

Abstract
Cyclosporine A and nonimmunosuppressive cyclophilin (Cyp) inhibitors such as Debio 025, NIM811, and SCY-635 block hepatitis C virus (HCV) replication in vitro. This effect was recently confirmed in HCV-infected patients where Debio 025 treatment dramatically decreased HCV viral load, suggesting that Cyps inhibitors represent a novel class of anti-HCV agents. However, it remains unclear how these compounds control HCV replication. Recent studies suggest that Cyps are important for HCV replication. However, a profound disagreement currently exists as to the respective roles of Cyp members in HCV replication. In this study, we analyzed the respective contribution of Cyp members to HCV replication by specifically knocking down their expression by both transient and stable small RNA interference. Only the CypA knockdown drastically decreased HCV replication. The re-expression of an exogenous CypA escape protein, which contains escape mutations at the small RNA interference recognition site, restored HCV replication, demonstrating the specificity for the CypA requirement. We then mutated residues that reside in the hydrophobic pocket of CypA where proline-containing peptide substrates and cyclosporine A bind and that are vital for the enzymatic or the hydrophobic pocket binding activity of CypA. Remarkably, these CypA mutants fail to restore HCV replication, suggesting for the first time that HCV exploits either the isomerase or the chaperone activity of CypA to replicate in hepatocytes and that CypA is the principal mediator of the Cyp inhibitor anti-HCV activity. Moreover, we demonstrated that the HCV NS5B polymerase associates with CypA via its enzymatic pocket. The study of the roles of Cyps in HCV replication should lead to the identification of new targets for the development of alternate anti-HCV therapies.
Hepatitis C virus (HCV)2 is the main contributing agent of acute and chronic liver diseases worldwide (1). Primary infection is often asymptomatic or associated with mild symptoms. However, persistently infected individuals develop high risks for chronic liver diseases such as hepatocellular carcinoma and liver cirrhosis (1). The combination of IFNα and ribavirin that serves as current therapy for chronically HCV-infected patients not only has a low success rate (about 50%) (2) but is often associated with serious side effects (2). There is thus an urgent need for the development of novel anti-HCV treatments (2).
The immunosuppressive drug cyclosporine A (CsA) was reported to be clinically effective against HCV (3). Controlled trials showed that a combination of CsA with IFNα is more effective than IFNα alone, especially in patients with a high viral load (4, 5). Moreover, recent in vitro studies provided evidence that CsA prevents both HCV RNA replication and HCV protein production in an IFNα-independent manner (6–10). CsA exerts this anti-HCV activity independently of its immunosuppressive activity because the nonimmunosuppressive Cyp inhibitors such as Debio 025, NIM811, and SCY-635 also block HCV RNA and protein production (9, 11–14). Unlike CsA, these molecules do not display calcineurin affinity and specifically inhibit the peptidyl-prolyl cis-trans-isomerase (PPIase) Cyps. Most importantly, recent clinical data demonstrated that Debio 025 dramatically decreased HCV viral load (3.6 log decrease) in patients coinfected with HCV and HIV (15). This 14-day Debio 025 treatment (1200 mg orally administered twice daily) was effective against the three genotypes (genotypes 1, 3, and 4) represented in the study. More recently, the anti HCV effect of Debio 025 in combination with peginterferon α 2a (peg-IFNα2a) was investigated in treatment-inexperienced patients with chronic hepatitis C. Debio 025 (600 mg administered once daily) in combination with peg-IFNα2a (180 μg/week) for 4 weeks induced a continuous decay in viral load that reached −4.61 ± 1.88 IU/ml in patients with genotypes 1 and 4 and −5.91 ± 1.11 IU/ml in patients with genotypes 2 and 3 at week 4 (16). The Debio 025 findings are critical because they suggest that Cyp inhibitors represent a novel class of anti-HCV agents. However, it remains unclear how these compounds control HCV replication. The fact that several recent studies using small RNA interference knockdown approaches suggest that Cyps are critical for the HCV life cycle (9, 17, 18) strongly implies that there is a direct or indirect link between the CsA- and CsA derivative-mediated inhibitory effect on HCV replication and host Cyps.
The discovery 20 years ago of the first cellular protein showing PPIase activity (19) was entirely unrelated to the discovery of CypA as an intracellular protein possessing a high affinity for CsA (20). It is only a few years later that Fischer et al. (21) demonstrated that the 18-kDa protein with PPIase activity and CypA represent a single unique protein. All Cyps contain a common domain of 109 amino acids, called the Cyp-like domain, which is surrounded by domains specific to each Cyp members and which dictates their cellular compartmentalization and function (22). Bacteria, fungi, insects, plants, and mammals contain Cyps, which all have PPIase activity and are structurally conserved (22). To date, 16 Cyp members have been identified, and 7 of them are found in humans: CypA, CypB, CypC, CypD, CypE, Cyp40, and CypNK (22).
Although there is a growing body of evidence that Cyps control HCV replication in human hepatocytes, a major disagreement currently exists on the respective roles of Cyp members in HCV replication. One study suggests that CypB, but not CypA, is critical for HCV replication (17), another suggests that CypA, but not CypB and CypC, is critical for HCV replication (18), and a third study suggests that three Cyps, CypA, B, and C, are all required for HCV replication (9). Thus, although it becomes evident that Cyps serve as HCV co-factors, their respective contributions and roles in the HCV life cycle remain to be determined. An understanding of the mechanisms that control the Cyp inhibitor-mediated anti-HCV effect is imperative because it will provide new alternate anti-HCV therapies and shed light on the still poorly understood early and late steps of the HCV life cycle.
EXPERIMENTAL PROCEDURES
Cells and Drugs
Huh7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics. CsA (Sigma) was prepared in dimethyl sulfoxide at 10 mg/ml and diluted in tissue culture medium for each experiment to 2.5 μm. Debio 025 (gift from Debiopharm, Lausanne, Switzerland) was prepared in ethanol at 10 mm and diluted further in tissue culture medium to 2 μm for each experiment.
