

Abstract
Bacterial lipopolysaccharide (LPS) is a key mediator in the development of Gram-negative septic shock, which is a major health problem. The effect of LPS on myeloid cells is mediated by a multicomplex receptor system in which CD14, a glycosylphosphatidylinositol-anchored glycoprotein, and Toll-like receptor 4 are the major players. We have found that incubation of macrophages with itraconazole (ICZ), an azole antifungal commonly used in humans, altered both the expression and glycosylation of CD14. This glycoprotein, which is endo H-resistant in untreated cells, becomes endo H-sensitive following ICZ treatment. The effect of ICZ on glycan processing was observed in all newly synthesized glycoproteins as indicated by incorporation of [2-3H]mannose. In addition, cells treated with ICZ increased surface concanavalin A (ConA) binding, corroborating an increase in high mannose surface glycoproteins. Although the glycosylation pattern of CD14 was altered, this glycoprotein was delivered to the cell surface or was secreted. Moreover, it appeared functional as demonstrated by the release of LPS-induced tumor necrosis factor-α under conditions specific for a CD14-mediated activation process. The effect of ICZ on glycosylation was not dependent on inhibition of the cholesterol biosynthetic pathway and was specific for this drug because other azole antifungals, such as ketoconazole and econazole, did not alter glycan processing. These results suggest a possible secondary effect of ICZ that impacts the processing of glyconjugates and may alter cellular function and homeostasis.
Bacterial lipopolysaccharide (LPS),2 or endotoxin, is a component of the outer membrane of Gram-negative bacteria and induces a robust inflammatory response. LPS is a trigger for Gram-negative sepsis, which is a major heath problem in the United States (1, 2). LPS is a pathogen-associated molecular pattern and is recognized by specific surface receptors on myeloid cells. The major LPS binding site on macrophages is CD14, which is a glycosylphosphatidylinositol-anchored, leucine-rich repeat glycoprotein (3). The recognition of LPS by CD14 requires that the bacterial product associates first with LPS-binding protein. Then, the complex of CD14 and LPS interacts with Toll-like receptor 4 and MD-2 to initiate the release of cytokines and other effector molecules (4). The importance of CD14 in LPS signaling is illustrated by the fact that it enhances the recognition of LPS up to 1000-fold (5). Moreover, CD14-deficient mice are LPS insensitive and resistant to septic shock (6). In addition to the glycosylphosphatidylinositol-anchored membrane CD14 (mCD14), a soluble variant (sCD14) of this glycoprotein is found in the extracellular space and circulation.
We have previously found that the expression of mCD14 in macrophages (Mϕs) is enhanced by lovastatin, which is a statin widely used for the treatment of hypercholesteremia. This elevation in CD14 expression was not correlated with a decrease in cellular cholesterol levels, but, rather, it was partially dependent on the depletion of geranylgeranylpyrophosphate (7), which is a non-sterol isoprenoid product of the cholesterol biosynthetic pathway. These observations illustrate that the statin-mediated inhibition of cholesterol biosynthesis has several additional side effects. This pathway is responsible for the synthesis of several important cellular products, such as the isoprenoid intermediates, which are involved in the post-translational modification of proteins. Consequently, the effect of statins on cellular physiology needs to be confirmed by using additional inhibitors for other steps in the cholesterol biosynthetic pathway. In an effort to understand if endogenously synthesized cholesterol affects CD14 expression, we used itraconazole (ICZ), which inhibits the cytochrome P450 (CYP450) enzyme 14α-demethylase, a key step in the conversion of lanosterol to cholesterol. This step in the cholesterol pathway is downstream of the isoprenoid intermediates, avoiding the inhibitory effects on isoprenoid moieties. We found that treatment of Mϕs with ICZ altered the processing of glycoproteins from high mannose to complex structures. Thus, cells treated with ICZ displayed an enrichment of high mannose glycoproteins on the surface. Moreover, this effect of ICZ was independent of the cholesterol pathway and unique to this drug. This dramatic alteration in glycosylation may have a tremendous impact on cellular function and may be pertinent to patients taking ICZ.
EXPERIMENTAL PROCEDURES
Materials
RAW 264.7 Mϕs were obtained from American Type Culture Collection and maintained in RPMI 1640 containing 10% fetal bovine serum and penicillin (50 IU/ml)/streptomycin (50 μg/ml). ICZ, econazole, ketoconazole, NB-598, MTT (thiazolyl blue tetrazolium bromide), lipoprotein-deficient fetal calf serum, 2,5-diphenyloxazole, and Escherichia coli LPS (serotype 026:B6, lot 023K4116) were obtained from Sigma. ICZ was also obtained from LKT Laboratories along with fluconazole. PNGase F, endo H, and sialidase were from New England BioLabs. Lovastatin was obtained from Calbiochem. TNF-α was measured with a mouse enzyme-linked immunosorbent assay (ELISA) kit from BIOSOURCE/Invitrogen. The CD14 antibody (clone rmC5–3) was from Pharmingen. The β-Actin antibody (monoclonal clone AC-15) was from Sigma. Horseradish peroxidase-conjugated concanavalin A was from Sigma. Fluorescein isothiocyanate-conjugated concanavalin A and α-methylmannoside were from EY Laboratories. Biotinylated Phaseolus vulgaris erythroagglutinin (PHA-E) and horseradish peroxidase avidin D were from Vector Laboratories. d-[2-3H]Mannose was from American Radiolabeled Chemicals, Inc. Complete mini EDTA-free protease inhibitor mixture pellets were obtained from Roche. RNA isolation was done with TRIzol reagent from Invitrogen.
