluble Au NPs as a PDT drug delivery agent, Pc 4 structure, and transmission electron microscopy image of the conjugates.
Experimental Section
Synthesis of PEGylated Au NPs Loaded with Pc 4
The synthesis of the Au NPs is based on the original synthesis described by Brust,18 with subsequent modifications as follows: 0.25 mmol of tetra-n-octylammonium bromide (TOAB) and 0.6 mmol of dodecylamine (DDA) were dissolved in 5 mL of toluene, and then a 0.53 mmol HAuCl4 solution (30% in HCl solution) was added. A 2 mmol cold NaBH4 aqueous solution was added into the organic phase and stirred vigorously for 2 h. The DDA–Au NPs were collected by precipitation in 40 mL of ethanol and then redispersed in 3 mL of chloroform. Next, HO-PEG-SH (MW = 5000) and the DDA-stabilized Au nanoparticles were mixed in chloroform and stirred overnight. The organic phase was washed twice with water and then evaporated under vacuum. The residues were washed three times by ethanol and then resuspended in chloroform. A 40-fold excess of Pc 4 (1.44 × 10−6 mol) compared to Au NPs was added into the above solution, which was then stirred for 2 days. After removal of the solvent under vacuum at room temperature and purification with 200 nm pore filters and centrifugal concentrators with 100 000 Da cutoff membranes, the PEGylated Au NP–Pc 4 conjugates were well dispersed in aqueous solvents.
Singlet Oxygen Quantum Yield Measurements
The singlet oxygen quantum yield ΦΔ was determined by decomposition of 1,3-diphenylisobenzofuran (DPBF) in ethanol; ΦΔ was correlated to the decay of the absorption of DPBF at 410 nm.19 The irradiation wavelength used to excite the sensitizers was 660 nm. As light source, we used a Clark MXR CPA laser to pump an optical parametric amplifier (OPA, Light Conversion), which produces visible light in the range of 450−11 000 nm as needed. The absorption spectra were measured by using a UV–vis spectrophotometer (Cary Bio50, Varian). Methylene blue (ΦΔ = 49%) was used as the reference compound.19
Transmission Electron Microscopy and Image Analysis
Transmission electron microscopy (TEM) images of Au NPs were obtained by using a JEOL JEM-1200 EX electron microscope. For the measurements, the TEM instrument was operated at an accelerating voltage of 80 kV. The number and size of the nanoparticles were analyzed with the software ImageJ. Over 1660 nanoparticles were counted, and an average size of 5.0 ± 2 nm was calculated on the basis of the Feret's diameter (upper limit diameter, as it measured the longest diameter of the counted particles).20
Dynamic Light Scattering Measurements
Dynamic light scattering (DLS) measurements were obtained on a BI-200SM laser light scattering goniometer with a BI 9000AT autocorrelator (Brookhaven Instruments Corp.). A He–Ne laser (632.8 nm, 15mW) was used to detect the scattering, and the detection angle was 90°. The duration was 30−60 min with a 200 μm pinhole. The CONTIN method was used to analyze the data. All the measurements were performed at 25 °C.
Determination of the Extinction Coefficient of PEGylated Au NPs
Assuming a spherical shape and a uniform face-centered-cubic structure, the extinction coefficient ε of Au NPs (mean diameter D ≈ 5.0 nm, based on TEM) was calculated on the basis of the total gold atom concentration, measured by inductively coupled plasma mass spectroscopy(ICP-MS),usingtheLambert–Beer law.21
Phase Transfer Study
One milliliter of PEGylated Au NP–Pc 4 aqueous solution was prepared in a 1 cm fluorescence cuvette and placed on a magnetic stirrer. An equal volume of toluene was slowly added into the cuvette and kept at room temperature. The transfer of Pc 4 into the organic phase was monitored by UV–vis absorption and fluorescence spectrophotometry (Varian Eclipse fluorescence spectrophotometer) for 4 h.
Fluorescence Imaging of Mice
The in vivo experiments were carried out at the Case Center for Imaging Research at Case Western Reserve University, Cleveland, OH. Male nude mice with tumors were anesthetized with Isoflurane. Multispectral fluorescent images were obtained using the Maestro In Vivo Imaging System (Cambridge Research and Instrumentation, Inc., Woburn, MA). Multispectral imaging data sets were acquired at a constant exposure of 500 ms in 2 nm increments. Spectral libraries were created by using an empty tube and a nude male mouse with tumor before treatment to obtain a background profile. The same animal was scanned every 15 min for 2 h before injection and after injection, and at 24 and 48 h after injection.