HCV RNA Replication
Ten micrograms of in vitro transcribed genomic Con1 RNA was electroporated into Huh-7 cells. At the indicated time points, intracellular HCV RNA was analyzed via reverse-transcription quantitative polymerase chain reaction and presented as genome equivalents/microgram total RNA as described previously (23). The primers for reverse-transcription quantitative polymerase chain reaction were: HCV, 5′-ATGGCGTTAGTATGAGTGTC-3′ (sense) and 5′-GGCATTGAGCGGGTTGATC-3′ (antisense); glyceraldehyde 3-phosphate dehydrogenase, 5′-GAAGGTGAAGGTCGGAGTC-3′ (sense), and 5′-GAAGATGGTGATGGGATTTC-3′ (antisense).
Small RNA Interference Knockdown
Annealed duplex siRNA oligonucleotides contained a 3′-dTdT overhand (Qiagen). siRNA target sequences were: AAGGGTTCCTGCTTTCACAGA for CypA; AAGGTGGAGAGAGCACCAAGACA for CypB; GTGACATCACCACTGGAGATG for CypC; AACCTGCTAAATTGTGCGTTA for CypD; and AATTCTCCGAACGTGTCACGT for control. The cells were transfected with 100 nm siRNA using Lipofectamine 2000 (Invitrogen). For effect of siRNA Cyp knockdown on HCV RNA replication, the cells were transfected with siRNA Cyp and then retransfected 24 h later. An HIV-1-based lentiviral vector was used to express all Cyp shRNA as described previously (24). The Cyp target sequences are the same as those indicated just above. Lentiviral particles production and transduction was conducted as described previously (24). Generation of stable Cyp knockdown cell lines was obtained after 3 weeks under puromycin (1 μg/ml) selection. All of the cell lines were tested for mycoplasm contamination, which may nonspecifically interfere with HCV replication. To restore CypA expression in Huh7 CypA knockdown cells, a CypA cDNA bearing silent mutations that render it nontargetable by the CypA shRNA was subcloned into pcDNA3 with HindIII and NotI sites to generate pcDNA3- resistant wild-type CypA, which contains an N-terminal HA tag. Using pcDNA3-resistant wild-type CypA as template, we engineered two plasmids carrying either the H126Q (pcDNA3-resistant H126Q CypA) or the R55A (pcDNA3- resistant R55A CypA) mutation in the hydrophobic pocket of CypA that disallows its isomerase activity (25).
Western Blotting
Parental or subgenomic HCV Con1-containing Huh7 cells (1 million) treated with or without siRNA or shRNA targeting Cyps were trypsinized and washed twice with 10 ml of sterile phosphate-buffered saline and lysed for 30 min on ice in 100 μl of lysis buffer (10 mm NaCl, 10 mm Tris, pH 7.4, 0.5% Nonidet P-40, 1× protease inhibitors). The lysates were cleared via centrifugation at 14,000 rpm for 10 min in a microcentrifuge. Supernatants (70 μl) were collected and protein concentration of cell lysates measured with a Coomassie-based Bio-Rad kit. The cell lysates were then subjected to Western blotting with antibodies to CypA (26), CypB (Zymed Laboratories Inc.), CypC (Protein Tech Group, Inc.), CypD (Calbiochem), NS5A (ViroStat), and NS5B (Alexis Biochemicals). Amido Black staining of the membranes confirmed that the loading of samples had been properly normalized. The cellular expression of resistant wild-type, H126Q, or R55A CypA proteins was verified by Western blotting using anti-HA antibodies (The Scripps Research Institute (TSRI), Antibody Core Facility).
Virus Infection and Replication
For HIV-1 infection, Cyp knockdown Huh7 cells lines were infected with HIV-1-GFP (NL4.3 virus encoding the GFP gene and pseudotyped with the vesicular stomatitis virus G envelope protein) (generous gift from C. Aiken and D. Gabuzda). Forty-eight hours post-infection, intracellular GFP levels were quantified by FACS. For HSV-1 infection, Cyp knockdown Huh7 cells lines were infected with HSV-1-GFP (K26GFP virus encoding the GFP gene) (generous gift from P. Desai). Forty-eight hours post-infection, intracellular GFP levels were quantified by FACS. For Dengue infection, Cyp knockdown Huh7 cells lines were infected with Dengue-2 (Dengue-2 16681) (generous gift from R. Kinney). Dengue-2 infection was examined using an intracellular FACS staining assay (IFSA). Briefly, IFSA was conducted as described previously (27, 28) with minor modifications. Three days post-infection, the cells were washed, trypsinized, resuspended in PBS at 1 × 106/ml, and fixed with 0.2% paraformaldehyde in PBS for 30 min on ice. The cells were washed, permeabilized in PBS containing 0.2% Tween for 15 min at 37 °C, washed, and resuspended in FACS buffer (PBS containing 2% fetal bovine serum). For intracellular staining, the cells (105) were incubated for 30 min at 4 °C with 10 μg/ml of isotype controls, mouse monoclonal anti-Dengue capsid 9A7 IgG (TSRI, Antibody Core Facility). Cell permeabilization was confirmed by staining cells with mouse anti-tubulin IgG (Santa Cruz Biotechnologies). The cells were washed, incubated with secondary phycoerythrin-conjugated anti-mouse IgG (10 μg/ml) for 30 min at 4 °C, washed again, resuspended in PBS, fixed in 2% paraformaldehyde, and stored at 4 °C until FACS analysis. For HCV replication, Cyp knockdown Huh7 cells lines were electroporated with 10 μg of in vitro transcribed genomic Con1 RNA. Seven days post-transfection, HCV infection was quantified by measuring intracellular NS5B levels by IFSA using anti-NS5B IgG (Alexis Biochemicals).
CypA Binding to HIV-1 Gag in Hepatocytes
CypA-HIV-1 Gag interaction was studied by examining the incorporation of host CypA into budding HIV-1 particles by Western blot. Briefly, HIV-1 particles were transiently expressed in Huh7 cells by GeneJuice (EMD Biosciences) transfection with a mixture of 5 μg of proviral HIV-1 (NL4.3) DNA together with 5 μg of CypA plasmids (pcDNA3-resistant wild-type, H126Q, and R55A plasmids). Viral supernatants, harvested 48 h post-transfection, were filtered through a 0.2-μm-pore size filter to remove cellular debris, pelleted through a sucrose cushion, standardized for HIV-1 capsid content by p24 enzyme-linked immunosorbent assay (Alliance, PerkinElmer), resuspended in 2× sodium dodecyl sulfate loading buffer, and subjected to Western blotting with antibodies directed against the HA tag.