Immunoblot Analysis
Following treatments, RAW 264.7 Mϕs were washed once in ice-cold PBS and scraped in ice-cold lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100) containing the complete mini EDTA-free protease inhibitor mixture from Roche. Protein concentration was determined using the BCA protein assay kit from Pierce. Cell lysates were mixed with 4× SDS-PAGE loading buffer and 50 μg of total protein was separated by SDS-PAGE using 10% slab gels. The separated proteins were transferred to nitrocellulose membranes and blocked in 5% milk in TBS, 0.1% T (100 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) for 1 h at room temperature. Blots were probed with the rat anti-mouse CD14 antibody (1:500) or the mouse β-Actin antibody (1:5000) in 5% milk TBS, 0.1% T at 4 °C overnight followed by three 10-min washes in TBS, 0.1% T at room temperature. Blots were then incubated with anti-rat or anti-mouse horseradish peroxidase antibodies (1:25,000) in 5% milk in TBS, 0.1% T for 2 h at room temperature. After three 10-min washes in TBS, 0.15% T, bands were detected by chemiluminescence using SuperSignal reagents from Pierce. Following detection, blots were stripped by washing 5 min in dH2O, 10 min in 0.2 m NaOH, and 5 min in dH2O.
Lectin Blotting and Flow Cytometry
Following treatments, RAW 264.7 Mϕs were washed once in ice-cold PBS and scraped in ice-cold lysis buffer containing the complete mini EDTA-free protease inhibitor mixture from Roche. Protein concentration was determined using the BCA protein assay kit from Pierce. Cell lysates were mixed with 4× SDS-PAGE loading buffer and 50 μg of total protein was separated by SDS-PAGE using 10% slab gels. The separated proteins were transferred to nitrocellulose membranes and blocked in 5% bovine serum albumin in TBS, 0.1% T. Blots were then incubated with 1 μg/ml horseradish peroxidase-conjugated concanavalin A (ConA) or 2.5 μg/ml biotinylated PHA-E overnight followed by four 10-min washes in TBS, 0.1% T. Following overnight incubation and washing, the biotinylated lectin was incubated for 1 h at room temperature with 2 μg/ml horseradish peroxidase avidin D in 5% bovine serum albumin in TBS, 0.1% T followed by four 10-min washes at room temperature in TBS, 0.1% T. Bands were detected by chemiluminescence using SuperSignal reagents from Pierce. Following detection, blots were stripped by washing 5 min in dH2O, 10 min in 0.2 m NaOH, and 5 min in dH2O, and re-probed for β-Actin.
For flow cytometry, RAW 264.7 Mϕs were treated with itraconazole (1 μm) for 16 h at which time cells were washed twice in ice-cold PBS. Approximately 1 million cells were incubated with 100 μg/ml fluorescein isothiocyanate/ConA for 15 min at room temperature followed by 3 washes in ice-cold PBS. Cells were then fixed in 1% paraformaldehyde in PBS and examined by flow cytometry. To determine specificity for ConA binding, cells were coincubated with α-methylmannoside.
RNA Isolation and Real Time PCR
RNA was isolated from RAW 264.7 Mϕs using TRIzol reagent according to the manufacturer's instructions. For real time PCR, RNA was reverse transcribed using the High Capacity cDNA Archive Kit from Applied Biosystems. The relative quantities of CD14 and Toll-like receptor 4 mRNA were quantified by real time reverse transcriptase-PCR using primers and probes developed by Applied Biosystems (TaqMan gene expression assays). The samples were each tested at least in triplicate and were normalized with glyceraldehyde-3-phosphate dehydrogenase mRNA. Data were analyzed using the comparative CT method and were confirmed by the standard curve method.
Isolation of Peritoneal Macrophages
Mice were anesthetized using isofluorane and sacrificed by cervical dislocation. Peritoneal Mϕs were isolated from C57BL6/J mice by peritoneal lavage using 5 ml of serum-free RPMI 1640 containing penicillin/streptomycin (medium A). Cells were collected by centrifugation and plated at a density of ∼1 × 106 cells/well in a 6-well Falcon tissue culture dish. Cells were allowed to adhere in medium A in a 37 °C, 5% CO2 incubator for 1.5 h and washed two times in PBS. The medium was replaced with RPMI 1640 containing 10% fetal bovine serum and penicillin/streptomycin (medium B). Subsequently, cells were stimulated in medium B for 16 h with 5 μm itraconazole. All animal procedures were part of protocols reviewed and approved by The Johns Hopkins Medical Institutions Animal Care and Use Committee.
ELISA
The concentration of TNF-α in culture supernatants was determined using mouse TNF-α ELISA kits according to the manufacturer's instructions. Culture supernatants were collected and frozen at −80 °C until analysis. After harvesting the supernatants, an MTT cytotoxicity assay was performed to assess viability and plating error. MTT (5 mg/ml) in PBS (filter sterilized) was diluted to 1.25 mg/ml in medium B, and 200 μl of this diluted solution was applied to the cells and incubated for 1 h. The MTT-containing medium was then removed and the formazan crystals were dissolved in 200 μl of DMSO plus 25 μl of Sorensen's buffer (0.1 m glycine, 0.1 m NaCl, pH 10.5)/well. The plate was then read in a microtiter plate reader at 560 nm.
Cell Surface Biotinylation
The Cell Surface Protein Biotinylation and Purification Kit from Pierce was used to isolate cell surface CD14 according to the manufacturer's instructions. Following cell lysis, an aliquot was removed for determination of total CD14. Following isolation, both total CD14 and cell surface CD14 were analyzed by Western blot analysis.
[2-3H]Mannose Labeling of N-Glycosylated Proteins
RAW 264.7 Mϕs were incubated with itraconazole (1 μm) for 16 h at which time cells were preincubated with low glucose RPMI 1640 (1 mm glucose) containing itraconazole (1 μm) for 30 min. 100 μCi of [2-3H]mannose (20–30 Ci/mmol) was then added for 7 h. Protein was harvested in lysis buffer containing protease inhibitors. One-third of the lysate was digested with PNGase F, and 1/3 was digested with endo H according to the manufacturer's instructions. Then, 75 μg of protein was separated on a 10% slab gel by SDS-PAGE. The gel was washed for 10 min in dH2O followed by two 20-min washes in DMSO. It was then incubated with 20% 2,5-diphenyloxazole in DMSO for 3 h at room temperature to enhance the tritium signal. The gel was then washed 4× 15 min in dH2O, dried, and exposed to film.