Results and Discussion
Spectroscopic Characterization
UV–vis and fluorescence spectra of the Au NP–Pc 4 conjugates in aqueous solution are shown in Figure 2. The spectra demonstrate that the Pc 4 molecules adsorb on the PEGylated Au NPs and are transported into the aqueous solution. Since Pc 4 is, by itself, completely insoluble in water, there is no measureable amount of Pc 4 in the aqueous phase, as determined by UV–vis and photoluminescence measurements, which can detect amounts as low as 10−7 M. The surface plasmon band of Au NPs is centered at 517 nm, which indicates no aggregation of the synthesized Au NPs. The fluorescence of the Pc in the conjugates was partially quenched relative to that of free Pc; however, it was still easily traceable by fluorescence imaging (Figure 2 and Figure 6, below). The well-dispersed Au NPs were also characterized by TEM and DLS. The average Au NP core diameter for this study was 5.0 nm (Figure 1), and the hydrodynamic diameter of these conjugates was 32 nm.
Figure 2.
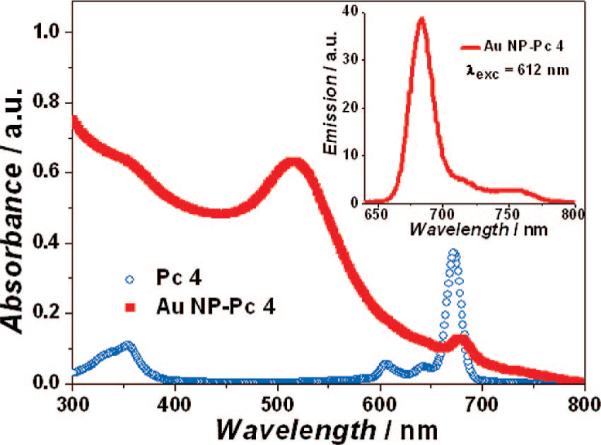
Absorption and emission spectra (inset) of PEGylated Au NP–Pc 4 conjugates in normal saline (0.9% NaCl, pH 7.2).
Figure 6.
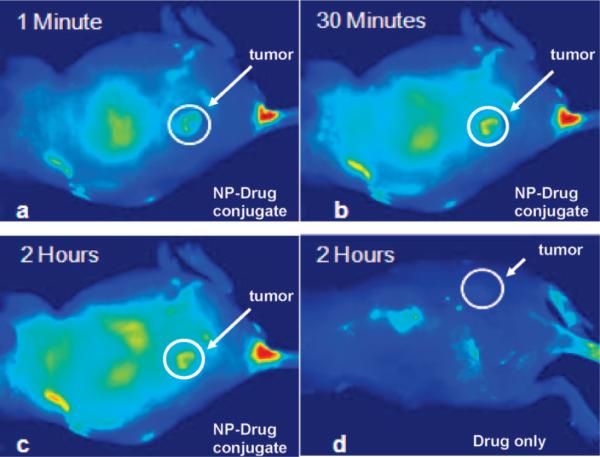
Fluorescence images of a tumor-bearing mouse after being injected with Au NP–Pc 4 conjugates in normal saline (0.9% NaCl, pH 7.2), (a) 1 min, (b) 30 min, and (c) 120 min after intravenous tail injection. Any bright signal is due to Pc 4 fluorescence, without which no fluorescence signals were detected from the mouse. (To reduce autofluorescence, the animal was fed a special diet for more than 2 weeks before the experiment.) Unprecedented delivery efficiency and accumulation rate of the drug in the tumor are monitored via the fluorescence increase in the tumor area (white circle). For comparison, a mouse that got only a Pc 4 formulation without the Au NP vector injected is shown in panel (d). No circulation of the drug in the body or into the tumor was detectable 2 h after injection without the Au NP as drug vector.
