

Abstract
G protein-coupled receptor (GPCR) kinases (GRKs) phosphorylate activated GPCRs and initiate their desensitization. Many prior studies suggest that activated GPCRs dock to an allosteric site on the GRKs and thereby stimulate kinase activity. The extreme N-terminal region of GRKs is clearly involved in this process, but its role is not understood. Using our recent structure of bovine GRK1 as a guide, we generated mutants of solvent-exposed residues in the GRK1 kinase domain that are conserved among GRKs but not in the extended protein kinase A, G, and C family and evaluated their catalytic activity. Mutation of select residues in strands β1 and β3 of the kinase small lobe, αD of the kinase large lobe, and the protein kinase A, G, and C kinase C-tail greatly impaired receptor phosphorylation. The most dramatic effect was observed for mutation of an invariant arginine on the β1-strand (∼1000-fold decrease in kcat/Km). These residues form a continuous surface that is uniquely available in GRKs for protein-protein interactions. Surprisingly, these mutants, as well as a 19-amino acid N-terminal truncation of GRK1, also show decreased catalytic efficiency for peptide substrates, although to a lesser extent than for receptor phosphorylation. Our data suggest that the N-terminal region and the newly identified surface interact and stabilize the closed, active conformation of the kinase domain. Receptor binding is proposed to promote this interaction, thereby enhancing GRK activity.
G protein-coupled receptor kinases (GRKs)2 are members of the protein kinase A (PKA), G, and C (AGC) family that phosphorylate Ser/Thr residues in the cytoplasmic loops and C termini of activated G protein-coupled receptors (GPCRs) (1). Receptor phosphorylation facilitates the binding of arrestin, which uncouples heterotrimeric G proteins, promotes receptor internalization, and activates arrestin-dependent signaling pathways (2, 3). Although playing a beneficial role in receptor desensitization, GRKs are implicated in a range of human diseases, including retinal degeneration, hypertension, heart failure, rheumatoid arthritis, opiate addiction, and various cancers (2, 4). Seven GRKs have been identified in mammals. They can be divided into the following three subgroups based on their sequence homology: GRK1 (GRK1 and GRK7), GRK2 (GRK2 and GRK3), and GRK4 (GRK4, GRK5, and GRK6). The primary structures of the three GRK subgroups are similar, consisting of tandem regulator of G protein signaling homology (RH) and kinase domains. Less conserved sequences involved in phospholipid membrane attachment are found at their C termini (Fig. 1A).
FIGURE 1.

GRK surface residues potentially important for GPCR phosphorylation. A, domain architecture of bGRK1. B, sequence alignment of regions from GRKs that were targeted in this study with other AGC kinases. Colored boxes map these regions back to the domain structure shown in A. Regions of the core kinase domain that contain residues conserved in the GRK subfamily, but not in the extended AGC kinase family, are highlighted in brown. Conserved residues of the AGC kinase C-tail are highlighted in green. Positions investigated in this study are indicated with asterisks. Only one PDB accession code for each kinase of known structure is shown in parentheses. Residue numbers correspond to those of bGRK1. The PXXP, turn, and hydrophobic (HF) motifs (highlighted in gray) are characteristic features found in most AGC kinase C-tails (22). Yeast, Saccharomyces cerevisiae; M.tb, Mycobacterium tuberculosis. C, ribbon diagram of the bGRK1535-H6·ATP complex. The model is a hybrid that contains all the ordered elements from the two unique chains resolved in the crystal structure (PDB accession number 3C4W), such that the observed N terminus and a nearly complete AST region of the kinase C-tail are displayed in a single model. The RH domain is colored gray, and the β-strands and α-helices of the core kinase domain are dark and light brown, respectively. The hinge region joining the kinase small and large lobes (between β5 and αD) is colored yellow. The N-terminal region and the AGC kinase C-tail are shown in green and the AST loop in cyan. The ATP molecule bound in the active site is shown as a stick model, and the two associated Mg2+ ions are colored black. D, conservation scores of GRKs mapped onto the surface of bGRK1. The area boxed in C is shown. The conservation scores were calculated by comparing the sequence conservation of residues from the core kinase domain of GRKs with those of the entire AGC family (see text). Residues are colored using a gradient, from magenta (more conserved in GRKs than the AGC kinases) to white (as conserved in GRKs as in AGC kinases) and to yellow (more variable in GRKs than in AGC kinases). The RH domain, which was not included in this calculation, is colored gray. Highest conservation among GRKs cluster at two sites, “site 1” and “site 2.” Site 1 corresponds to a region of the small lobe left exposed by the shorter AST loop found in GRKs relative to other AGC kinases. The AST region of protein kinase B (PDB accession number 1O6K) is superimposed for comparison (blue ribbon).
All eukaryotic protein kinases, including GRKs, contain a core catalytic domain of ∼250 amino acids that can be divided into two subdomains, called the small (or N) and large (or C) lobes. The small lobe consists of a five-stranded β-sheet (β1–5) and a conserved helix, αC, whereas the large lobe is mostly α-helical. The active site is located at their interface, with the nucleotide-binding pocket formed primarily by the small lobe and the phosphoacceptor-binding site primarily by the large lobe. In their active conformation, kinases position the hydroxyl group of the phosphoacceptor substrate in the proper orientation with respect to the γ-phosphate of ATP via a network of interactions formed by conserved structural elements from both lobes. Control of this network often underlies the molecular basis for allosteric regulation of protein kinase activity (5–9).
In GRKs, this allosteric regulation appears to be mediated by interactions with activated GPCRs. Steady-state kinetics indicate that the Km values of receptor substrates are in the micromolar range, whereas those of peptide substrates, even those derived from receptors, are in the millimolar range (10–13). Moreover, the catalytic efficiency for peptide phosphorylation by GRKs is much lower than that for receptor phosphorylation, and it can be enhanced in the presence of activated receptors (11, 12, 14). Thus, in addition to the peptide phosphoacceptor-binding site of the large lobe, an additional allosteric receptor-docking site appears to be required to promote catalytic activity in GRKs.
The molecular basis for how GRKs interact with activated GPCRs is poorly understood. In vitro, GRKs show little specificity among GPCRs, requiring only that the receptor be in an activated conformation. For example, although GRK1 is the predominant kinase expressed in rod outer segments, GRK1, GRK2, and GRK5 all phosphorylate bovine rhodopsin in a light-dependent manner with comparable catalytic efficiencies (15–17). Therefore, it seems likely that GRKs have a common molecular mechanism for the recognition of activated GPCRs. The region of GRKs most strongly linked to efficient receptor phosphorylation is the highly conserved N-terminal region, which is unique to the GRK subfamily and predicted to form an α-helix (Fig. 1B). Deletion of this region in GRK1, -2, or -5 abolishes receptor phosphorylation (18–20). Additionally, the binding of antibodies (18) or of recoverin (21) to the GRK1 N-terminal region inhibits receptor phosphorylation. In GRK5, it has also been suggested that the N terminus plays a role in phospholipid interactions (20). Another region that is likely involved in the allosteric regulation of GRKs is the AGC kinase C-terminal tail (C-tail), which is an extension of the kinase core domain and often plays a regulatory role in AGC kinases (22–24) (Fig. 1, B and C). The central segment of the C-tail, termed the active site tether (AST), contributes residues to the active site and is only well ordered in kinase domain structures that are in conformations resembling the active state.