2-Aminobenzamide HPLC Analysis
Glycolipids were removed from cell pellets by two extractions each sequentially with chloroform/methanol (2:1), (1:1), (1:2), followed by one extraction with ethanol. Extracts were resuspended in buffer (0.1 m Tris, pH 7.4, 1% SDS), dialyzed and digested with trypsin/PNGase F. Glycopeptides were released using Sep-Pak® C18 and PGC Hypersep Hypercarb columns. 100 μg of N-linked glycans were derivatized using 2-aminobenzamide, and 30 μg were subjected to HPLC analysis. This analysis was performed by the University of California, San Diego, Glycoconjugate Analysis Core.
Analysis of Sterols by Gas Chromatography
Following treatment, confluent 60-mm plates of RAW 264.7 Mϕs were washed twice in ice-cold PBS and scraped in 1 ml of ice-cold PBS. A 100-μl aliquot of each sample was removed to measure protein levels and cells were pelleted at 800 × g for 5 min. Cell pellets were mixed with 9 ml of methanol and transferred to glass tubes. As an internal standard, 5 μg of ergosterol was added followed by the addition of 4.5 ml of 60% KOH. Samples were vortexed and saponified in a 75 °C shaking water bath for 2 h. After cooling, 4 ml of petroleum ether was added and samples were vortexed. Following separation, the top layer was transferred to a new glass tube and dried under nitrogen. Sterols were resuspended in heptane and analyzed by gas chromatography.
RESULTS
Itraconazole Treatment Results in Altered Glycosylation and Increased Expression of CD14
RAW 264.7 Mϕs were treated with ICZ (1 μm) for 16 h in media containing 10% fetal bovine serum. The electrophoretic mobility of CD14 was altered as visualized by Western blotting following treatment with ICZ (Fig. 1a, lanes 1 and 2). Because CD14 is a glycoprotein with four potential N-linked glycosylation sites, the change in electrophoretic mobility of CD14 after ICZ treatment could be due to a modification in the glycosylation pattern. To test this possibility, samples of cells treated or not with ICZ were digested with PNGase F, which removed all N-linked oligosaccharides. The products were analyzed by Western blotting using an antibody against CD14. Samples from both the control and ICZ-treated cells showed the same pattern following PNGase F digestion, suggesting that, indeed, the alteration in CD14 mobility was the result of altered N-linked glycosylation after ICZ treatment (Fig. 1a, lanes 3 and 4). Digestion with endo H, which cleaves high mannose oligosaccharides, demonstrated that CD14 was completely sensitive to endo H after ICZ treatment, whereas it was mainly resistant in untreated cells (Fig. 1b). Consistent with the endo H sensitivity after ICZ treatment, CD14 was also resistant to digestion with sialidase, confirming the lack of complex glycans containing sialic acid resides (Fig. 1c). In addition to an alteration in N-linked glycosylation of CD14 after treatment with ICZ, the expression of this glycoprotein was increased at the protein level (2.9-fold) as well as the mRNA level (2.8-fold) after treatment with the drug (Fig. 1, d and e).
FIGURE 1.

Itraconazole treatment results in altered glycosylation and increased expression of CD14. RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h and lysed. Lysates were digested or not with PNGase F (a and b), endo H (b), or sialidase (c) followed by detection of CD14 by Western blotting. Notice that the electrophoretic migration of CD14 after PNGase F or endo H digestion is slightly different, which is consistent with the fact that the first enzyme removes all sugars from the glycoproteins, whereas endo H leaves a GlcNac residue. d, CD14 protein levels were quantitated by densitometry of the Western blot bands and corrected by β-Actin levels. The results represent the average of three separate samples (p < 0.001, t test). e, CD14 mRNA levels were quantitated by real time PCR, corrected by glyceraldehyde-3-phosphate dehydrogenase levels, and normalized to control. The results represent the average of three separate samples (p < 0.001, t test).
N-Linked glycosylation is initiated by the addition of a precursor oligosaccharide containing dolichol to asparagine residues in target proteins. Dolichol is synthesized from farnesylpyrophosphate, an isoprenoid product of the cholesterol biosynthetic pathway. It is therefore possible that the transfer of the precursor oligosaccharide to proteins is inhibited in the presence of ICZ. Murine CD14 has four potential sites for N-linked glycosylation, three of which are known to be utilized (8, 9). The number of utilized N-linked glycosylation sites in CD14 in the presence or absence of ICZ was evaluated by partial digestion with endo H. Four distinct bands were identified following partial digestion with endo H in ICZ-treated cells (Fig. 2), indicating that all sites are modified and correspond to unprocessed glycans.
FIGURE 2.

All glycosylation sites are utilized in the presence of itraconazole. RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h and lysed. The lysates were digested with increasing amounts of endo H followed by detection of CD14 by Western blotting (arrows indicate migration of the partially digested glycoprotein).
CD14 Was Present on the Cell Surface and Released as a Soluble Variant after Treatment of Macrophages with ICZ
One possible explanation for the altered glycosylation of CD14 in ICZ-treated Mϕs is that ICZ inhibits the trafficking of CD14 along the secretory pathway. RAW 264.7 Mϕs were treated with ICZ (1 μm) for 16 h and then surface proteins were biotinylated, separated by affinity chromatography on immobilized streptavidin, and the presence of CD14 was detected by Western blotting. Similar levels of surface CD14 were observed in cells treated or not with ICZ (28.1 versus 28.6%, Fig. 3a). In addition, there were no differences in the release of sCD14 from cells treated or not with ICZ (Fig. 3b). Glycosylation of CD14 that was released from cells after ICZ treatment was also altered, as demonstrated by differences in electrophoretic mobility in comparison with untreated cells. Stimulation of ICZ-treated cells with LPS (100 ng/ml) resulted in an increase of both membrane-bound and soluble CD14 levels, both with altered glycosylation (Fig. 3b). These results suggest that the traffic of CD14 was not affected by treatment with ICZ.