Pc 4 Load and Stability of the Au NPs
Pc 4 can attach to the Au NPs surface through N–Au bonding by the terminal amine group on the Pc 4 axial ligand.22 Assuming a horizontal position of the Pc ring system on the Au NP surface, simple geometrical considerations show that a spherical area of 5.0 nm diameter NP can host a maximum of ∼100 Pc molecules (1.6 nm2) at complete coverage. It is crucial to balance drug load and solubility in water. We performed a series of experiments with different Pc 4-to-Au NP ratios. After being stirred for 24 h, the solution with>30% Pc on the Au NPs changed color, indicating aggregation of the Au NPs (Figure 3). This process was monitored and analyzed by UV–vis absorption spectrometry. It indicated that, when Pc 4 occupied more than 30% of the NP surface, the Au NPs started to aggregate quicker, and the shelf life of the solutions was reduced.
Figure 3.

Photograph of Au NP dispersions, which aggregated when the concentration of Pc 4 was increased in chloroform. Vial 1 was pure Au NPs as control. From vial 2 to vial 5, the [Pc]/[Au NP] concentration ratios were 20:1, 40:1, 50:1, and 60:1, respectively.
Au NPs have little absorption at 670 nm, while Pc 4 has its maximum absorption at this wavelength. In order to quantify the final Pc 4 concentration in the conjugates solution, we obtained a series of [Pc 4]/[Au NP] absorption spectra to study how much the Au NPs absorption added to the absorption of Pc 4 at 670 nm. The concentrations of Au NPs were kept constant, and Pc 4 solutions with defined concentrations were added in different volumes. While the optical density of Pc 4 at 670 nm was in the range of 0.1−0.8, the absorption of Au NPs was negligible. The concentration of Pc 4 per Au NP was then calculated on the basis of the Beer–Lambert law. On the basis of the extinction coefficients of the Au NPs (1.5 × 107 M−1 cm−1) and Pc 4 (2.7 × 10 7 M−1 m−1 in chloroform), one can estimate that there are ∼30 Pc molecules per Au NP.
The N–Au bond is relatively weak (6 kcal/mol) compared to the S–Au bond (47 kcal/mol).23 In order to get stable Au NP–Pc 4 conjugates in water, the randomly coiled PEG ligands on the NP surface are excellent candidates, since they not only provide a steric repulsion to separate Au NPs but also stabilize the Pc 4 through van der Waals interactions and protect the drug from the aqueous phase. These together provide a cage-type protection effect for the Pc molecule in a spatial sense, on the basis of which the lipophilic drug molecules can be transported to the site of action. These PEGylated Au NP–Pc 4 conjugates were stable in aqueous solution at least for 6 months. Using PEG molecules alone as the control showed that they could not transport the hydrophobic PDT drug into the aqueous solution.
Singlet Oxygen Quantum Yield
The solution containing the photosensitizer was stored in a 1 cm cuvette and saturated with O2 in the dark. Next, 9.1 × 10−8 mol DPBF solution was added, and the mixture was stirred on a magnetic stirrer at room temperature. The final volume in the cell was maintained at 2 mL, and the overall optical density of the solution was kept below 1.5, to be able to rely on the Beer–Lambert law (linear proportionality between absorbance and concentration). The laser power was ≤5 mW to ensure about 5% DPBF decay per 10 s irradiation interval. The experiment was repeated three times for each sensitizer.
| (1) |
| (2) |
ΦΔ was calculated on the basis of eqs 1 and 2.24 Superscript S and R indicate the sample and reference compound, respectively. Ia is defined as the total amount of light absorbed by the sensitizers. A is the corresponding absorbance at irradiation wavelength. After the data were plotted as –ln[DPBF]/[DPBF]0 versus irradiation time t, straight lines were obtained for the sensitizers, and the slope for each compound was obtained after fitting with a linear function (correlation coefficient R>0.99946), as shown in Figure 4. The 1O2 quantum yield (ΦΔ) of Pc 4 in ethanol was ∼50%. For Pc 4 on PEGylated Au NPs, a 1O2 quantum yield of ΦΔ = 35% was measured.
Figure 4.