To date, crystal structures representing each GRK subgroup have been reported, i.e. bovine GRK1 (bGRK1), bovine GRK2 (bGRK2), and human GRK6 (hGRK6) (25–29). Although most of these structures were co-crystallized in the presence of ATP or nucleotide analogs, none adopted the closed, active conformation exhibited by nucleotide-bound PKA (30), and the AST region of their AGC kinase C-tails were either partially or totally disordered. Similarly, the N-terminal region important for receptor phosphorylation was only observed in one crystal structure, namely that of one chain of the bGRK1·ATP complex. Thus, the regions believed to be most important for receptor interaction were largely disordered in these structures, leaving the molecular basis for how GPCRs interact with GRKs unclear. Because the kinase domains in these structures appear to be otherwise competent for catalysis, it is expected that activated GPCRs bind in a manner that promotes full kinase domain closure. Interactions with negatively charged lipids in the cell membrane are also expected to play a role in this transition (20, 31, 32).
In this study, we used recently determined structures of bGRK1 as a template to identify surface residues of the kinase domain that are conserved in GRKs but not in the extended AGC family. Biochemical characterization of site-directed mutants of these residues in bGRK1 identified a continuous surface on the small lobe and the AGC kinase C-tail that is critical for GRK activation by GPCRs. The residue whose mutation showed the largest effect on receptor phosphorylation is nearly as important as the N-terminal region, and the analogous residue is also critical in the other two GRK subgroups, represented by bGRK2 and hGRK6. Comparison with other AGC kinases reveals that this surface is uniquely available for protein-protein interactions in the GRK subfamily. A model for activation that involves cooperative interactions between the N-terminal region and the kinase domain is presented.
EXPERIMENTAL PROCEDURES
Reagents
[γ-32P]ATP was purchased from MP Biomedicals and GE Healthcare. Peptide substrates DDEASTTVSKTETSQVARRR (peptide C) and RRREEEEESAAA (RESA) were synthesized by solid-phase synthesis, and their concentrations in solution were determined by quantitative amino acid analysis at the Protein Structure Facility, University of Michigan Medical Center, Ann Arbor. Dark-adapted bovine retinas were purchased from W. L. Lawson Co., Lincoln, NE. A pcDNA3.1 vector bearing the coding sequence for human β2-adrenergic receptor (hβ2AR) with an N-terminal influenza hemagglutinin (HA) tag was a gift from Drs. Marc Caron and Larry Barak (Duke University). A pcDNA3 vector bearing the coding sequence for bGRK2 was a gift from Dr. Jeffrey Benovic (Thomas Jefferson University). Anti-GRK2 monoclonal antibodies C5/1 and R11E1 were gifts from Dr. Martin Oppermann (Georg-August-University, Germany).
Modeling
Structure figures were prepared using PyMOL.
Production of GRK Mutants
The QuikChange site-directed mutagenesis kit (Stratagene) was used to introduce mutations into a construct that expresses bGRK1 C-terminally truncated at residue 535 and hexahistidine (H6)-tagged (bGRK1535-H6) (29). An N-terminal deletion mutant (ΔN19) of bGRK1 was generated by cloning the cDNA sequence corresponding to amino acids 20–535 between the BamHI and SalI sites of pFastBacDualH6. All mutations were verified by DNA sequencing. Mutants were expressed in High Five insect cells using the Bac-to-Bac system (Invitrogen) and purified as described previously (29). Briefly, a metal-chelating affinity chromatography (nickel-nitrilotriacetic acid, Qiagen) was followed by cation exchange chromatography (Source 15S, GE Healthcare). Some mutants were further purified by size-exclusion chromatography (Superdex 200, GE Healthcare). The purity of each bGRK1535-H6 variant was >90% as judged by SDS-PAGE.
Full-length bGRK2 and residues 1–531 of hGRK6, whose C-terminal end is analogous to the last visible residue in the crystal structure of bovine GRK1, were cloned into the pFastBacDualH6 vector between the BamHI and SalI sites to create vectors that express bGRK2FL-H6 and hGRK6531-H6, respectively. The bGRK2FL-H6 (R195A) and hGRK6531-H6 (R190A) mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene). Proteins were purified in the same manner as bGRK1535-H6.
All GRK proteins were stored in small aliquots at −80 °C, and their concentrations were determined by absorbance at 280 nm using estimated molar extinction coefficients of 63,830 m−1 cm−1 for bGRK1535-H6, 71,280 m−1 cm−1 for bGRK2FL-H6, and 59,820 m−1 cm−1 for hGRK6531-H6.
Rhodopsin Phosphorylation Assay
Urea-washed bovine rod outer segments (bROS) were purified as described previously (33). Steady-state kinetics were conducted at saturating (0.5–2 mm) [γ-32P]ATP, 0–80 μm urea-washed bROS, and 0.03–0.2 μm GRK variants in 100 mm HEPES-NaOH, pH 7.5, 0.15 m NaCl, 10 mm MgCl2, and 1 mm EDTA (Buffer A). The reaction was initiated by addition of ATP and then incubated at room temperature (22–26 °C). Small aliquots (3–5 μl) of the reaction mixture were removed at 4–5 time points within the first 5 min of the reaction where a linear relationship holds, and less than 10% of substrate was consumed. The reactions were quenched with SDS-PAGE sample loading buffer and analyzed by SDS-PAGE. Dried gels were exposed to a storage phosphor screen, and phosphorylated rhodopsin was quantified using a Typhoon 9410 imager (GE Healthcare). Initial rates were calculated from the slope of phosphorylated rhodopsin production at various time points and fitted to the Michaelis-Menten equation to obtain kcat and Km (bROS) using Prism version 4.0a.
In Vivo Receptor Phosphorylation Assays
bGRK2-R195A and N-terminal deletion (bGRK2-ΔN16) were generated in the pcDNA3-bGRK2 vector, and the hβ2AR-Y326A mutant was generated in the pcDNA3.1-hβ2AR vector. HEK293T cells (6-cm dish, ∼50% confluent) were transiently co-transfected with 1 μg of HA-hβ2AR-Y326A pcDNA3.1 and 1 μg of pcDNA3 of bGRK2-WT, -R195A, -ΔN16, or empty vector using FuGENE 6 (Roche Applied Science). After ∼44 h of transfection, cells were treated ±1 μm isoproterenol for 20 min at 37 °C. After being washed with ice-cold phosphate-buffered saline, the cells were immediately lysed in 200 μl of RIPA lysis buffer containing 50 mm Tris-HCl, pH 7.5, 0.15 m NaCl, 1% Triton X-100, 1% sodium cholate, 0.1% SDS, 10 mm NaF, and protease inhibitor mixture (1836145001, Roche Applied Science) and phosphatase inhibitor mixtures (sc-45045 and sc-45065, Santa Cruz Biotechnology) at the manufacturers' recommended concentrations on ice. The lysate was then spun down at 16,000 × g for 15 min. 5 or 10 μl of the soluble fraction was analyzed by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and then probed with mouse anti-GRK2 monoclonal antibody (C5/1) (10 μg/ml) and rabbit anti-actin polyclonal antibodies (Cytoskeleton AAN01) (1:1000 dilution). The rest of the soluble fraction was incubated with 1 μg of anti-HA monoclonal antibody 16B12 (Covance MMS-101P) and ∼10 μl of protein A-Sepharose (16-156, Upstate) at 4 °C overnight. The beads were washed with cold phosphate-buffered saline three times and then resuspended in a total of ∼40 μl of SDS-PAGE sample loading buffer. ∼10 μl was then analyzed by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed with either mouse anti-β2AR monoclonal antibody R11E1 (5–10 μg/ml) or rabbit anti-Ser(P)-355,356-β2AR polyclonal antibodies (1:500 dilution) (sc-16719-R, Santa Cruz Biotechnology). Horseradish peroxide (HRP)-conjugated secondary antibodies were used, and the Western blots were visualized by film after being exposed for 5–30 min.