FIGURE 3.

CD14 is present on the cell surface and released as a soluble variant in itraconazole-treated macrophages. a, RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h followed by biotinylation and purification of cell surface proteins (Pierce Cell Surface Biotinylation kit). Fractions of both total lysates and cell surface proteins were analyzed by Western blot for CD14. Bands were quantitated by densitometry and % cell surface CD14 was calculated. b, RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h followed by stimulation with 100 ng/ml LPS for additional amounts of time. Both lysates and supernatants were collected and CD14 was analyzed by Western blot.
Itraconazole Treatment Results in Altered Lectin Binding to Macrophage Glycoproteins
ConA is a lectin that recognizes mannose moieties, whereas PHA-E recognizes complex carbohydrates. RAW 264.7 Mϕs were treated with ICZ (1 μm) for 16 h, lysed, and the glycoprotein patterns were analyzed by lectin blot analysis with ConA and PHA-E. Increased binding of ConA was observed in samples from ICZ-incubated cells as compared with control cells. ConA binding was blocked by coincubation of the lectin with α-methylmannoside (Fig. 4A). This result suggests an increase in the content of high mannose glycans after ICZ treatment. Consistent with this observation, a decrease in binding of PHA-E was observed in samples from ICZ-treated cells with respect to control cells (Fig. 4B), indicating a reduction of complex carbohydrates on glycoproteins in ICZ-treated cells. In addition, lysates obtained from primary C57BL/6J peritoneal Mϕs that were treated with ICZ (5 μm) for 16 h in culture conditions displayed increased binding of ConA with respect to untreated cells (Fig. 4C). To determine whether the increased binding of ConA also occurs with cell surface glycoproteins, RAW 264.7 Mϕs were treated with ICZ (1 μm) for 16 h, stained with fluorescein isothiocyanate-conjugated ConA, and examined by flow cytometry. An increase in ConA cell surface binding was observed after ICZ treatment as compared with untreated cells, and ConA binding was specific as demonstrated by competition with α-methylmannoside (Fig. 4C).
FIGURE 4.

Itraconazole treatment results in altered lectin binding to macrophage proteins. RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h followed by ConA in the presence or absence of α-methylmannoside (A) or PHA-E (B) lectin blot analysis. Blots were stripped and re-probed for β-Actin as a loading control. Primary C57BL/6J peritoneal Mϕs were treated with itraconazole (5 μm) or the respective carrier (DMSO) for 16 h followed by ConA lectin blot analysis. The blot was stripped and re-probed for β-Actin as a loading control (C). RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h followed by staining with fluorescein isothiocyanate-conjugated ConA (100 μg/ml) in the presence or absence of α-methylmannoside (α-MM) (1 mg/ml) and analyzed by flow cytometry (D).
No Complex Modified Carbohydrates Were Produced in the Presence of ICZ
To determine whether the effect of ICZ on glycosylation was global, RAW 264.7 Mϕs were treated with ICZ (1 μm) for 16 h, and cells were pulse-labeled with [2-3H]mannose (10). All newly synthesized N-linked glycoproteins in the presence of ICZ treatment were sensitive to endo H digestion, whereas glycoproteins from untreated cells were resistant to endo H (Fig. 5). This observation indicates that the processing from high mannose to complex structures is blocked by treatment with ICZ. These findings were further investigated by 2-aminobenzamide HPLC analysis of total glycans isolated from cells treated or not with ICZ. The presence of high mannose oligosaccharides was determined by comparison with high mannose standards as well as the pattern of ribonuclease B. The levels of mannose structures, such as Man 5 and 6, were elevated in samples obtained from ICZ-treated cells in comparison with untreated cells (Fig. 6A, see arrows 2 and 3). After mannosidase digestion, the level of Man 3 was higher in samples of cells treated with ICZ than in control cells (Fig. 6B, see inset). The limitation of the 2-aminobenzamide HPLC analysis is that it reflects the total pattern of glycoproteins from cells, including newly synthesized proteins as well as glucoconjugates with a long half-life. Only newly synthesized glycoproteins are affected by ICZ treatment, whereas glucoconjugates with a long half-life are not due to the limited incubation time with the drug. Nevertheless, this analysis, in combination with lectin binding and metabolic labeling, supports the assumption that ICZ treatment has an impact, directly or indirectly, on glycoprotein processing.
FIGURE 5.

No complex-modified carbohydrates are produced in the presence of itraconazole. RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h in complete media. Cells were then incubated in low glucose (1 mm) media containing itraconazole (1 μm) for 30 min followed by the addition of 100 μCi of [2-3H]mannose for an additional 7 h. Fractions of each lysate was digested with PNGase F or endo H followed by SDS-PAGE. The gel was treated with 20% 2,5-diphenyloxazole in DMSO to enhance the tritium signal. After exposure, the gel was washed in DMSO and subsequently stained with Coomassie Blue to assess loading.
FIGURE 6.