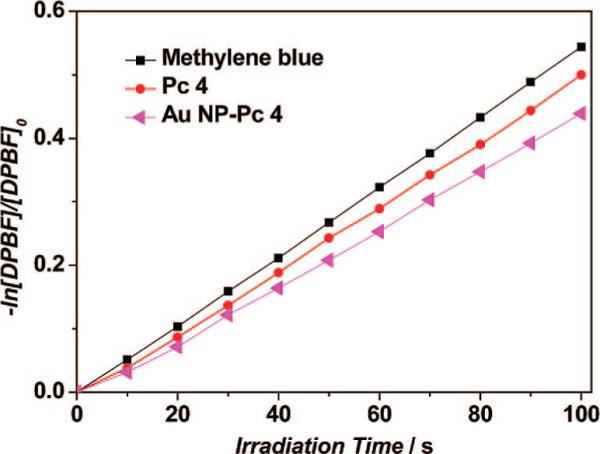
Photodecomposition of DPBF by 1O2 after irradiation of methylene blue, Pc 4, and Au NP–Pc 4 conjugates in ethanol (monitoring the maximum absorption of DPBF at 410 nm).
Phase Transfer Studies
PDT of Pc 4 was shown to take place in or at cell membranes.4b Therefore, it is particularly interesting to study the phase transfer efficiency of these conjugates in vitro before they are applied in vivo.25 A toluene–water phase was used as the model to monitor the release of the drug into a hydrophobic environment. Since Pc 4 preferentially locates in apolar sites,26 such as lipid membranes, the toluene–water system provides a simple and direct way to monitor the release dynamics, as illustrated in Figure 5.
Figure 5.
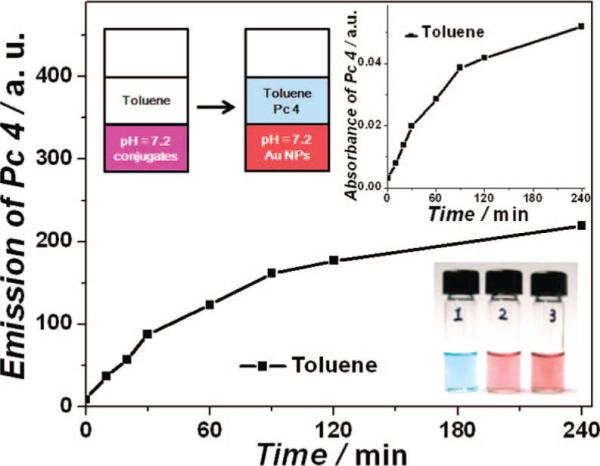
Phase transfer study of the conjugates at room temperature and color photographs of Pc 4 in toluene (1), Au NPs (2), and Au NP–Pc 4 conjugates (3) in normal saline (0.9% NaCl, pH 7.2) phases.
Pc 4 was slowly released from the Au NP surface. By monitoring the absorption and emission properties of the toluene phase over time, we observed an increase in the Pc 4 concentration, and drug release was very fast within the first 2 h. Only a trace amount of PEGylated Au NPs (2 × 10−10 M) was detected in the toluene phase, which was only 1% of the NP concentration in the aqueous phase (1.7 × 10−8 M). It implies that the adsorption of Pc 4 to the water-soluble Au NPs is reversible and that Pc 4 can be transferred into a lipophilic environment when applied in vivo. On the other hand, the PEGylated Au NPs themselves do not transfer into the organic phase but strongly prefer the aqueous phase once they are hydrated. In vitro cell viability using 10−6 M conjugate solution was greater than 95% after incubation overnight in the dark. Irradiation at>500 nm light caused selective photokilling of the cells with>90% efficiency.
In Vivo Drug Delivery
The successful phase-transfer studies in addition to the excellent biocompatibility of the conjugates inspired us to investigate this Au NP drug delivery vector for in vivo PDT. In general, Pc 4 is administrated in vivo through intravenous injection, and it takes about 2 days after injection in order to get the maximum drug distribution in the target tumor. The distribution of the PDT drugs can be detected by measuring their intrinsic fluorescence. In vivo study of mice revealed that the Pc 4 drug attached to PEGylated Au NPs was instantaneously transported through the blood vessels and accumulated at the tumor site within 2 h (Figure 6). Once Pc 4 is delivered and released from the Au NPs, the singlet oxygen yield is the same as for free Pc 4 without using the Au NP vector. Thus, the Pc 4–NP conjugate functions as a protected Pc 4 that is being released in the presence of a lipophilic environment, e.g., cell membranes, while being retained in the tumor tissue.