Nucleotide Binding Assays
The dissociation constant of the WT bGRK1535-H6·ADP complex was measured on a MicroCal PC-isothermal titration calorimeter. 10 μm bGRK1535-H6-WT was placed in the sample cell and titrated by 15 injections of 20 μl of 100 μm ADP in 20 mm HEPES-NaOH, pH 7.5, 0.15 m NaCl, and 5 mm MgCl2 (Buffer B) at 25 °C. The data were analyzed using Origin software to obtain KA, ΔH, and ΔS values.
The dissociation constants for bGRK1535-H6·ADP variant complexes were measured using a competition fluorescence polarization assay. The dissociation constant of BODIPY® TR ADP (Invitrogen) was first determined using 0.02 μm BODIPY® TR ADP and 0–6.4 μm bGRK1535-H6 variants in Buffer B at λex = 575 nm and λem = 620 nm. Samples were incubated in the dark at room temperature for 5–10 min before the millipolarization (mP) was recorded. For background subtraction, 20 mm EDTA was then added to the wells, and the mP was recorded again after 5–10 min of incubation in the dark. Data were fitted to Equation 1,
 |
where mPmax is the maximal mP, [P]t is the total protein concentration, and KD is the dissociation constant.
Competition assays were conducted using 0.02 μm BODIPY® TR ADP, 1 μm of each bGRK1535-H6 protein, and 0–10 μm ADP in Buffer B at room temperature. The data were fitted to a competition model accounting for ligand depletion as shown in Equations 2 and 3,
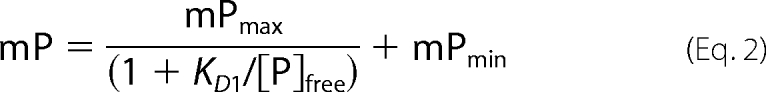 |
where [L2]t is the total ADP concentration; KD1 is the dissociation constant of BODIPY® TR ADP; [P]free is the free protein concentration; mPmin is the minimal mP; and b = [P]t + [L2]t + KD2, where KD2 is the dissociation constant of ADP.
Peptide Phosphorylation Assay
The activity of GRKs for peptide substrates was measured by filter binding assays using either peptide C, which corresponds to the C terminus of bovine rhodopsin and contains its nascent phosphorylation sites (11), or RESA, an acidic peptide substrate with a single phosphorylation site (10). Three Arg residues located at the ends of these peptides facilitate their binding to phosphocellulose paper. The reactions contained 1–10 μm GRK variant, 100 μm [γ-32P]ATP, and 0–16 mm peptide in Buffer A. Reactions were incubated at room temperature for 30–270 min, and small aliquots (∼5 μl) were quenched by addition of 15–30 μl of 15% trichloroacetic acid at various time points. Samples were pelleted in a microcentrifuge at 13,000 rpm for 10 min, and their supernatants were applied to P-81 paper (2.5-cm circles; Whatman) and washed with 2 ml of 75 mm H3PO4 at least 10–15 times (total ∼20–30 ml). Filters were then air-dried, and their radioactivity was quantified by liquid scintillation counting. Initial rates were calculated from the slope of phosphorylated peptide production and fitted to the Michaelis-Menten equation to obtain apparent kcat (kcat,app) and Km (Km,app) values using Prism. These values are only apparent because the reactions were not performed under steady-state conditions, in that the total product concentration was less than the total enzyme concentration.
RESULTS
The most complete atomic model for a GRK is the structure of the bGRK1·ATP complex (PDB accession number 3C4W), in which we were able to trace structural elements that were unresolved in prior structures of bGRK2 and hGRK6 (29). Presumably due to the relatively closed conformation of the GRK1 kinase domain and to the presence of well ordered ATP in the active site, portions of the AST region of the AGC kinase C-tail were ordered. The best ordered portion was the “AST loop” (residues 472–477 of the AST region, colored in cyan in Fig. 1C), which adopts a loop conformation very similar to that observed in other AGC kinase domains (30, 34). The interactions at the base of the AST loop in this bGRK1 structure are almost identical to those in the structure of the PKB·AMP-PNP complex (34). A hydrogen bond is formed between a tyrosine hydroxyl and a backbone amide nitrogen located at the beginning of the loop, and an aromatic side chain from αD in the large lobe stacks against the peptide bond that immediately follows the AST loop (Fig. 1, C and D). The AST loop, however, is four residues shorter in GRK1 than in PKB, and as a result a region of the GRK small lobe adjacent to the hinge is more solvent-exposed than in structures of either PKA or PKB (Fig. 1D). In the sole GRK structure wherein the N terminus was observed, residues 11–20 form a helix that packs adjacent to this exposed surface of the small lobe (Fig. 1, C and D). Given the location of this surface near the hinge of the kinase domain and to the observed N terminus, we hypothesized that it could be either directly or indirectly involved in docking with receptors (29).
We also anticipated that if there were residues on the surface of the GRK kinase domain that were important for GPCR docking, then they should be conserved in GRKs but not in the broader AGC family. To test this hypothesis, we analyzed unique sequences from 44 GRKs and 542 AGC kinases, including GRKs (on-line Protein Kinase Resource) (35). Both ClustalW (36) and MUSCLE (37) were used to create alignments. Only the core kinase domain spanning from β1 of the small lobe to αI of the large lobe (bGRK1 residues 187–445) (Fig. 1C) was used for comparisons between the GRK subfamily and the AGC family. The AGC kinase C-tail was not included because the low sequence homology of this region made alignments unreliable. Outputs from ClustalW and MUSCLE were then analyzed using ConSurf to calculate the conservation score for each residue (38, 39). The results were then mapped onto a composite model of the bGRK1 structure (Fig. 1D).
Several selection criteria were applied to identify the residues of most interest. First, an amino acid position had to have a negative normalized score (conserved) in the GRK subfamily and a positive normalized score (variable) in the broader AGC kinase family. Second, the absolute difference of these scores had to be greater than 0.5. Finally, these criteria had to be met in both ClustalW and MUSCLE alignments. A total of 24 residues met all of these criteria, including Phe190, Arg191, Arg195, Gln205, Ala208, Thr209, Gly210, Lys211, Leu212, Lys221, Arg222, Lys224, Arg225, Arg226, Met233, Ser249, Ala251, Lys257, Asn268, Asn278, Gly393, Glu400, Val410, and Phe416 (bGRK1 numbering). Seven residues, Val192, His275, Tyr277, Gly285, Glu394, Glu397, and Arg404, met both criteria in only one alignment and one criterion in the other. Interestingly, most of these residues cluster at one of two sites on the small lobe (Fig. 1, B and D). The first is on a continuous surface formed by the β1–β3-strands (site 1) and the other as a cluster of positively charged residues primarily from the αB-helix (site 2). Site 1 was of most interest with regards to receptor coupling, as it corresponded to the same surface of the small lobe left exposed by the relatively short AST loop of GRKs. Site 2, on the other hand, seemed more likely to be involved in interacting with an autophosphorylation site (the “turn motif”) of the AGC kinase C tail (Fig. 1B) or perhaps with acidic substrates in the polypeptide-binding site. A total of 10 residues from site 1 that were not playing an obvious structural role in bGRK1 were selected for subsequent mutagenesis and kinetic analysis, including Phe190, Arg191, Val192, Arg195, Gln205, Gly210, Leu212, Asn268, Tyr274, and His275 (Fig. 1B). Because the AST region of the AGC kinase C-tail is in close proximity to site 1, we also selected six residues from this region (Val476, Asn480, Gln482, Asp483, Val484, and Phe487) for functional analysis (Fig. 1B). For a control, we also generated a truncation mutant of bGRK1 that lacked the N-terminal 19 amino acids (ΔN19), as this protein is not expected to phosphorylate GPCRs. All mutations were made in the background of the bGRK1535-H6 protein and purified to homogeneity from baculovirus-infected insect cells (29).