High mannose oligosaccharides are increased in itraconazole-treated cells. RAW 264.7 Mϕs were incubated with ICZ (1 μm) or the respective carrier (DMSO) for 16 h at which time glycolipids were removed by sequential extractions with chloroform/methanol (2:1), (1:1), (1:2), followed by one extraction with ethanol. Extracts were then dialyzed and digested with trypsin/PNGase F. Glycopeptides were released and N-linked glycans were derivatized using 2-aminobenzamide and subjected to HPLC analysis. High mannose standards as well as ribosilase B were used to identify the high mannose oligosaccharide moieties (A). Arrows and numbers represent the different high mannose oligosaccharide moieties: 1, Man 4; 2, Man 5; 3, Man 6; 4, Man 7; 5, Man 8; 6, Man 9. Samples were treated with mannosidase and also examined by 2-aminobenzamide HPLC; insets display mannosidase treatment products (B). The analysis was performed by the University of California, San Diego Glycotechnology Core Facility.
Alterations in CD14 Glycosylation Are Independent of the Cholesterol Pathway in ICZ-treated Macrophages
To understand the cause of the altered glycosylation following ICZ treatment, the levels of different sterols were quantitated by gas chromatography in samples obtained from control or ICZ-treated (1 μm, 16 h) Mϕs. No significant change in cholesterol levels was detected between samples from ICZ-treated and non-treated Mϕs (Table 1). However, an increase in lanosterol was observed in samples from ICZ-treated cells, which is consistent with the step in the cholesterol pathway that is blocked by this drug (Table 1). To test whether this accumulation of lanosterol could be responsible for the alteration in glycosylation of CD14 after ICZ treatment, Mϕs were coincubated with ICZ (1 μm) and lovastatin (10 μm) or NB-598 (30 μm) for 16 h. Lovastatin and NB-598 competitively inhibit hydroxymethylglutaryl-CoA reductase and squalene epoxidase, respectively. Because these two enzymes are upstream of lanosterol in the pathway, the build-up of lanosterol should be blocked under these conditions. Indeed, coincubation with these drugs reverted the build-up of lanosterol observed in the presence of ICZ alone (Table 1). However, they did not block the alteration in CD14 observed after ICZ treatment (Fig. 7). These results indicate that the accumulation of lanosterol is not responsible for the alteration of CD14 glycosylation. Moreover, treatment with NB-598 (30 μm) or incubation of cells in 5% lipoprotein-deficient serum for 16 h resulted in a decrease of cholesterol levels as compared with untreated cells (Table 1). These conditions of reduced cellular cholesterol levels did not alter glycosylation of CD14 ((Fig. 7) data not shown for 5% lipoprotein-deficient serum), suggesting that depletion of cholesterol is not responsible for the effect of ICZ on glycosylation.
TABLE 1.
Sterol analysis of RAW 264.7 macrophages
Cells were treated with itraconazole (1 μm), lovastatin (10 μm), NB-598 (30 μm), 5% lipoprotein-deficient serum or itraconazole (1 μm) + lovastatin (10 μm) or NB-598 (30 μm) or the respective carrier (DMSO) for 16 h. Sterols were saponified by incubation of cell pellets in MeOH, 60% KOH (2:1), extracted and analyzed by gas chromatography. Ergosterol was added as an internal standard, and samples were corrected by both ergosterol for extraction and protein levels for input.
| Treatment | Cholesterol | Lanosterol |
|---|---|---|
| Control | 11.42 ± 1.12 | |
| Carrier | 13.17 ± 1.42 | |
| Itraconazole (1 μm) | 11.69 ± 2.45 | 0.63 ± 0.16 |
| Lovastatin (10 μm) | 12.31 ± 1.14 | |
| NB-598 (30 μm) | 8.84 ± 0.83a | |
| Itraconazole + lovastatin | 11.17 ± 1.20 | |
| Itraconazole + NB-598 | 7.83 ± 0.22a | |
| 5% LPDS | 7.29 ± 0.45a |
a p < 0.05 versus control and carrier by t test.
FIGURE 7.
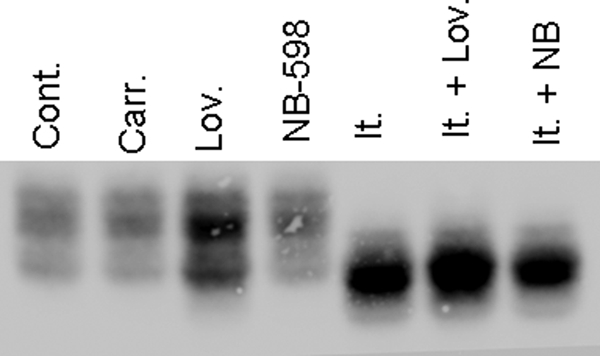
Alterations in CD14 glycosylation are independent of the cholesterol pathway in itraconazole-treated macrophages. RAW 264.7 Mϕs were treated with itraconazole (1 μm), lovastatin (10 μm), NB-598 (30 μm), or itraconazole (1 μm) + lovastatin (10 μm) or NB-598 (30 μm) or the respective carrier (DMSO) for 16 h and lysed. Lysates were separated (equal amount of protein) by SDS-PAGE, and the presence of CD14 was analyzed by Western blotting.
Other Azoles Do Not Affect Glycosylation of CD14
We next investigated whether the effect of ICZ on glycosylation was specific for this azole. Cells were treated with ketoconazole or econazole following similar protocols to the one used for ICZ. Both treatments raised lanosterol levels without depleting cholesterol levels (Table 2), and neither azole had an effect on glycosylation of CD14 (Fig. 8). These results indicate that the change in glycosylation observed in CD14 after ICZ treatment is likely due to a mechanism that is not common to all azoles.
TABLE 2.
Sterol analysis of macrophages treated with ketoconazole and econazole
RAW 264.7 Mϕs were treated with ketoconazole (1 μm), econazole (10 μm), or the respective carrier (DMSO) for 16 h. Sterols were saponified by incubation of cell pellets in MeOH, 60% KOH (2:1), extracted and analyzed by gas chromatography. Ergosterol was added as an internal standard, and samples were corrected by both ergosterol for extraction and protein levels for input.
| Treatment | Cholesterol | Lanosterol |
|---|---|---|
| Econazole carrier | 13.17 ± 1.42 | 0 |
| Econazole (10 μm) | 12.74 ± 1.12 | 0.72 ± 0.07 |
| Ketoconazole carrier | 14.00 ± 1.89 | 0 |
| Ketoconazole (1 μm) | 15.26 ± 1.32 | 0.66 ± 0.07 |
FIGURE 8.