Co-immunoprecipitations
Parental Huh7 cells (3 million) were co-transfected with NS5B-Myc (5 μg of DNA) and wild-type CypA-HA or H126Q CypA-HA (5 μg of DNA) plasmids in the presence or absence of Debio 025 (2 μm). Three days post-transfection, the cells were collected and lysed. The cell lysates (1 ml) were precleared for 1 h with 20 μl of agarose beads. Co-immunoprecipitation procedures were conducted according to the manufacturer's instructions (Pierce HA tag IP/Co-IP kit). Bound material was eluted and analyzed by Western blotting using anti-HA and anti-Myc IgG (Santa Cruz Biotechnology).
RESULTS
Analysis of the Respective Contribution of Cyp Members to HCV Replication by Transient Small RNA Interference
Previous studies suggested that at least three members of the Cyp family, CypA, CypB, and CypC, modulate HCV replication (9, 17, 18). We thus examined whether HCV exploits all of these Cyp members or rather a unique Cyp member to efficiently replicate in human hepatocytes. To address this issue, we knocked down each of these Cyps by transient siRNA interference and examined the effect of the Cyp knockdown on HCV replication. Specifically, Huh7 cells containing the subgenomic HCV Con1 replicon (genotype 1b) were transfected with siRNAs that target either luciferase (control siRNA), CypA (CypA siRNA), CypB (CypB siRNA), CypC (CypC siRNA), or CypD (CypD siRNA). To avoid siRNA toxicity, the cells were washed 24 h post-transfection. Seven days post-transfection, the cells were collected and lysed. To ensure that the siRNA treatments did not nonspecifically influence growth and viability of transfected hepatocytes, the cells were counted and analyzed for trypan blue uptake 7 days post-transfection. We exclusively analyzed the lysates of cells, which gave numbers of viable cells comparable with those of control siRNA-treated cells. The cell lysates were standardized for protein content and analyzed for Cyp content by Western blot using antibodies directed against CypA, CypB, CypC, or CypD.
The expression of each Cyp was profoundly reduced by siRNAs Cyp compared with siRNA control (Fig. 1A), demonstrating the efficacy of the siRNA treatments. Moreover, siRNA Cyp treatments were specific because each siRNA Cyp treatment did not alter the expression of the other Cyps (Fig. 1A). For example, the siRNA, which targeted CypA, did not influence CypB, CypC, or CypD expression (Fig. 1A). Importantly, transient CypB, CypC, or CypD knockdown did not significantly influence HCV protein expression. Indeed, NS5A and NS5B levels in siRNA CypB-, CypC-, and CypD-treated hepatocytes were similar to those of control siRNA-treated cells (Fig. 1B), suggesting that these Cyps play no or a minor role in HCV protein expression. In sharp contrast, NS5A and NS5B levels were profoundly diminished in hepatocytes treated with the siRNA CypA (Fig. 1B), demonstrating that CypA, rather than CypB, CypC, and CypD, plays a major role in HCV protein expression.
FIGURE 1.

Respective contribution of Cyp members to HCV replication. A, Huh7 cells containing the subgenomic HCV Con1 replicon were transfected with an irrelevant control siRNA or siRNAs that target CypA (siRNA CypA), CypB (siRNA CypB), CypC (siRNA CypC), or CypD (siRNA CypD). The cells were washed 24 h post-transfection. Seven days post-transfection, the cells were collected an dialyzed. To ensure that the siRNA treatments did not nonspecifically influence growth and viability of transfected hepatocytes, the cells were counted and analyzed for trypan blue uptake at the time of collection. The cell lysates were standardized for protein content and analyzed for Cyp content by Western blot using antibodies directed against CypA, CypB, CypC, or CypD. B, same as A except that cell lysates were analyzed for HCV protein expression using antibodies directed against NS5A and NS5B. C, naïve Huh7 cells were electroporated with 10 μg of in vitro transcribed genomic Con1 RNA. Twenty-four hours post-HCV RNA electroporation, the cells were transfected with siRNA Cyp and then retransfected 24 h later. At the indicated time points, intracellular HCV RNA was analyzed via reverse-transcription quantitative polymerase chain reaction and presented as genome equivalents (GE)/μg of total RNA.
We then asked whether CypA, but not other Cyp members, is also critical for HCV RNA replication. To address this issue, naïve Huh7 cells were electroporated with in vitro transcribed genomic Con1 RNA. One day post-electroporation, the cells were transfected with control or siRNA Cyp as described above. Viral RNA replication was monitored for 8 days by analyzing intracellular HCV RNA via reverse-transcription quantitative polymerase chain reaction as described previously (23). We verified that the expression of CypA, CypB, CypC, and CypD was knocked down via the Cyp siRNA treatment 8 days post-electroporation (data not shown). We found that the HCV RNA replication in hepatocytes treated with siRNA CypC and CypD was comparable with that in siRNA control-treated cells, whereas the viral RNA replication in siRNA CypB was only slightly diminished (Fig. 1C). In contrast, viral RNA replication was profoundly attenuated in the siRNA CypA-treated cells (Fig. 1C). Altogether these data suggest that CypA, but not CypB, CypC, and CypD, represents an essential host factor for both HCV RNA replication and HCV protein expression.