To test the catalytic efficiency of bGRK1535-H6 mutants against a receptor, we used bROS as the substrate (Fig. 2, A and B, and Table 1). The kcat and Km (bROS) values for bGRK1535-H6 were 3.5 min−1 (57 nmol/min/mg) and 4.7 μm, respectively, at room temperature. The Km value is comparable with the reported value (4 μm) for wild-type GRK1, but the kcat value is lower than the reported number of ∼700 nmol/min/mg at 30 °C (10). However, an ∼2.5–3-fold lower rate is observed for kcat at room temperature, and we also included 0.15 m NaCl in the buffer, which lowers the kcat by another ∼3-fold (data not shown). Although inclusion of salt affects the reactivity, we felt it was more appropriate to keep the ionic strength as close to physiological as possible, as well as constant for all of our reactions.
FIGURE 2.

Receptor phosphorylation by GRK mutants. A, bGRK1535-H6 R191A mutant is severely impaired in its ability to phosphorylate receptors. Shown is the Michaelis-Menten plot (Vi/[E]) of bROS phosphorylation by bGRK1535-H6 (closed squares) and the R191A mutant (open squares). Kinetic parameters for these and other bGRK1 variants are listed in Table 1. B, bGRK1535-H6 R191A mutant is nearly as defective in bROS phosphorylation as the ΔN19 truncation. Shown is the Michaelis-Menten plot (Vi) of bROS phosphorylation by bGRK1535-H6 R191A (open squares) and ΔN19 (open circles) at 2 μm, and the background reaction in the absence of GRK (closed triangles). The R191A mutant shows no curvature up to 40 μm bROS, and the ΔN19 truncation shows no product formation above background. C, residues most critical for bROS phosphorylation cluster in or adjacent to site 1 on the small lobe. The structure is in the same orientation and uses the same color scheme as Fig. 1C. Side chains of residues targeted by site-directed mutagenesis in this study are shown as spheres, which are colored in a gradient according to the fold decrease of their catalytic efficiency for bROS compared with WT from white (no change) to magenta (severely impaired). Residues whose mutation led to a >10-fold decrease in catalytic efficiency (kcat/Km) are labeled with their fold decrease. D, bGRK2-R195A and -ΔN16 mutants are defective in receptor phosphorylation in vivo. HEK293T cells were transiently co-transfected with vectors expressing HA-hβ2AR-Y326A and either bGRK2-WT, -R195A, -ΔN16, or empty vector for ∼44 h. Immunoprecipitations and immunoblots were performed and analyzed after the cells were treated ±1 μm isoproterenol for 20 min. Only bGRK2-WT was able to phosphorylate agonist-stimulated receptor. This figure shows representative immunoblots from one of two independent experiments.
TABLE 1.
Kinetic parameters of GRK variants for bROS phosphorylation
Steady-state kinetics were conducted at saturating ATP using 0–80 μm urea-washed bROS and 0.03–0.2 μm GRK mutants in 100 mm HEPES-NaOH, pH 7.5, 0.15 m NaCl, 10 mm MgCl2, and 1 mm EDTA (Buffer A). The reaction was initiated by addition of ATP and then incubated at room temperature (22–26 °C). Values shown represent means ± S.E. of the nonlinear regression fit calculated from three to six independent experiments.
| GRK mutants | kcat | Km | kcat/Km | -Fold decrease (kcat/Km) |
|---|---|---|---|---|
| min−1 | μm | μm−1·min−1 | ||
| bGRK1-WT | 3.5 ± 0.3 | 4.7 ± 1.6 | 0.73 ± 0.20 | 1 |
| Kinase small lobe | ||||
| F190A | 0.64 ± 0.04 | 9.4 ± 1.9 | 0.069 ± 0.029 | 11 |
| F190Y | 3.2 ± 0.4 | 4.8 ± 2.0 | 0.67 ± 0.22 | 1.1 |
| F190H | 2.4 ± 0.1 | 2.3 ± 0.6 | 1.0 ± 0.2 | 0.71 |
| R191Aa | ≥0.032 | ≥47 | 0.00069 ± 0.00004 | 1100 |
| R191Ka | ≥0.017 | ≥33 | 0.00051 ± 0.00006 | 1400 |
| V192A | 2.0 ± 0.2 | 2.6 ± 1.0 | 0.77 ± 0.24 | 0.91 |
| R195A | 1.2 ± 0.1 | 1.9 ± 0.8 | 0.61 ± 0.21 | 1.2 |
| Q205A | 1.7 ± 0.1 | 2.0 ± 0.6 | 0.85 ± 0.20 | 0.83 |
| G210E | 0.87 ± 0.06 | 5.4 ± 1.0 | 0.16 ± 0.02 | 4.5 |
| L212A | 0.51 ± 0.08 | 7.7 ± 3.9 | 0.066 ± 0.025 | 11 |
| L212M | 3.5 ± 0.7 | 2.2 ± 0.7 | 1.6 ± 0.4 | 0.45 |
| N268A | 1.9 ± 0.3 | 3.1 ± 2.1 | 0.64 ± 0.36 | 1.1 |
| Kinase large lobe | ||||
| Y274A | 0.20 ± 0.04 | 12 ± 6 | 0.016 ± 0.005 | 45 |
| Y274F | 4.4 ± 0.2 | 2.4 ± 0.6 | 1.8 ± 0.4 | 0.40 |
| H275A | 1.8 ± 0.2 | 2.1 ± 1.1 | 0.87 ± 0.37 | 0.83 |
| AGC kinase C-tail | ||||
| V476A | 0.26 ± 0.05 | 9.8 ± 5.5 | 0.027 ± 0.011 | 27 |
| N480A | 2.1 ± 0.2 | 2.3 ± 1.1 | 0.89 ± 0.36 | 0.83 |
| Q482A | 2.4 ± 0.2 | 2.6 ± 0.7 | 0.96 ± 0.22 | 0.77 |
| D483A | 2.1 ± 0.7 | 6.7 ± 5.7 | 0.31 ± 0.17 | 2.4 |
| V484A | 0.32 ± 0.06 | 9.5 ± 5.5 | 0.034 ± 0.014 | 21 |
| F487A | 2.0 ± 0.3 | 4.3 ± 3.0 | 0.46 ± 0.26 | 1.6 |
| N-terminal region | ||||
| ΔN19 | NDb | NDb | NDb | >3900b |
| bGRK2-WT | 28 ± 5 | 47 ± 14 | 0.60 ± 0.08 | 1 |
| R195A | 0.030 ± 0.011 | 45 ± 31 | 0.00066 ± 0.00020 | 910 |
| hGRK6-WT | 0.026 ± 0.006 | 20 ± 11 | 0.0013 ± 0.0004 | 1 |
| R190A | NDc | NDc | NDc | >100c |
| Uncatalyzed reaction (Vi/[S])d | 0.00037 ± 0.00004 min−1 | |||
| Dark reaction (Vi)e | 0.00025 ± 0.00009 μm/min | |||
a No curvature was observed up to 40 μm bROS.
b ND means not detectable. No product formation above the background up to 2 μm GRK was observed. The minimum fold decrease of kcat/Km was calculated based on the uncatalyzed reaction.
c ND means not detectable. No product formation above the background up to 30 μm GRK was observed. The minimum fold decrease of kcat/Km was calculated based on the uncatalyzed reaction.
d Uncatalyzed reaction was measured in the absence of GRK. Initial rates show a linear relationship to the substrate concentration. The number shown is the slope of the linear fit.
e Dark reactions were measured at 0.2 μm GRK1535-H6-WT, R191A, and ΔN19. Initial rates are independent of substrate concentration or GRK variants. The number shown is the averaged initial rates from 12 measurements.