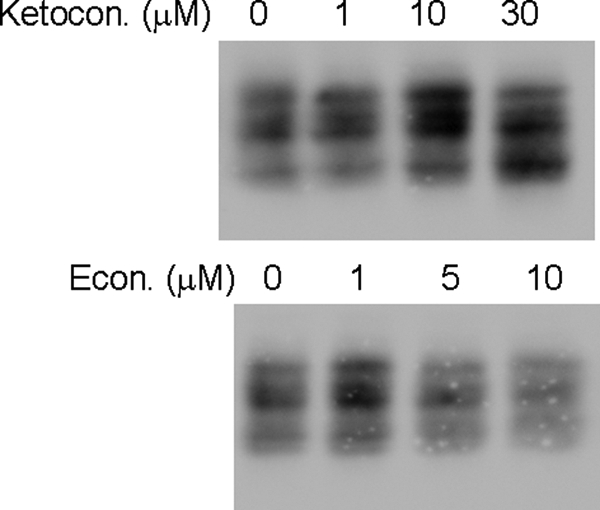
Altered glycosylation is not an effect of all azoles. RAW 264.7 Mϕs were treated with different doses of ketoconazole and econazole or the respective carrier (DMSO) for 16 h and lysed. Lysates (equal amount of protein) were separated by SDS-PAGE and the presence of CD14 was analyzed by Western blotting.
Itraconazole Treatment Results in Increased TNF-α Levels following LPS Stimulation
Finally, we tested whether the altered glycosylated CD14 was functional. RAW 264.7 Mϕs were incubated with ICZ (1 μm) for 16 h followed by stimulation with LPS at a concentration of 1 or 100 ng/ml for 5 h. Following LPS stimulation, the extracellular medium was collected and assayed for TNF-α by ELISA. ICZ treatment resulted in increased TNF-α levels as compared with control cells at 1 ng/ml LPS, but this effect was not observed at a concentration of 100 ng/ml LPS (Fig. 9). Low doses of LPS, such as 1 ng/ml, are specific for the stimulation of macrophages via CD14 (7), whereas large doses of LPS involve other receptors, such as Toll-like receptor 4. The increase in the release of LPS-induced TNF-α at lower concentrations of LPS in ICZ-treated cells is consistent with the increase in CD14 levels observed after incubation with this drug. This observation suggests that despite the alteration in CD14 glycosylation, this glycoprotein seems to be functional.
FIGURE 9.

Itraconazole treatment results in increased TNF-α levels following LPS stimulation. RAW 264.7 Mϕs were treated with itraconazole (1 μm) or the respective carrier (DMSO) for 16 h, then cells were incubated with LPS at two concentrations, 1 or 100 ng/ml LPS, for 5 h. The extracellular medium was collected, and TNF-α levels were determined by ELISA. TNF-α levels were corrected by the number of cell in the well by the MTT assay (*, p = 0.009 and **, p = 0.04 by t test).
DISCUSSION
ICZ is an azole antifungal agent widely used in humans. Although this drug has a broad spectrum of activity, it is particularly useful in the treatment of Aspergillus (11). ICZ inhibits fungal CYP450 oxidase-mediated synthesis of ergosterol, resulting in cell growth arrest. In mammals, ICZ is an inhibitor of the CYP450 enzyme lanosterol 14α-demethylase, which is involved in the conversion of lanosterol to cholesterol. In the present study, we found that treatment of murine Mϕs with ICZ at pharmacological doses resulted in a dramatic alteration in the processing of newly synthesized N-linked oligosaccharides. Thus, treatment with ICZ resulted in an increase in high mannose structures. The preponderance of high mannose glycans and lack of conversion into complex structures was demonstrated by sensitivity to endo H digestion of newly synthesized glycoproteins after incorporation of [2-3H]mannose in cells treated with ICZ. In addition, the levels of high mannose glycan were elevated in cells treated with ICZ in comparison with control cells. Moreover, cells treated with ICZ displayed an elevated content of high mannose glycoconjugates on the surface as demonstrated by ConA binding.
Surface glycoconjugates have been shown to play critical roles in the immune response, in particular during T-cell differentiation (12). Thus, changes in the pattern of cell surface glycoproteins have been observed during the maturation of thymocytes (13). Naïve (immature) CD8+ cortical thymocytes, which typically undergo an addition of sialic acid to the O-linked glycan Galβ1–3GalNacαThr/Ser during maturation, showed an enhancement of apoptosis, resulting in a reduction in their population in sialyltransferase (ST3Gal-I/ST3Gal-I)-null mice (14). The absence of fucosyltransferase (FucT-VII) resulted in defects in selectin-mediated T-cell trafficking (15). Alterations in N-linked glycosylation have also been linked to alterations in the immune response, in particular with autoimmunity. Ablation of N-acetylglucosaminyltransferase V (Mgat5) results in a lack of candidate structures for polylactosamine and increased T-cell receptor signaling due to enhanced T-cell receptor clustering (16), resulting in autoimmune disease. It was recently demonstrated that lack of polylactosamine (a fundamental structure in both N- and O-glycans) in β1,3-N-acetylglucosaminyltransferase (β3GnT2)-deficient mice resulted in increased T-cell activation, hyperproliferation upon B-cell receptor stimulation, and enhanced activation of macrophages in response to endotoxin (17). Finally, a dramatic remodeling of N-glycans on the surface of CD4 and CD8 T-cells has been demonstrated following activation of these cells, which is due to alterations in the expression of key glycosyltransferases (18). Mice deficient in α-mannosidase II, which displays reduced expression of complex N-glycans, develop autoimmune disease similar to human systemic lupus erythematosus (19). This effect has recently been attributed to the presence of immune stimulatory mannose ligands for innate immune lectin receptors (20). We showed that ICZ, at clinically relevant concentrations (21), resulted in a change of surface glycoproteins from complex to high mannose. Thus, it is possible that prolonged treatment with this drug may also result in the occurrence of autoimmune disease in patients.