Demonstration of the Exclusive Contribution of CypA to HCV Replication by Stable Small RNA Interference
The introduction of siRNA into cultured cells provides a fast and efficient means of knocking down gene expression. However, siRNA has been shown to be effective for only short term gene inhibition in certain mammalian cells. Supporting this notion, we found that although Cyp expression was dramatically reduced 5–7 days after siRNA transfection, Cyp expression recovered 9 or 10 days post-siRNA transfection (data now shown). Even a slight rebound in Cyp protein expression may interfere with the interpretation of the data. To avoid this issue, we conducted experiments similar to those described above but using shRNA to stably silence Cyp gene expression. We constructed plasmids encoding shRNA that target CypA, CypB, or CypC. As above, we constructed as control a plasmid that encodes an shRNA that targets luciferase. Plasmids were packaged into HIV-1-based particles pseudotyped with the vesicular stomatitis virus G envelope to permit entry into and infection of hepatocytes. The advantage of using the HIV-1-based vector is that the DNA encoding the shRNA will be stably integrated into the host chromosomes of the hepatocytes. Parental Huh7 cells were exposed to the HIV-1-based particles for 24 h, cultured under puromycin selection for 3 or 4 weeks, and analyzed for Cyp content by Western blot. As expected, CypA, CypB, and CypC levels in shRNA Cyp-transduced cells were severely reduced compared with control shRNA-treated cells (Fig. 2A).
FIGURE 2.

HCV, like HIV-1, requires host CypA to fully replicate in human cells. A, naïve Huh7 cells were transduced with an vesicular stomatitis virus G-pseudotyped HIV-1-based vector containing an irrelevant control shRNA or shRNAs that target CypA, CypB, or CypC. Transduced cells were selected for 7 weeks under puromycin (1 μg/ml). Stable cell lines were analyzed for CypA, CypB, and CypC content by Western blotting. B, stable Cyp-KD cell lines were electroporated with 10 μg of in vitro transcribed genomic Con1 RNA. At the indicated time points, intracellular HCV RNA was analyzed via reverse-transcription quantitative polymerase chain reaction and presented as genome equivalents (GE)/μg of total RNA. C, top panels, Cyp-KD cells lines were exposed to cell-free vesicular stomatitis virus G-HIV-1-GFP (left panel) or HSV-1-GFP (right panel) at a multiplicity of infection of 0.1. Forty-eight hours post-infection, the percentage of GFP-positive cells was quantified by FACS. Bottom left panel, cells were exposed to cell-free Dengue-2 (multiplicity of infection of 0.1). Three days post-infection, Dengue-2 infection was quantified by measuring the amounts of intracellular Dengue-2 capsid using an IFSA. Bottom right panel, Cyp-KD cells lines were electroporated with 10 μg of in vitro transcribed genomic Con1 RNA. Seven days post-transfection, HCV infection was quantified by measuring the amounts of intracellular HCV NS5B. The data are expressed as percentages of infected cells (GFP-, capsid-, or NS5B-positive cells) by fixing arbitrary infection of parental cells at 100. The results are representative of three independent experiments.
We then tested the Cyp knockdown (KD) cell lines for their capacities to support HCV RNA replication as described above. Importantly, the HCV RNA replication was only profoundly reduced in the stable CypA-KD cell line (Fig. 2B), further suggesting that CypA, but not CypB, CypC, and CypD, is essential for HCV replication. It is important to note that we did not observe differences in growth between the cell lines (data not shown), suggesting that these particular Cyps do not play a significant role in Huh7 cell division and multiplication.
To further demonstrate the specificity of the CypA requirement for HCV replication, CypA, CypB, and CypC knockdown and control cell lines were exposed to various viruses including HIV-1, HSV-1, the flavivirus Dengue and HCV. Infectivity was scored by measuring the intracellular levels of GFP for both HIV-1 and HSV-1, levels of capsid for Dengue, and levels of NS5B for HCV. GFP levels were significantly reduced in HIV-1-exposed CypA knockdown cells compared with control, CypB- and CypC-KD cells (Fig. 2C, top left panel). This is in accordance with previous studies suggesting that HIV-1 requires CypA to optimally infect human cells (29, 30). All of the cell lines exposed to HSV-1 or Dengue expressed similar levels of GFP (HSV-1) and capsid (Dengue) (Fig. 2C, top right and bottom left panels, respectively), suggesting that HSV-1 and Dengue do no require any of these Cyps to infect human cells. Importantly, NS5B levels were dramatically reduced in the CypA knockdown cells compared with control, CypB and CypC knockdown cells (Fig. 2C). This further suggests that HCV, like HIV-1, specifically exploits CypA to infect and replicate in human cells, more specifically in hepatocytes.
HCV Requires the Isomerase Activity of CypA to Replicate in Human Hepatocytes
The peptide bond generally exists in two relatively stable isomeric forms: cis and trans (31). The ribosome synthesizes peptide bonds in the lower energy state trans peptide bond form, which is sterically favored, and whose side chains are 180 degrees opposite each other (32). However, bonds preceding each proline (peptidyl-prolyl bonds) also occur in the cis form in both unfolded and native proteins, with the side chains adjacent to each other (32). The isomerization to the cis form is required for both de novo protein folding, protein restructuring, and refolding processes following cellular membrane traffic. Spontaneous cis/trans-isomerization of peptidyl-prolyl bonds is a slow process at room temperature that does not require free energy. Thus, this isomerization represents a rate-limiting step in the refolding of chemically denatured proteins (34).
We thus asked whether CypA promotes HCV replication via its isomerase activity. To address this issue, we created a CypA mutant deprived of its isomerase activity. Specifically, we replaced the histidine located at position 126 in the hydrophobic pocket of CypA by a glutamine, creating the H126Q CypA mutant. Importantly, this mutation diminishes CypA isomerase activity by more than 99% compared with wild-type CypA (25). To determine whether HCV requires the isomerase activity of CypA to replicate, we had to transfect the H126Q CypA mutant into the CypA-KD cells and asked if HCV replication can be rescued. To introduce the H126Q CypA mutant into the CypA-KD cell line, we had to generate the H126Q mutation into the context of an shRNA escape CypA plasmid. We thus generated one plasmid encoding the wild-type shRNA escape CypA and another encoding the H126Q shRNA escape CypA. CypA-KD cells were transfected with wild-type and H126Q shRNA escape CypA plasmids and tested for their capacities to support HCV RNA replication as described above. Importantly, the introduction of the wild-type shRNA escape CypA into the CypA-KD cells restored HCV RNA replication at levels similar to those observed in parental Huh7 cells (Fig. 3A). This rescue not only demonstrates the specificity of the CypA knockdown, but it also further demonstrates the importance of CypA in HCV replication. More importantly, the introduction of the H126Q shRNA escape CypA into the CypA-KD cells did not restore viral RNA replication (Fig. 3A). Note that the cellular levels of H126Q CypA were similar to those of wild-type CypA (Fig. 3B). Importantly, we obtained similar data for the R55A CypA mutant (data not shown), which is also deprived of isomerase activity (25). These findings strongly suggest that the isomerase activity of CypA is essential for HCV replication in human hepatocytes.