On the kinase small lobe, the R191A mutant showed the most dramatic effect with a decrease in kcat/Km of more than 1000-fold (Fig. 2, A and B, and Table 1). Arginine is invariant at this position in GRKs. Mutation of structurally adjacent residues (F190A and L212A) also exhibited >10-fold decreases in catalytic efficiency. The Y274A mutant in αD of the kinase large lobe and the V476A and V484A mutants in the AGC kinase C-tail showed 20–50-fold decreases in kcat/Km. The remainder of the mutations showed <5-fold decreases in their kcat/Km values (Table 1), indicating that their side chains do not significantly contribute to the process of receptor phosphorylation. They are likely conserved for other reasons. As expected, the activity of the ΔN19 truncation was severely compromised, and its activity could not be determined under the assay conditions (up to 2 μm enzyme) because there was no observed product formation above the background rate (Fig. 2B). Based on this rate, the catalytic efficiency for the ΔN19 mutant is decreased by at least 3900-fold.
We anticipated that proteins with defects in receptor docking should exhibit higher Km values. This was true for the R191A mutant, whose initial rates show no curvature in the Michaelis-Menten plot up to 40 μm ROS. However, most of the bGRK1535-H6 mutants exhibited a far more profound effect on kcat than Km, suggesting that the primary defect is in kinase domain closure or other steps critical for the phosphotransfer reaction. When the fold decrease in catalytic efficiency for each mutant was mapped onto the bGRK1 structure, residues showing significantly impaired receptor phosphorylation form a surface that spans across the site 1 on the small lobe, the AGC kinase C-tail, and the large lobe (Fig. 2C).
Because GRKs are promiscuous in their ability to phosphorylate a variety of GPCRs, we anticipated that conversion of residues in GRK1 that are important for receptor phosphorylation to their equivalents in other GRKs should generate proteins with activity close to wild type. For example, bGRK1-Phe190 is Tyr in GRK4, -5, and -6 and His in GRK2 and -3; bGRK1-Leu212 is Met in other GRKs, and bGRK1-Tyr274 is Phe in GRK4, -5, -6, and -7 (Fig. 1B). As expected, the F190Y, F190H, L212M, and Y274F mutants of bGRK1535-H6 retained catalytic efficiencies similar to those of wild type (Table 1).
Thus far, our data strongly suggested that site 1 and adjacent residues in the AGC kinase C-tail were important for GPCR phosphorylation. To confirm that this region is also important to other GRKs, we introduced mutations equivalent to bGRK1-R191A into bGRK2 and hGRK6 (R195A and R190A, respectively). To simplify protein purification, we first generated C-terminal H6-tagged versions of full-length bGRK2 (bGRK2FL-H6) and residues 1–531 of hGRK6 (hGRK6531-H6). Indeed, bGRK2FL-H6 R195A and hGRK6531-H6 R190A exhibited substantially decreased values of kcat/Km compared with their unmodified kinase equivalents (Table 1). For bGRK2FL-H6, this corresponded to an ∼900-fold decrease in catalytic efficiency. For hGRK6531-H6 R190A, the reactivity was too low to measure accurately (no product formation above the background rate using up to 30 μm enzyme). Based on this background rate, the activity of hGRK6531-H6 R190A is decreased at least 100-fold.
In PKA and some other AGC kinases, the residue equivalent to bGRK1-Arg191 is lysine (Fig. 1A), a conservative substitution that preserves charge. In PKA, this residue (Lys47) forms a salt bridge with a residue in the AGC kinase C-tail, implying a role in stabilizing kinase domain closure. To test whether a Lys substitution at this position could preserve the activity of GRKs, we created the bGRK1-R191K mutant. Strikingly, this mutant was as inactive as bGRK1535-H6 R191A (Table 1), indicating that Arg191 is not simply involved in a charge-charge interaction with GPCRs or phospholipid head groups. It is more likely that the guanidinium group of arginine is required for its function, such as via a specific hydrogen bond network or stacking interaction.
To confirm that this invariant arginine residue also plays a role in receptor phosphorylation in cells, we measured in vivo phosphorylation of the hβ2AR-Y326A mutant by overexpressed bGRK2-WT, -R195A, and -ΔN16 (equivalent to bGRK1-ΔN19) mutants in the presence or absence of the agonist isoproterenol. The hβ2AR-Y326A mutant, which is compromised in its ability to be phosphorylated by endogenous GRKs, was used in the assay to observe the effects from overexpressed GRK2 (40–42). HEK293T cells were transiently co-transfected with a vector expressing N-terminally HA-tagged hβ2AR-Y326A and either a vector expressing either bGRK2-WT, -R195A, or -ΔN16, or vector alone. As shown in Fig. 2D, only bGRK2-WT was able to phosphorylate the receptor in the presence of the agonist isoproterenol. For detection, we used an antibody specific for phosphorylation at Ser355 and Ser356 of the hβ2AR, positions that are specifically phosphorylated by GRKs (43) and that are critical for the receptor desensitization and internalization mediated by β-arrestin (44, 45). Thus, both bGRK2-R195A and -ΔN16 exhibit defects in receptor phosphorylation that will profoundly affect regulation of hβ2ARs in cells.
Although our structure of bGRK1 suggests that the residues we mutated in site 1 do not appear to directly contribute to the active site, we wanted to confirm that the defects observed for these mutants did not result from perturbation of the active site. We therefore compared the dissociation constants of bGRK1535-H6 variants for ADP (Fig. 3 and Table 2). We first used isothermal titration calorimetry, which revealed the KD of the bGRK1535-H6-WT·ADP complex to be 0.89 μm (Fig. 3A). Nucleotide binding was enthalpically driven, with ΔH and TΔS values of −15.7 ± 0.3 and −7.5 ± 0.6 kcal/mol, respectively. Because this method requires large amounts of protein, nucleotide binding to each of the bGRK1535-H6 mutants was instead measured using a competitive fluorescence polarization assay. In good agreement with the calorimetry result, we measured a KD of 0.90 μm for the bGRK1535-H6·ADP complex (Fig. 3B). The KDvalue measured for each of the bGRK1535-H6 mutants for ADP was similar to that observed for bGRK1535-H6 (0.7–1.0 μm), indicating that the impaired ability of site 1 mutants to phosphorylate receptors is not because of defects in nucleotide binding.
FIGURE 3.

Measurement of the KD of the bGRK1535-H6·ADP complex. A, isothermal titration calorimetry; B, competition fluorescence polarization. The KD value of BODIPY® TR ADP for each bGRK1535-H6 protein was first measured, and then the KD of unlabeled ADP for each bGRK1535-H6 mutant was determined by competition displacement of BODIPY® TR ADP. Both assays indicated that the KD of the bGRK1535-H6·ADP complex is 0.9 μm. The KD values for selected bGRK1535-H6 variants are listed in Table 2.
TABLE 2.