Location of the PDT Drug
As can be seen in Figure 6, the PDT drugs accumulated in the tumor site through a passive targeting process. The Au NP–Pc 4 conjugates were injected intravenously into the mouse's tail. PEG is one the very few capping materials that does not interact with serum proteins and can improve the circulation of drugs in the blood.11 When the PEGylated Au NP vector circulates in the body, it can escape uptake by RES. The entrapment of Au NPs by serum proteins was previously studied.27 The random coiled PEG molecules on Au NPs can sterically hinder the reaction with proteins in cell culture media or blood. The drug loaded on the PEGylated Au NP can be shielded from the serum proteins and can be delivered to the target tumor efficiently, as shown in Figure 6. Furthermore, since tumors lack an effective lymphatic system, the Au NP–Pc 4 conjugates can accumulate in tumors due to the inherent leaky tumor neovasculature, which enhances the permeability and retention of the nanosized particles. Pc 4 fluorescence was observed in the whole body of the mouse, with some localization in the lung and in the kidneys, as usual for phthalocyanines.17
Highly Efficient Delivery
A mouse's heart rate is extremely high (e.g., 600 beats per min), and its blood circulation is much faster than that of humans.28 This caused the PDT drug to circulate through the whole body and accumulate in the tumor within minutes. This accumulation effect in the tumor was clearly apparent immediately after injection (Figure 6a). Compared to conventional PDT drug delivery in vivo, PEGylated Au NPs accelerated the Pc 4 administration by about 2 orders of magnitude (Figure 6a,d). No apparent side effects were observed in PDT-treated mice. After treatment, the tumors became necrotic within 1 week after PDT, followed by shrinkage of the tumor size, which indicates the effect of the therapy.
Conclusions
In conclusion, a highly efficient drug vector for PDT drug delivery was developed by synthesizing PEGylated Au NP conjugates with a reversible PDT drug adsorption. This delivery mode greatly improved the transport of the drug to the tumor relative to conventional drug administration. The presented system is of unique versatility and provides a measurable benchmark, since it allows highly efficient drug delivery, quantitative monitoring of the delivery process, and cancer therapy with, to date, no noticeable toxicity to the animals or side effects. Delivery via the PEGylated Au NP vector can, in principle, be further specified for active targeting of tumor sites by conjugating receptor-specific targeting moieties onto the NP surface. Any future improvements of this PDT drug delivery process can thereby easily be monitored and quantified. The localization of the drug within cells is currently under study.
Acknowledgment
We thank Prof. M. E. Kenney for providing the Pc 4 sample. This research is supported by CWRU (PRI Award, PIs Fei and Burda), NSF (CHE-0239688, PI Burda), and NIH/NCI (CA120536, PI Fei).
References
- 1.Dolmans DEJGJ, Fukumura D, Jain RK. Nature Rev. Cancer. 2003;3:380. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 2.Allen CM, Sharman WM, Van Lier JEJ. Porphyrins Phthalocyanines. 2001;5:161. [Google Scholar]
- 3.Castano AP, Mroz P, Hamblin MR. Nature Rev. Cancer. 2006;6:535. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Teiten M, Marchal S, D'Hallewin MA, Guillemin F, Bezdetnaya L. Photochem. Photobiol. 2003;78:9. doi: 10.1562/0031-8655(2003)078<0009:ppsame>2.0.co;2. [DOI] [PubMed] [Google Scholar]; b Oleinick NL, Morris RL, Belichenko I. Photochem. Photobiol. Sci. 2002;1:1. doi: 10.1039/b108586g. [DOI] [PubMed] [Google Scholar]