FIGURE 3.

HCV requires the isomerase activity of CypA to replicate in hepatocytes. A, parental or CypA knockdown Huh7 cell lines were electroporated with 10 μg of in vitro transcribed genomic Con1 RNA. Twenty-four hours post-HCV RNA electroporation, the cells were transfected with shRNA-resistant wild-type or H126Q CypA-HA in the presence or absence of the Cyp inhibitors CsA (2.5 μm) or Debio 025 (2 μm). At the indicated time points, intracellular HCV RNA was analyzed via reverse-transcription quantitative polymerase chain reaction and presented as genome equivalents (GE)/μg of total RNA. B, the inability of the H126Q CypA mutant to bind to HIV-1 Gag in hepatocytes was examined by measuring amounts of CypA incorporated into released particles. Huh7 cells were co-transfected with HIV-1 together with wild-type (WT) or H126 CypA-HA in the presence or absence of Cyp inhibitors CsA (10 μm) or Debio 025 (2 μm). Forty-eight post-transfection, both transfected cells and released virions were analyzed for CypA content by Western blotting using anti-HA antibodies. The cell lysates were standardized for protein content, whereas virions were standardized for HIV-1 capsid content by p24 enzyme-linked immunosorbent assay.
The hydrophobic pocket of CypA not only contains the residues vital for the isomerase activity of CypA, it also contains the residues responsible for the binding of CypA to its, to date, unique known viral ligand, the structural polyprotein HIV-1 Gag. One can thus envision that the H126Q mutation fails to rescue HCV activity in hepatocytes because the mutation not only blocks the enzymatic activity of CypA, but it also precludes CypA binding to its still unidentified HCV ligand. Therefore, we asked whether the H126Q mutation, which abolishes the enzymatic activity of CypA, also prevents the binding of CypA to HIV-1 Gag within human hepatocytes. CypA binding to HIV-1 Gag was never examined in hepatocytes. To address this issue, we measured amounts of CypA incorporated via Gag into HIV-1 particles released from human hepatocytes. As controls, the hepatocytes were treated with CsA or Debio 025, which, by binding to the hydrophobic pocket of CypA, prevents CypA-Gag interaction (30, 35). Specifically, Huh7 cells were co-transfected with HIV-1 together with wild-type or H126Q CypA-HA in the presence or absence of CsA or Debio 025. Forty-eight post-transfection, both transfected cells and released virions were analyzed for CypA content by Western blotting using anti-HA antibodies.
In the absence of any treatment, HIV-1 particles released from hepatocytes contain significant amounts of wild-type CypA (Fig. 3B, top panel), demonstrating that CypA-Gag interactions also occur in human hepatocytes. Virions released from hepatocytes treated with either CsA or Debio 025 contain minimal amounts of CypA (Fig. 3B, top panel), suggesting that the two compounds, by binding to the hydrophobic pocket of CypA, interfere with CypA-Gag interactions in hepatocytes. Although the H126Q CypA mutant was efficiently expressed in transfected hepatocytes (Fig. 3B, bottom panel), it was not incorporated into released virions (Fig. 3B, top panel), suggesting that the H126Q CypA mutation in the hydrophobic pocket of CypA alters both the enzymatic activity of CypA and the binding capacity of CypA to its viral ligand in hepatocytes. Thus, the inability of the H126Q CypA mutant to support HCV replication may arise from either its inability to isomerizes peptidyl-prolyl bonds, its inability to bind to its viral or host ligand, or both. It is also important to note that the inability of the overexpressed isomerase-deficient H126Q CypA mutant to restore HCV replication argues against the possibility that CypA plays a more important role in HCV replication than other Cyp members simply because of its superabundance in a cell. Overexpression of CypB or CypC in the CypA knockdown cells did not rescue HCV replication either, further supporting the notion that the CypA requirement for HCV is specific (data not shown).
HCV NS5B Polymerase Associates with CypA via Its Enzymatic Pocket
A previous study presented data suggesting that CypA binds to NS5B (18). We thus asked whether the isomerase pocket of CypA is critical for the interaction between host CypA and the HCV NS5B polymerase. To address this issue, hepatocytes were co-transfected with Myc-tagged NS5B and HA-tagged wild-type or H126Q CypA plasmids in the presence or absence of the Cyp inhibitor Debio 025. Three days post-transfection, the cells were collected and lysed. The cell lysates were then used for co-immunoprecipitation experiments. First, we confirmed that wild-type CypA associates with HCV NS5B (Fig. 4). Importantly, the Cyp inhibitor prevents NS5B-CypA association (Fig. 4). Most importantly, we found that the isomerase-deficient CypA, although well expressed in transfected cells, fails to associate with NS5B (Fig. 4). This demonstrates for the first time that the enzymatic pocket of CypA is critical for the contact between host CypA and the viral NS5B polymerase.
FIGURE 4.
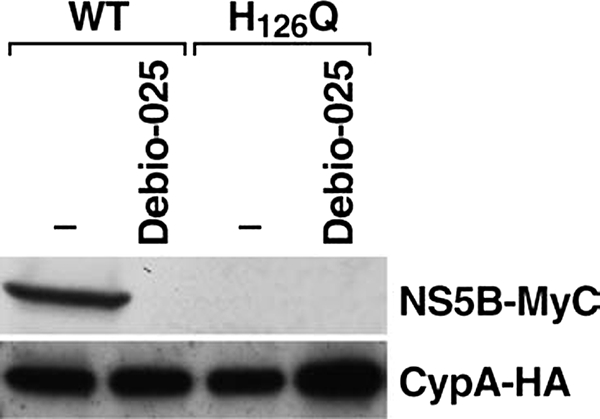
The HCV NS5B polymerase associates with CypA via its enzymatic pocket. Parental Huh7 cells (3 million) were co-transfected with NS5B-Myc (5 μg of DNA) and wild-type (WT) CypA-HA or H126Q CypA-HA (5 μg of DNA) in the presence or absence of the Cyp inhibitor Debio 025 (2 μm). Three days post-transfection, the cells were collected and lysed. The cell lysates were precleared with agarose beads. CypA-NS5B association was assessed by co-immunoprecipitation using the Pierce HA tag Co-IP kit. Bound material was eluted and analyzed by Western blotting using anti-HA and anti-Myc antibodies. The results are representative of three independent transfections.