The effect of ICZ on glycosylation could not be correlated with a decrease in cellular cholesterol levels, because reduction of this sterol by treatment with NB-598 or incubation in lipoprotein-deficient serum did not affect glycoprotein processing. Moreover, the increase in high mannose glycans was not the product of lanosterol build-up, secondary to inhibition 14α-demethylase, as demonstrated by coincubation of ICZ with either lovastatin or NB-598, which are inhibitors of hydroxymethylglutaryl-CoA reductase and squalene epoxidase, respectively. Treatment with other azoles, such as ketoconazole or econazole, did not alter glycosylation processing, suggesting that the effect of ICZ is not due to the inhibition of another CYP450 enzyme, such as epoxygenase (22). Taken together, these results suggest that the effect of ICZ on glycosylation is distinctive and independent of the cholesterol biosynthetic pathway.
ICZ has been shown to have unique effects on other systems as well. ICZ, but not ketoconazole, was found to demonstrate synergy with resident peritoneal murine macrophages in killing Blastomyces dermatitidis (23). ICZ and its derivative hydroxyitraconazole, but not econazole, fluconazole, miconazole, or ketoconazole were also shown to induce morphological changes in macrophages as determined by light and scanning electron microscopy (24). Finally, ICZ has recently been shown to inhibit angiogenesis in human endothelial cells, an effect that was unique to ICZ among all the azoles tested (25). It is possible that these observed effects of ICZ may be due to its ability to alter glycosylation and therefore function of cellular proteins.
We observed a significant accumulation of high mannose glycans, in particular Man 5 and 6, in cells treated with ICZ in comparison to untreated cells. Because Man 5 and 6 precede the addition of GlcNAc by GlcNAc transferase I, we speculate that ICZ may directly or indirectly influence the activity of this glycosyltransferase. However, we cannot discard the possibility that ICZ may affect the transport of UDP-GlcNAc across the Golgi membrane or mannosidase II activity. Changes in calcium levels may also be responsible for the effect of ICZ on glycan processing. ICZ has been shown to directly inhibit 5′-lipoxygenase, blocking the production of leukotriene B4, and subsequently interfering with store-operated uptake of calcium by activated neutrophils (26). Calcium has been demonstrated to be required for optimal processing of glycoproteins by the endoplasmic reticulum, whereas depletion of manganese inhibits O-linked and N-linked protein glycosylation in mammalian cells (27–29). Thus, the precise site of ICZ action responsible for the alteration of glycoprotein processing remains to be elucidated.
In addition to altered glycosylation, we also found that CD14 expression was increased on both the mRNA and protein levels in the presence of ICZ. This increase in CD14 expression was correlated with an elevation of TNF-α production at low LPS concentrations, which are specific for CD14-dependent macrophage activation. Lack of polylactosamine, a complex N- and O-glycan modification, in β3GnT2-deficient mice resulted in peritoneal macrophages with higher CD14 expression and enhanced responses to LPS. Polylactosamine was not detected on receptors for LPS, including CD14, by lectin microarrays (17). It is therefore possible that lack of polylactosamine on some other macrophage protein(s) results in up-regulation of CD14 expression. This is a possible explanation for the increased expression of CD14 observed following ICZ treatment of RAW 264.7 Mϕs, because ICZ should also deplete polylactosamine.
Although ICZ is extensively used with great safety, a small risk of liver and congestive heart failure has been observed. However, this situation could change with the prolonged use of this drug or in the case of immunocompromised patients, such as patients receiving organ transplants or suffering from AIDS. In fact, a common cause of death in immunocompromised patients is invasive aspergillosis (30). Treatment with azoles, such as ICZ for this condition, could be potentially harmful. Thus, prolonged treatment with ICZ could result in a dramatic alteration of surface glycoprotein patterns, which is likely to affect cellular function, homeostasis, and the immune response. Consequently, the long-term effect of ICZ on the immune system should be investigated.
Acknowledgments
We thank Dr. Roger Reeves (JHU) for helpful comments regarding the performed experiments. We also thank Natasha Naidu from the UCSD Glycobiology Core for glycoconjugate analysis and Dr. Ajit Varki (UCSD) for help in the interpretation of the results. The help from Dr. Daniel Vazquez (UCSD), Dr. David Cauvi (UCSD), and Dr. Diego Nino (UCSD) is also recognized. The editorial assistance of Molly Wofford was highly appreciated.
This work was supported, in whole or in part, by National Institutes of Health Grant NIGMS Grants GM 073825 and GM05078.
- LPS
- lipopolysaccharide
- ICZ
- itraconazole
- TNF
- tumor necrosis factor
- PBS
- phosphate-buffered saline
- DMSO
- dimethyl sulfoxide
- MTT
- thiazolyl blue tetrazolium bromide
- ELISA
- enzyme-linked immunosorbent assay
- PHA-E
- Phaseolus vulgaris erythroagglutinin
- ConA
- concanavalin A
- endo H
- endoglycosidase H
- PNGase F
- peptide:N-glycosidase
- HPLC
- high pressure liquid chromatography
- Mϕs
- macrophage.