Dissociation constants for bGRK1·ADP complexes
Isothermal titration calorimetry (ITC) was conducted by placing 10 μm bGRK1535-H6 in the sample cell and titrating with 15 injections of 20 μl of 100 μm ADP in 20 mm HEPES-NaOH, pH 7.5, 0.15 m NaCl, and 5 mm MgCl2 (Buffer B) at 25 °C. Competition fluorescence polarization (FP) assays were conducted using 0.02 μm BODIPYTM-ADP, 1 μm of each bGRK1535-H6 protein, and 0–10 μm ADP in Buffer B at room temperature. Values of fluorescence polarization experiments shown represent mean ± S.D. of three independent experiments.
| bGRK1 | KD, FP | KD, ITC |
|---|---|---|
| μm | μm | |
| WT | 0.90 ± 0.19 | 0.89 ± 0.05 |
| F190A | 0.80 ± 0.35 | |
| R191A | 0.90 ± 0.35 | |
| L212A | 0.69 ± 0.23 | |
| Y274A | 0.97 ± 0.23 | |
| V476A | 0.86 ± 0.13 | |
| V484A | 0.90 ± 0.27 | |
| ΔN19 | 0.68 ± 0.20 |
Although residues in site 1 and adjacent regions on bGRK1 were clearly involved in phosphorylation of activated bROS, it was not known whether these residues are directly or indirectly involved in binding GPCRs. We hypothesized that if the sole function of a surface residue was to dock with activated receptors, then mutation of this residue should not affect the catalytic efficiency of peptide phosphorylation. We therefore measured the reactivity of our bGRK1535-H6 variants for nonreceptor substrates. For this study, we used a known GRK peptide substrate (peptide C), which contains residues 330–346 from the C terminus of bovine rhodopsin. The apparent kcat and Km (peptide C) for bGRK1535-H6-WT was 0.29 h−1 and 0.44 mm, respectively, at room temperature (Table 3 and Fig. 4). Interestingly, all bGRK1535-H6 mutants that showed decreased catalytic efficiency for bROS phosphorylation also displayed 10–50-fold decreased catalytic efficiency for peptide phosphorylation. Use of another GRK acidic peptide substrate (RESA) gave similar results (data not shown), indicating the decreased reactivity is not unique to peptide C. bGRK2FL-H6 R195A and hGRK6531-H6 R190A also exhibited impaired reactivity for peptides (data not shown), indicating that the function of this invariant arginine is shared in all three GRK subgroups. We also showed that bGRK1535-H6 mutants that bear tolerated amino acid substitutions (F190Y, F190H, L212M, and Y274F) could phosphorylate peptide substrates similarly to their wild-type counterparts (data not shown).
TABLE 3.
Kinetic parameters for peptide C phosphorylation
The reactions contained 1–10 μm GRK variant, 100 μm ATP, and 0–16 mm peptide in Buffer A. Values shown represent means ± S.E. of the nonlinear regression fit calculated from 40 (WT) or 8 (mutants) data points at various peptide substrate concentrations.
| bGRK1 | kcat,app | Km,app | kcat/Km,app | -Fold decrease(kcat/Km,app) |
|---|---|---|---|---|
| h−1 | mm | mm−1 h−1 | ||
| WT | 0.29 ± 0.05 | 0.44 ± 0.21 | 0.66 ± 0.22 | 1 |
| F190A | 0.020 ± 0.007 | 0.88 ± 0.61 | 0.023 ± 0.009 | 29 |
| R191A | 0.018 ± 0.005 | 1.2 ± 0.6 | 0.015 ± 0.004 | 44 |
| R191K | 0.033 ± 0.007 | 1.4 ± 0.5 | 0.023 ± 0.004 | 29 |
| L212A | 0.016 ± 0.002 | 0.41 ± 0.15 | 0.038 ± 0.010 | 17 |
| Y274A | 0.022 ± 0.005 | 1.5 ± 0.6 | 0.015 ± 0.003 | 44 |
| V476A | 0.025 ± 0.004 | 0.73 ± 0.24 | 0.033 ± 0.007 | 20 |
| V484A | 0.019 ± 0.005 | 0.89 ± 0.47 | 0.021 ± 0.006 | 31 |
| ΔN19 | 0.027 ± 0.008 | 0.53 ± 0.39 | 0.052 ± 0.025 | 12 |
FIGURE 4.

Nonreceptor (peptide) phosphorylation. Michaelis-Menten plots (Vi/[E]) are shown for peptide C phosphorylation either by bGRK1535-H6-WT (closed squares), by the R191A mutant (open squares), or by the ΔN19 truncation (open circles). Both the R191A and ΔN19 variants are compromised in their ability to phosphorylate peptide C. Kinetic parameters are listed in Table 3.
Because others have reported that deletion of the N terminus from the GRK does not affect its catalytic activity toward soluble substrates (19, 20), we also tested the ΔN19 truncation in our peptide phosphorylation assay. Surprisingly, we found that the catalytic efficiency of this protein was also defective. Because we expect that the phosphorylation of both peptide and GPCR substrates requires the kinase to adopt a similarly closed, active conformation, these results indicate that the N-terminal region and the residues we identified in site 1, the AGC kinase C-tail, and the large lobe are all involved in the allosteric transition of GRKs from an inactive, open conformation to the active, closed form. However, it is not yet clear which residues from these elements might also be involved in direct contact with GPCRs.
DISCUSSION
A central theme of protein kinase activation is to allow proper positioning of the γ-phosphate of ATP with respect to the phosphoacceptor hydroxyl of a protein substrate. Because the principal binding sites for these substrates reside on separate domains, kinase activation often entails proper orientation of the small and large lobes of the kinase domains through modulation of allosteric sites, either directly through covalent modification or indirectly through the repositioning of structural elements that block or alter key features of the active site. Phosphorylation of the so-called activation loop in the large lobe is commonly observed in AGC and other protein kinases, allowing this loop to undergo a conformational change that helps to order the catalytic machinery. This does not occur in GRKs because their analogous loops do not contain phosphorylation sites and, despite this, these loops are well ordered in all GRK structures determined thus far. Another mechanism commonly utilized by protein kinases is to control the position of the αC-helix, which bears an invariant glutamate that forms an ion pair with an invariant lysine on the β3-strand in the active state. This regulatory strategy is also not used by GRKs because this ion pair pre-exists in all structures of GRKs determined so far, and structures of GRKs bound to nucleotides reveal that these residues make their expected interactions with nucleotide ligands.
A hallmark feature of the AGC family is the AGC kinase C-tail, which bears additional phosphorylation sites called the “turn” and “hydrophobic” motifs. Phosphorylation at these sites is believed to contribute to stabilization of the closed state of the kinase (46). For example, phosphorylation of the hydrophobic motif of PKBβ and PKCθ helps to anchor the motif to the small lobe and to configure the small lobe in an active conformation (23, 47). Such an activation mechanism is not evident for GRKs, as their analogous hydrophobic motifs do not contain phosphorylation sites and are well ordered in all structures thus far. In PKA, an important Phe side chain located within the AST region contacts the adenine ring of ATP. When mutated to alanine, activity is lost (48). The residues equivalent to this position in GRKs are smaller (Ala or Cys). When this Ala is substituted with Phe in bGRK1, no effect on bROS phosphorylation was observed (data not shown). Thus, the C tails of GRKs do not have all of the same regulatory and structural properties found in those of other AGC kinases, in line with the poor sequence conservation of this region between GRKs and the rest of the AGC family. Although it is not yet clear how the C-tail is involved in the allosteric activation of GRKs, its role is likely significant given that the ordering of the AST region appears to correlate with the relative degree of kinase domain closure and that it is in close proximity to the kinase hinge and active site.