- 5.Konan YN, Gurny R, Alléman E. J. Photochem. Photobiol. B. 2002;66:89. doi: 10.1016/s1011-1344(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 6.a Derycke ASL, deWitte PAM. Adv. Drug Delivery Rev. 2004;56:17. doi: 10.1016/j.addr.2003.07.014. [DOI] [PubMed] [Google Scholar]; b van Nostrum CF. Adv. Drug Delivery Rev. 2004;56:9. doi: 10.1016/j.addr.2003.07.013. [DOI] [PubMed] [Google Scholar]; c Regehly M, Greish K, Rancan F, Maeda H, Bohm F, Roder B. Bioconjugate Chem. 2007;18:494. doi: 10.1021/bc060158u. [DOI] [PubMed] [Google Scholar]; d Roy I, Ohulchanskyy TY, Pudavar HE, Bergey EJ, Oseroff AR, Morgan J, Dougherty TJ, Prasad PN. J. Am. Chem. Soc. 2003;125:7860. doi: 10.1021/ja0343095. [DOI] [PubMed] [Google Scholar]
- 7.Hone DC, Walker PI, Evans-Gowing R, FitzGerald S, Beeby A, Chambrier I, Cook MJ, Russell DA. Langmuir. 2002;18:2985. [Google Scholar]
- 8.Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Small. 2005;1:325. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 9.Daniel M-C, Astruc D. Chem. Rev. 2004;104:293. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 10.a Wuelfing WP, Gross SM, Miles DT, Murray RW. J. Am. Chem. Soc. 1998;120:12696. [Google Scholar]; b Liu Y, Shipton MK, Ryan J, Kaufman ED, Franzen S, Feldheim DL. Anal. Chem. 2007;79:2221. doi: 10.1021/ac061578f. [DOI] [PubMed] [Google Scholar]
- 11.Greenwald RB, Choe YH, McGuire J, Conover CD. Adv. Drug Delivery Rev. 2003;55:217. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 12.a Blättler TM, Pasche S, Textor M, Griesser HJ. Langmuir. 2006;22:5760. doi: 10.1021/la0602766. [DOI] [PubMed] [Google Scholar]; b Bearinger JP, Terrettaz S, Michel R, Tirelli N, Vogel H, Textor M, Hubbell JA. Nat. Mater. 2003;2:259. doi: 10.1038/nmat851. [DOI] [PubMed] [Google Scholar]
- 13.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Drug Delivery. 2004;11:169. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 14.Paciotti GF, Kingston DGI, Tamarkin L. Drug Dev. Res. 2006;67:47. [Google Scholar]
- 15.Oleinick NL, Antunez AR, Clay ME, Rihter BD, Kenney ME. Photochem. Photobiol. 1993;57:242. doi: 10.1111/j.1751-1097.1993.tb02282.x. [DOI] [PubMed] [Google Scholar]
- 16.Detty MR, Gibson SL, Wagner SJ. J. Med. Chem. 2004;47:3897. doi: 10.1021/jm040074b. [DOI] [PubMed] [Google Scholar]
- 17.Fei B, Wang H, Meyers JD, Feyes DK, Oleinick NL, Duerk JL. Lasers Surg. Med. 2007;39:723. doi: 10.1002/lsm.20576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman RJ. Chem. Soc., Chem. Comm. 1994;7:801. [Google Scholar]; b Duan H, Nie S. J. Am. Chem. Soc. 2007;129:2412. doi: 10.1021/ja067727t. [DOI] [PubMed] [Google Scholar]
- 19.Spiller W, Kliesch H, Wöhrele D, Hackbarth S, Röder B, Schnurpfeil G. J. Porphyrins Phthalocyanines. 1998;2:145. [Google Scholar]
- 20.Shimmin RG, Schoch AB, Braun PV. Langmuir. 2004;20:5613. doi: 10.1021/la036365p. [DOI] [PubMed] [Google Scholar]
- 21.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 22.Samia ACS, Chen X, Burda C. J. Am. Chem. Soc. 2003;125:15736. doi: 10.1021/ja0386905. [DOI] [PubMed] [Google Scholar]
- 23.Di Felice R, Selloni A. J. Chem. Phys. 2004;120:4906. doi: 10.1063/1.1645789. [DOI] [PubMed] [Google Scholar]
- 24.Musil Z, Zimcik P, Miletin M, Kopecky K, Petrik P, Lenco J. J. Photochem. Photobiol. A. 2007;186:316. doi: 10.1111/j.1751-1097.2007.00193.x. [DOI] [PubMed] [Google Scholar]
- 25.Hong R, Han G, Fernandez JM, Kim B.-j., Forbes NS, Rotello VM. J. Am. Chem. Soc. 2006;128:1078. doi: 10.1021/ja056726i. [DOI] [PubMed] [Google Scholar]
- 26.Morris RL, Azizuddin K, Lam M, Berlin J, Nieminen AL, Kenney ME, Samia ACS, Burda C, Oleinick NL. Cancer Res. 2003;63:5194. [PubMed] [Google Scholar]
- 27.a Homola J. Chem. Rev. 2008;108:462. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]; b Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat. Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qian Xi., Peng X, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S. Nat. Biotechnol. 2008;26:83. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 28.James JF, Hewett TE, Robbins J. Circ. Res. 1998;82:407. doi: 10.1161/01.res.82.4.407. [DOI] [PubMed] [Google Scholar]