DISCUSSION
It is well established that host proteins highly regulate the viral life cycles. Because cellular chaperones and enzymes control the correct folding of host proteins, one could assume that they also control the correct folding of viral proteins. This assumption is now apparently proven to be correct for two prime human pathogens: HIV-1 and HCV. In 1993, Luban et al. (35) using the yeast two-hybrid system identified for the first time the interaction between CypA and HIV-1 Gag. One year later, two independent studies elegantly demonstrated that CypA-Gag interactions are critical for HIV-1 replication in human cells (29). Specifically, Thali et al. (30) showed that CsA, by preventing CypA-Gag interactions, inhibits HIV-1 infection. Moreover, Franke et al. (29) showed that the introduction of mutations in the CypA-binding region of Gag also decreases HIV-1 infection of human cells. Further supporting the notion that HIV-1 requires CypA to replicate in human cells, several studies demonstrated that CypA knock-out or knockdown human cells poorly support HIV-1 replication (24, 36, 37). Similarly to HIV-1, we present here several lines of evidence that CypA is also required for HCV replication. We showed that both transient and stable small RNA interferences, which specifically target CypA, profoundly hamper HCV RNA replication as well as HCV protein expression. In contrast to previous studies (9, 17), we did not observe a significant contribution to HCV replication of other Cyp members including CypB, CypC, and CypD. Although we do not have any clear explanation for these apparent divergent results, one cannot exclude the possibility that the use of different cells, HCV strains, or replication systems somehow modulates the respective contribution of Cyp members to HCV replication. It is critical to emphasize that during the course of our study, two independent studies from the Tang (18) and the Bartenschlager (38) laboratories obtained similar results, which convincingly showed that CypA, but not CypB and CypC, is an essential factor for HCV infection. Thus, to date, the findings of three independent studies including ours, all using a stable shRNA knockdown approach, converge to the same conclusion, which is that CypA serves as a major host co-factor for HCV replication. A couple of observations may explain why HCV preferentially exploits CypA rather than CypB and CypC. One is that CypA is 10- and 100-fold more abundant in a cell than CypB and CypC, respectively (22, 39). Another is that CypA, which resides in the cytosol, is more appropriately located to interact with the HCV replication complex than CypB and CypC, which reside in the lumen of the endoplasmic reticulum (22).
Does CypA assist HCV replication as a peptidyl isomerase? To date, auxiliary and essential biochemical functions can be attributed to Cyps. The auxiliary function is characterized by polypeptide sequestration using extended catalytic subsites of the enzyme, whereas catalysis essentially requires direct participation of active site residues (40). We demonstrated here that when we mutate residues that reside in the hydrophobic pocket of CypA (histidine in position 126 and arginine in position 55) where the proline-containing peptide substrates bind, the resulting CypA mutants fail to restore HCV replication. The simplest hypothesis for the mechanism of action of CypA in the HCV life cycle is that it catalyzes a trans to cis- or a cis to trans-isomerization of a peptidyl-prolyl bond in a viral or host protein critical for HCV replication. The observation that Cyp inhibitors (Debio 025, NIM811, and SCY-635) that neutralize PPIase activity but that are not immunosuppressive also block HCV replication is consistent with this hypothesis. In Drosophila melanogaster, the CypA homolog, called NinaA, forms a stable and specific complex with the Rh1 isoform of rhodopsin. The formation of this complex is essential for the transit of the visual pigment through the endoplasmic reticulum (41, 42). Interestingly, an elegant study by Schmid and colleagues (43) showed that a proline serves as a molecular timer in the infection of Escherichia coli by the filamentous phage fd. The phage infection is activated by the disassembly of two domains of its gene-3-protein, which is located at the phage tip. A proline (Pro213) located in the hinge between the two gene-3-protein domains serves as a timer for the infective state. The timer is switched on by cis-to-trans and switched off by the unusually slow trans-to-cis-isomerization of the Gln212–Pro213 peptide bond. Importantly, the switching rate and the phage infectivity are determined by the local sequence surrounding Pro213 and can be tuned by mutagenesis (43). Another hypothesis is that the PPIase activity and the auxiliary polypeptide sequestration function of CypA are both required for its function in HCV replication. Indeed, mutagenesis of NinaA failed to identify a mutant, which distinguishes the isomerase from the polypeptide sequestration activity of CypA (41). Similarly, we showed here that mutations, which disrupt the isomerase activity of CypA, also disrupt the capacity of CypA to bind HIV-1 Gag in hepatocytes. This is in accordance with the work of Luban and co-workers (44), who showed that all mutations that neutralize CypA enzymatic activity also preclude CypA incorporation into HIV-1.
The mechanisms of action of CypA in HCV replication remain to be unraveled. A current hypothesis is that Cyps, by interacting with the nonstructural protein 5B (NS5B), increase the affinity of the polymerase to the viral RNA and therefore enhance HCV replication (45). This hypothesis is supported by the fact that two studies showed that HCV resistance to CsA (39, 46) resulted in the emergence of mutations in NS5B. Moreover, Yang et al. (18) presented convincing pulldown data showing that CypA binds NS5B. These data are in accordance with our co-immunoprecipitation data, which showed that the isomerase pocket of CypA serves as a binding site for the NS5B polymerase. This finding demonstrates for the first time that there is a direct correlation between NS5B binding to the isomerase pocket of CypA and HCV replication. These new findings are important because they suggest that CypA, by catalyzing a trans to cis or a cis to trans isomerization of a peptidyl-prolyl bond within NS5B, enhances HCV replication. Interestingly, recent resistance mapping studies suggest that Cyp inhibition may also act on the HCV NS5A protein (46–48). Thus, further work is required to determine whether NS5A, NS5B, both, or another viral protein represent(s) the true ligand(s) for CypA.