REFERENCES
- 1.Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. ( 2001) Crit. Care Med. 29, 1303– 1310 [DOI] [PubMed] [Google Scholar]
- 2.Annane D., Bellissant E., Cavaillon J. M. ( 2005) Lancet 365, 63– 78 [DOI] [PubMed] [Google Scholar]
- 3.Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. ( 1990) Science 249, 1431– 1433 [DOI] [PubMed] [Google Scholar]
- 4.Cohen J. ( 2002) Nature 420, 885– 891 [DOI] [PubMed] [Google Scholar]
- 5.Moore K. J., Andersson L. P., Ingalls R. R., Monks B. G., Li R., Arnaout M. A., Golenbock D. T., Freeman M. W. ( 2000) J. Immunol. 165, 4272– 4280 [DOI] [PubMed] [Google Scholar]
- 6.Haziot A., Ferrero E., Köntgen F., Hijiya N., Yamamoto S., Silver J., Stewart C. L., Goyert S. M. ( 1996) Immunity 4, 407– 414 [DOI] [PubMed] [Google Scholar]
- 7.Frey T., De Maio A. ( 2007) Mol. Med. 13, 592– 604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J. I., Lee C. J., Jin M. S., Lee C. H., Paik S. G., Lee H., Lee J. O. ( 2005) J. Biol. Chem. 280, 11347– 11351 [DOI] [PubMed] [Google Scholar]
- 9.Meng J., Parroche P., Golenbock D. T., McKnight C. J. ( 2008) J. Biol. Chem. 283, 3376– 3384 [DOI] [PubMed] [Google Scholar]
- 10.Varki A. ( 1994) Methods Enzymol. 230, 16– 32 [DOI] [PubMed] [Google Scholar]
- 11.Zonios D. I., Bennett J. E. ( 2008) Semin. Respir. Crit. Care Med. 29, 198– 210 [DOI] [PubMed] [Google Scholar]
- 12.Lowe J. B. ( 2001) Cell 104, 809– 812 [DOI] [PubMed] [Google Scholar]
- 13.De Maio A., Lis H., Gershoni J. M., Sharon N. ( 1986) Cell. Immunol. 99, 345– 353 [DOI] [PubMed] [Google Scholar]
- 14.Priatel J. J., Chui D., Hiraoka N., Simmons C. J., Richardson K. B., Page D. M., Fukuda M., Varki N. M., Marth J. D. ( 2000) Immunity 12, 273– 283 [DOI] [PubMed] [Google Scholar]
- 15.Malý P., Thall A., Petryniak B., Rogers C. E., Smith P. L., Marks R. M., Kelly R. J., Gersten K. M., Cheng G., Saunders T. L., Camper S. A., Camphausen R. T., Sullivan F. X., Isogai Y., Hindsgaul O., von Andrian U. H., Lowe J. B. ( 1996) Cell 86, 643– 653 [DOI] [PubMed] [Google Scholar]
- 16.Demetriou M., Granovsky M., Quaggin S., Dennis J. W. ( 2001) Nature 409, 733– 739 [DOI] [PubMed] [Google Scholar]
- 17.Togayachi A., Kozono Y., Ishida H., Abe S., Suzuki N., Tsunoda Y., Hagiwara K., Kuno A., Ohkura T., Sato N., Sato T., Hirabayashi J., Ikehara Y., Tachibana K., Narimatsu H. ( 2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15829– 15834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Comelli E. M., Sutton-Smith M., Yan Q., Amado M., Panico M., Gilmartin T., Whisenant T., Lanigan C. M., Head S. R., Goldberg D., Morris H. R., Dell A., Paulson J. C. ( 2006) J. Immunol. 177, 2431– 2440 [DOI] [PubMed] [Google Scholar]
- 19.Chui D., Sellakumar G., Green R., Sutton-Smith M., McQuistan T., Marek K., Morris H., Dell A., Marth J. ( 2001) Proc. Natl. Acad. Sci. U.S.A. 98, 1142– 1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green R. S., Stone E. L., Tenno M., Lehtonen E., Farquhar M. G., Marth J. D. ( 2007) Immunity 27, 308– 320 [DOI] [PubMed] [Google Scholar]
- 21.Kageyama S., Masuya M., Tanaka I., Oka K., Morita K., Tamaki S., Tsuji K., Katayama N., Sugimoto H., Kagawa Y., Kojima M., Shiku H. ( 1999) J. Infect. Chemother. 5, 213– 216 [DOI] [PubMed] [Google Scholar]
- 22.Roman R. J. ( 2002) Physiol. Rev. 82, 131– 185 [DOI] [PubMed] [Google Scholar]
- 23.Brummer E., Bhagavathula P. R., Hanson L. H., Stevens D. A. ( 1992) Antimicrob. Agents Chemother. 36, 2487– 2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ochi H., Ishijima S. A., Abe S., Tameike A., Yamaguchi H., Osumi M. ( 1998) FEMS Immunol. Med. Microbiol. 20, 181– 189 [DOI] [PubMed] [Google Scholar]
- 25.Chong C. R., Xu J., Lu J., Bhat S., Sullivan D. J., Jr., Liu J. O. ( 2007) ACS Chem. Biol. 2, 263– 270 [DOI] [PubMed] [Google Scholar]
- 26.Steel H. C., Tintinger G. R., Theron A. J., Anderson R. ( 2007) Clin. Exp. Immunol. 150, 144– 150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dürr G., Strayle J., Plemper R., Elbs S., Klee S. K., Catty P., Wolf D. H., Rudolph H. K. ( 1998) Mol. Biol. Cell 9, 1149– 1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufman R. J., Swaroop M., Murtha-Riel P. ( 1994) Biochemistry 33, 9813– 9819 [DOI] [PubMed] [Google Scholar]
- 29.Kuznetsov G., Brostrom M. A., Brostrom C. O. ( 1993) J. Biol. Chem. 268, 2001– 2008 [PubMed] [Google Scholar]
- 30.Marques S. A., Hozumi S., Camargo R. M., Carvalho M. F., Marques M. E. ( 2008) Med. Mycol. 46, 725– 728 [DOI] [PubMed] [Google Scholar]