Prior to this study, the region of GRKs most strongly implicated in receptor-dependent phosphorylation was the extreme N terminus. This region could potentially play many roles, including receptor or phospholipid binding, allosteric modulation of the kinase domain, or some combination thereof. It is possible that the N-terminal region serves as the principal docking site for the receptor, and then their complex docks with a second site on the kinase domain to trigger kinase domain closure. Alternatively, the N-terminal region can interact with the kinase domain and together form a receptor-docking site. In either case, there must be residues on the surface of the kinase domain that are critical for GRK activation by receptors.
We combined structure-based and bioinformatics approaches to locate hot spots for receptor coupling on the surface of the kinase domain. Subsequently, we showed that Phe190, Arg191, and Leu212 on the small lobe, Tyr274 on the large lobe, and Val476 and Val484 on the AGC kinase C-tail are important for receptor phosphorylation. All of these residues are solvent-exposed in crystal structures and, after mutation, retain the ability to bind ADP with affinities similar to wild type. When mapped onto the bGRK1 structure, these residues constitute a continuous surface with the most critical residue, Arg191, located near its center (Fig. 2C). We showed that residues equivalent to bGRK1-Arg191 in other subgroups of GRKs are critical both in vitro and in vivo. Additionally, substitution of residues physically adjacent to bGRK1-Arg191 with amino acids found at corresponding positions in other GRKs retains wild-type catalytic efficiency. In a parallel study, mutation of residues Tyr281, Val477, and Ile485 of bGRK2 (equivalent to Tyr274, Val476, and Val484 of bGRK1, respectively), also show decreased catalytic efficiency for both receptor and peptide phosphorylation (49).
There are several possible mechanisms for how the residues we identified might be involved in kinase activation. First, they can simply make direct contacts with receptors. Second, they can collaborate with the N-terminal region to form a receptor-docking site. Third, they can be involved in the mechanism of kinase domain closure that is allosterically induced by GPCR binding. Because all of the positions we identified have defects in both GPCR and peptide phosphorylation, these residues cannot solely be involved in GPCR binding, ruling out the first model. However, this observation alone cannot rule out either the second or third models. Tyr274 on αD of the kinase large lobe is likely only involved in kinase domain closure. In nucleotide-bound bGRK1, the aromatic ring of Tyr274 stacks against the peptide bond formed between Tyr477 and Ala478 in the AST. It is therefore expected that this interaction contributes structurally to domain closure and to the stability of the active conformation.
It is somewhat surprising that the ΔN19 mutant of bGRK1 also exhibited a decreased reactivity for peptide phosphorylation. This is in contrast to previous studies, wherein similar truncations did not display obvious defects in soluble substrate phosphorylation, although steady-state kinetics were not evaluated in these cases (19, 20). Our data strongly suggest that this N-terminal region plays a role in kinase domain closure, even independently of receptors, because kinase domain closure must take place in order for either receptor or peptide phosphorylation to occur. We favor a model in which the N-terminal region docks with the adjacent key surface (site 1) of the kinase domain, where it may also interact with the AST, and thus drive closure of the kinase domain (Fig. 5). This model is appealing in that the cumulative interactions are concentrated near the hinge of the kinase domain and thus could help stabilize the kinase in its closed state. GPCRs likely bind to solvent-exposed structural elements in this closed configuration, thereby increasing substrate affinity by bringing its phosphoacceptor sites in proximity to the active site, while at the same time stabilizing the active GRK conformation. Such interactions are expected to enhance specificity and catalytic efficiency. Given the high sequence conservation among GRKs in their N-terminal regions and in the key surface residues we identified on the kinase domain, this model would represent a universal mechanism for GRK activation.
FIGURE 5.

Proposed model for GRK activation. In the absence of either nucleotide or protein substrate, GRK adopts an open conformation with a disordered N terminus (shown in multiple orientations as a semi-transparent rod) and a poorly ordered AST region (dotted green tube). Based on crystal structures of GRK1, the kinase domain becomes partially closed, and the AST becomes partially ordered upon ATP binding, but the N-terminal region remains disordered. Several events must then occur for the phosphotransfer reaction to occur, including docking of the receptor (or other protein substrate), interaction of the N-terminal region with the kinase domain, binding of the phosphoacceptor region of the substrate to the large lobe, and kinase domain closure. However, we cannot yet discern the order of these events. The interaction of an activated GPCR with its docking site, which is likely formed by the N terminus in complex with the kinase domain, is expected not only to bring phosphoacceptor sites in either its third cytoplasmic loop or C terminus in close proximity to the active site, but also to stabilize the closed, active conformation of the kinase domain. The diagram is not meant to imply an ordered sequential mechanism where ATP binds before the protein/peptide substrate.
In summary, we have identified novel residues on the kinase domain of GRKs that constitute a continuous surface critical for receptor phosphorylation. Our data are consistent with a model wherein the N-terminal region, both lobes of the kinase domain, and the AST region of the AGC kinase C-tail act in concert to stabilize the active state of GRKs. Our work, along with a parallel receptor-docking study in GRK2 (49), provides a framework by which we can further explore the molecular basis for the recognition of activated GPCRs by GRKs.
Acknowledgments
We thank Dr. Mohamed Aittaleb for help with in vivo receptor phosphorylation assays, Dr. Rowena Matthews for use of the MicroCal PC-isothermal calorimeter, the Center for Chemical Genomics at the Life Sciences Institute for use of the PHERAstar plate reader, and Dr. Jason Gestwicki for use of the SpectraMax M5 microplate reader.
This work was supported, in whole or in part, by National Institutes of Health Grants HL086865 and HL071818 (to J. J. G. T.).
- GRK
- G protein-coupled receptor kinase
- PKA
- protein kinase A
- AGC
- protein kinase A, G, and C
- GPCR
- G protein-coupled receptor
- RH
- regulator of G protein signaling homology
- C-tail
- C-terminal tail
- AST
- active site tether
- bGRK1
- bovine GRK1
- bGRK2
- bovine GRK2
- hGRK6
- human GRK6
- peptide C
- DDEASTTVSKTETSQVARRR
- RESA
- RRREEEEESAAA
- H6
- hexahistidine
- bROS
- bovine rod outer segments
- mP
- millipolarization
- PKB
- protein kinase B
- WT
- wild type
- PDB
- Protein Data Bank
- β2-AR
- β2-adrenergic receptor
- h
- human
- b
- bovine
- HA
- hemagglutinin
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate.