Although both HIV-1 and HCV exploit the abundant cytosolic CypA, it is likely that the immunophilin acts at distinct steps of their viral life cycles. HIV-1 requires CypA post-entry (early events) (49). It is currently thought that target cell CypA, by interacting with the HIV-1 core delivered into the cytosol of infected cells, protects these cores from an antiviral activity present in human cells (50–53). The identity of the anti-HIV-1 factor counteracted by CypA remains unknown. In contrast to HIV-1, HCV requires CypA before budding (late events). CypA likely does not act on the HCV core, like HIV-1, because HCV replicon (no core expressed) replication is also CypA-dependent. Nevertheless, further studies are required to determine whether CypA acts at several steps of the HCV life cycle. Although CypA apparently acts at distinct steps of the HIV-1 and HCV life cycles, one cannot exclude the possibility that CypA via its hydrophobic pocket assists these two prime human pathogens by accomplishing the same task such as peptidyl-prolyl cis-trans-isomerization or another unknown task. Given that the Cyp inhibitor Debio 025 exhibits a remarkable anti-HCV activity in patients (15, 16), it is imperative to understand at a molecular level how this novel class of anti-HCV agents, Cyp inhibitors, block HCV replication.
Acknowledgments
We thank J. Kuhns for secretarial assistance. We thank C. Aiken and D. Gabuzda for providing the NL4.3-GFP plasmid. We thank P. Desai and R. Kinney for providing the HSV-1-GFP (K26GFP) and Dengue-2 16681 viruses. We thank R. Bartenschlager for providing the HCV Con1 plasmid. We thank Debiopharm for providing Debio 025. We thank Gunter Fischer and Raf Crabbé for careful reading of the manuscript.
This is Publication 19940-IMM from the Department of Immunology and Microbial Science of the Scripps Research Institute.
- HCV
- hepatitis C virus
- CsA
- cyclosporine A
- Cyp
- cyclophilin
- IFN
- interferon
- PPIase
- peptidyl-prolyl cis-trans-isomerase
- siRNA
- small interfering RNA
- HA
- hemagglutinin
- shRNA
- small hairpin RNA
- HIV-1
- human immunodeficiency virus, type 1
- FACS
- fluorescence-activated cell sorter
- IFSA
- intracellular FACS staining assay
- PBS
- phosphate-buffered saline
- HSV-1
- herpes simplex virus, type 1
- GFP
- green fluorescent protein.
REFERENCES
- 1.Pawlotsky J. M. ( 2006) Hepatology 43, S207– S220 [DOI] [PubMed] [Google Scholar]
- 2.Hayashi N., Takehara T. ( 2006) J. Gastroenterol. 41, 17– 27 [DOI] [PubMed] [Google Scholar]
- 3.Akiyama H., Yoshinaga H., Tanaka T., Hiruma K., Tanikawa S., Sakamaki H., Onozawa Y., Wakita T., Kohara M. ( 1997) Bone Marrow Transplant. 20, 993– 995 [DOI] [PubMed] [Google Scholar]
- 4.Inoue K., Sekiyama K., Yamada M., Watanabe T., Yasuda H., Yoshiba M. ( 2003) J. Gastroenterol. 38, 567– 572 [DOI] [PubMed] [Google Scholar]
- 5.Inoue K., Yoshiba M. ( 2005) Transplant. Proc. 37, 1233– 1234 [DOI] [PubMed] [Google Scholar]
- 6.Goto K., Watashi K., Murata T., Hishiki T., Hijikata M., Shimotohno K. ( 2006) Biochem. Biophys. Res. Commun. 343, 879– 884 [DOI] [PubMed] [Google Scholar]
- 7.Ishii N., Watashi K., Hishiki T., Goto K., Inoue D., Hijikata M., Wakita T., Kato N., Shimotohno K. ( 2006) J. Virol. 80, 4510– 4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma S., Boerner J. E., TiongYip C., Weidmann B., Ryder N. S., Cooreman M. P., Lin K. ( 2006) Antimicrob. Agents Chemother. 50, 2976– 2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakagawa M., Sakamoto N., Enomoto N., Tanabe Y., Kanazawa N., Koyama T., Kurosaki M., Maekawa S., Yamashiro T., Chen C. H., Itsui Y., Kakinuma S., Watanabe M. ( 2004) Biochem. Biophys. Res. Commun. 313, 42– 47 [DOI] [PubMed] [Google Scholar]
- 10.Watashi K., Hijikata M., Hosaka M., Yamaji M., Shimotohno K. ( 2003) Hepatology 38, 1282– 1288 [DOI] [PubMed] [Google Scholar]
- 11.Coelmont L., Kaptein S., Paeshuyse J., Vliegen I., Dumont J. M., Vuagniaux G., Neyts J. ( 2009) Antimicrob. Agents Chemother. 53, 967– 976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopkins S., Scorneaux B., Huang Z., Murray M. G., Harris R. ( 2008) 59th Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, California, October 31–November 1, 2008, Abstr. 1814, AASLD, Alexandria, VA [Google Scholar]
- 13.Mathy J. E., Ma S., Compton T., Lin K. ( 2008) Antimicrob. Agents Chemother. 52, 3267– 3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paeshuyse J., Kaul A., De Clercq E., Rosenwirth B., Dumont J. M., Scalfaro P., Bartenschlager R., Neyts J. ( 2006) Hepatology 43, 761– 770 [DOI] [PubMed] [Google Scholar]
- 15.Flisiak R., Horban A., Gallay P., Bobardt M., Selvarajah S., Wiercinska-Drapalo A., Siwak E., Cielniak I., Higersberger J., Kierkus J., Aeschlimann C., Grosgurin P., Nicolas-Métral V., Dumont J. M., Porchet H., Crabbé R., Scalfaro P. ( 2008) Hepatology 47, 817– 826 [DOI] [PubMed] [Google Scholar]
- 16.Flisiak R., Dumont J. M., Crabbé R. ( 2007) Expert Opin. Invest. Drugs 16, 1345– 1354 [DOI] [PubMed] [Google Scholar]
- 17.Watashi K., Ishii N., Hijikata M., Inoue D., Murata T., Miyanari Y., Shimotohno K. ( 2005) Mol. Cell. 19, 111– 122 [DOI] [PubMed] [Google Scholar]
- 18.Yang F., Robotham J. M., Nelson H. B., Irsigler A., Kenworthy R., Tang H. ( 2008) J. Virol. 82, 5269– 5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang K., Schmid F. X., Fischer G. ( 1987) Nature 329, 268– 270 [DOI] [PubMed] [Google Scholar]
- 20.Handschumacher R. E., Harding M. W., Rice J., Drugge R. J., Speicher D. W. ( 1984) Science 226, 544– 547 [DOI] [PubMed] [Google Scholar]
- 21.Fischer G., Wittmann-Liebold B., Lang K., Kiefhaber T., Schmid F. X. ( 1989) Nature 337, 476– 478 [DOI] [PubMed] [Google Scholar]
- 22.Wang P., Heitman J. ( 2005) Genome Biol. 6, 226– 231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapadia S. B., Brideau-Andersen A., Chisari F. V. ( 2003) Proc. Natl. Acad. Sci. U. S. A. 100, 2014– 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S., Asparuhova M., Brondani V., Ziekau I., Klimkait T., Schümperli D. ( 2004) Nucleic Acids Res. 32, 3752– 3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zydowsky L. D., Etzkorn F. A., Chang H. Y., Ferguson S. B., Stolz L. A., Ho S. I., Walsh C. T. ( 1992) Protein Sci. 1, 1092– 1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saphire A. C., Bobardt M. D., Gallay P. A. ( 1999) EMBO J. 18, 6771– 6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao C. L., Wu M. C., Chiu Y. H., Lin J. L., Wu Y. C., Yueh Y. Y., Chen L. K., Shaio M. F., King C. C. ( 2001) J. Clin. Microbiol. 39, 3672– 3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin N. C., Pardo J., Simmons M., Tjaden J. A., Widjaja S., Marovich M. A., Sun W., Porter K. R., Burgess T. H. ( 2006) J. Virol. Methods 134, 74– 85 [DOI] [PubMed] [Google Scholar]
- 29.Franke E. K., Yuan H. E., Luban J. ( 1994) Nature 372, 359– 362 [DOI] [PubMed] [Google Scholar]
- 30.Thali M., Bukovsky A., Kondo E., Rosenwirth B., Walsh C. T., Sodroski J., Göttlinger H. G. ( 1994) Nature 372, 363– 365 [DOI] [PubMed] [Google Scholar]
- 31.Schiene C., Fischer G. ( 2000) Curr. Opin. Struct. Biol. 10, 40– 45 [DOI] [PubMed] [Google Scholar]
- 32.Hübner D., Drakenberg T., Forsén S., Fischer G. ( 1991) FEBS Lett. 284, 79– 81 [DOI] [PubMed] [Google Scholar]
- 33.Deleted in proof
- 34.Bang H., Fischer G. ( 1991) Biomed. Biochim. Acta 50, S137– S142 [PubMed] [Google Scholar]
- 35.Luban J., Bossolt K. L., Franke E. K., Kalpana G. V., Goff S. P. ( 1993) Cell 73, 1067– 1078 [DOI] [PubMed] [Google Scholar]
- 36.Braaten D., Luban J. ( 2001) EMBO J. 20, 1300– 1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sokolskaja E., Sayah D. M., Luban J. ( 2004) J. Virol. 78, 12800– 12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaul A., Stauffer S., Schmitt J., Pertel T., Luban J., Bartenschlager R. ( 2008) 15th International Symposium on Hepatitis C Virus & Related Viruses, San Antonio, Texas, October 5–9, 2008, Abstr. 218 [Google Scholar]
- 39.Robida J. M., Nelson H. B., Liu Z., Tang H. ( 2007) J. Virol. 81, 5829– 5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer G., Wawra S. ( 2006) Mol. Microbiol. 61, 1388– 1396 [DOI] [PubMed] [Google Scholar]
- 41.Colley N. J., Baker E. K., Stamnes M. A., Zuker C. S. ( 1991) Cell 67, 255– 263 [DOI] [PubMed] [Google Scholar]
- 42.Stamnes M. A., Shieh B. H., Chuman L., Harris G. L., Zuker C. S. ( 1991) Cell 65, 219– 227 [DOI] [PubMed] [Google Scholar]
- 43.Eckert B., Martin A., Balbach J., Schmid F. X. ( 2005) Nat. Struct. Mol. Biol. 12, 619– 623 [DOI] [PubMed] [Google Scholar]
- 44.Braaten D., Ansari H., Luban J. ( 1997) J. Virol. 71, 2107– 2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rice C. M., You S. ( 2005) Hepatology 42, 1455– 1458 [DOI] [PubMed] [Google Scholar]
- 46.Fernandes F., Poole D. S., Hoover S., Middleton R., Andrei A. C., Gerstner J., Striker R. ( 2007) Hepatology 46, 1026– 1033 [DOI] [PubMed] [Google Scholar]
- 47.Goto K., Watashi K., Inoue D., Hijikata M., Shimotohno K. ( 2007) 14th International Symposium on Hepatitis C viruses, Glasgow, United Kingdom, September 9–13,2008, P235 [Google Scholar]
- 48.Weidmann B., Puyang X., Poulin D., Mathy J. E., Ma S., Anderson L. J., Fujimoto R., Compton T. G., Lin K. ( 2008) 15th International Symposium on Hepatitis C Virus & Related Viruses, San Antonio, Texas, October 5–9, 2008, Abstr. 315 [Google Scholar]
- 49.Braaten D., Franke E. K., Luban J. ( 1996) J. Virol. 70, 3551– 3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bieniasz P. D. ( 2004) Nat. Immunol. 5, 1109– 1115 [DOI] [PubMed] [Google Scholar]
- 51.Cullen B. R. ( 2003) Nat. Med. 9, 1112– 1113 [DOI] [PubMed] [Google Scholar]
- 52.Luban J. ( 2007) J. Virol. 81, 1054– 1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Towers G. J. ( 2007) Retrovirology 4, 40– 45 [DOI] [PMC free article] [PubMed] [Google Scholar]