REFERENCES
- 1.Palczewski K., Benovic J. L. ( 1991) Trends Biochem. Sci. 16, 387– 391 [DOI] [PubMed] [Google Scholar]
- 2.Pitcher J. A., Freedman N. J., Lefkowitz R. J. ( 1998) Annu. Rev. Biochem. 67, 653– 692 [DOI] [PubMed] [Google Scholar]
- 3.Krupnick J. G., Benovic J. L. ( 1998) Annu. Rev. Pharmacol. Toxicol. 38, 289– 319 [DOI] [PubMed] [Google Scholar]
- 4.Métayé T., Gibelin H., Perdrisot R., Kraimps J. L. ( 2005) Cell. Signal. 17, 917– 928 [DOI] [PubMed] [Google Scholar]
- 5.Nolen B., Taylor S., Ghosh G. ( 2004) Mol. Cell 15, 661– 675 [DOI] [PubMed] [Google Scholar]
- 6.Huse M., Kuriyan J. ( 2002) Cell 109, 275– 282 [DOI] [PubMed] [Google Scholar]
- 7.Gold M. G., Barford D., Komander D. ( 2006) Curr. Opin. Struct. Biol. 16, 693– 701 [DOI] [PubMed] [Google Scholar]
- 8.Goldsmith E. J., Akella R., Min X., Zhou T., Humphreys J. M. ( 2007) Chem. Rev. 107, 5065– 5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellicena P., Kuriyan J. ( 2006) Curr. Opin. Struct. Biol. 16, 702– 709 [DOI] [PubMed] [Google Scholar]
- 10.Onorato J. J., Palczewski K., Regan J. W., Caron M. G., Lefkowitz R. J., Benovic J. L. ( 1991) Biochemistry 30, 5118– 5125 [DOI] [PubMed] [Google Scholar]
- 11.Palczewski K., Buczyłko J., Kaplan M. W., Polans A. S., Crabb J. W. ( 1991) J. Biol. Chem. 266, 12949– 12955 [PubMed] [Google Scholar]
- 12.Chen C. Y., Dion S. B., Kim C. M., Benovic J. L. ( 1993) J. Biol. Chem. 268, 7825– 7831 [PubMed] [Google Scholar]
- 13.Kim C. M., Dion S. B., Benovic J. L. ( 1993) J. Biol. Chem. 268, 15412– 15418 [PubMed] [Google Scholar]
- 14.McCarthy N. E., Akhtar M. ( 2002) Biochem. J. 363, 359– 364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palczewski K., McDowell J. H., Hargrave P. A. ( 1988) Biochemistry 27, 2306– 2313 [DOI] [PubMed] [Google Scholar]
- 16.Kim C. M., Dion S. B., Onorato J. J., Benovic J. L. ( 1993) Receptor 3, 39– 55 [PubMed] [Google Scholar]
- 17.Kunapuli P., Onorato J. J., Hosey M. M., Benovic J. L. ( 1994) J. Biol. Chem. 269, 1099– 1105 [PubMed] [Google Scholar]
- 18.Palczewski K., Buczyłko J., Lebioda L., Crabb J. W., Polans A. S. ( 1993) J. Biol. Chem. 268, 6004– 6013 [PubMed] [Google Scholar]
- 19.Yu Q. M., Cheng Z. J., Gan X. Q., Bao G. B., Li L., Pei G. ( 1999) J. Neurochem. 73, 1222– 1227 [DOI] [PubMed] [Google Scholar]
- 20.Noble B., Kallal L. A., Pausch M. H., Benovic J. L. ( 2003) J. Biol. Chem. 278, 47466– 47476 [DOI] [PubMed] [Google Scholar]
- 21.Chen C. K., Inglese J., Lefkowitz R. J., Hurley J. B. ( 1995) J. Biol. Chem. 270, 18060– 18066 [DOI] [PubMed] [Google Scholar]
- 22.Kannan N., Haste N., Taylor S. S., Neuwald A. F. ( 2007) Proc. Natl. Acad. Sci. U. S. A. 104, 1272– 1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J., Cron P., Thompson V., Good V. M., Hess D., Hemmings B. A., Barford D. ( 2002) Mol. Cell 9, 1227– 1240 [DOI] [PubMed] [Google Scholar]
- 24.Hauge C., Antal T. L., Hirschberg D., Doehn U., Thorup K., Idrissova L., Hansen K., Jensen O. N., J⊘rgensen T. J., Biondi R. M., Frödin M. ( 2007) EMBO J. 26, 2251– 2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lodowski D. T., Barnhill J. F., Pyskadlo R. M., Ghirlando R., Sterne-Marr R., Tesmer J. J. ( 2005) Biochemistry 44, 6958– 6970 [DOI] [PubMed] [Google Scholar]
- 26.Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., Tesmer J. J. ( 2003) Science 300, 1256– 1262 [DOI] [PubMed] [Google Scholar]
- 27.Lodowski D. T., Tesmer V. M., Benovic J. L., Tesmer J. J. ( 2006) J. Biol. Chem. 281, 16785– 16793 [DOI] [PubMed] [Google Scholar]
- 28.Tesmer V. M., Kawano T., Shankaranarayanan A., Kozasa T., Tesmer J. J. ( 2005) Science 310, 1686– 1690 [DOI] [PubMed] [Google Scholar]
- 29.Singh P., Wang B., Maeda T., Palczewski K., Tesmer J. J. ( 2008) J. Biol. Chem. 283, 14053– 14062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madhusudan, Akamine P., Xuong N. H., Taylor S. S. ( 2002) Nat. Struct. Biol. 9, 273– 277 [DOI] [PubMed] [Google Scholar]
- 31.Pitcher J. A., Fredericks Z. L., Stone W. C., Premont R. T., Stoffel R. H., Koch W. J., Lefkowitz R. J. ( 1996) J. Biol. Chem. 271, 24907– 24913 [DOI] [PubMed] [Google Scholar]
- 32.DebBurman S. K., Ptasienski J., Benovic J. L., Hosey M. M. ( 1996) J. Biol. Chem. 271, 22552– 22562 [DOI] [PubMed] [Google Scholar]
- 33.Papermaster D. S. ( 1982) Methods Enzymol. 81, 240– 246 [DOI] [PubMed] [Google Scholar]
- 34.Yang J., Cron P., Good V. M., Thompson V., Hemmings B. A., Barford D. ( 2002) Nat. Struct. Biol. 9, 940– 944 [DOI] [PubMed] [Google Scholar]
- 35.Smith C. M., Shindyalov I. N., Veretnik S., Gribskov M., Taylor S. S., Ten Eyck L. F., Bourne P. E. ( 1997) Trends Biochem. Sci. 22, 444– 446 [DOI] [PubMed] [Google Scholar]
- 36.Thompson J. D., Higgins D. G., Gibson T. J. ( 1994) Nucleic Acids Res. 22, 4673– 4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgar R. C. ( 2004) Nucleic Acids Res. 32, 1792– 1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. ( 2005) Nucleic Acids Res. 33, W299– 302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armon A., Graur D., Ben-Tal N. ( 2001) J. Mol. Biol. 307, 447– 463 [DOI] [PubMed] [Google Scholar]
- 40.Ferguson S. S., Ménard L., Barak L. S., Koch W. J., Colapietro A. M., Caron M. G. ( 1995) J. Biol. Chem. 270, 24782– 24789 [DOI] [PubMed] [Google Scholar]
- 41.Whalen E. J., Foster M. W., Matsumoto A., Ozawa K., Violin J. D., Que L. G., Nelson C. D., Benhar M., Keys J. R., Rockman H. A., Koch W. J., Daaka Y., Lefkowitz R. J., Stamler J. S. ( 2007) Cell 129, 511– 522 [DOI] [PubMed] [Google Scholar]
- 42.Tran T. M., Jorgensen R., Clark R. B. ( 2007) Biochemistry 46, 14438– 14449 [DOI] [PubMed] [Google Scholar]
- 43.Seibold A., Williams B., Huang Z. F., Friedman J., Moore R. H., Knoll B. J., Clark R. B. ( 2000) Mol. Pharmacol. 58, 1162– 1173 [DOI] [PubMed] [Google Scholar]
- 44.Vaughan D. J., Millman E. E., Godines V., Friedman J., Tran T. M., Dai W., Knoll B. J., Clark R. B., Moore R. H. ( 2006) J. Biol. Chem. 281, 7684– 7692 [DOI] [PubMed] [Google Scholar]
- 45.Krasel C., Zabel U., Lorenz K., Reiner S., Al-Sabah S., Lohse M. J. ( 2008) J. Biol. Chem. 283, 31840– 31848 [DOI] [PubMed] [Google Scholar]
- 46.Newton A. C. ( 2003) Biochem. J. 370, 361– 371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Z. B., Chaudhary D., Olland S., Wolfrom S., Czerwinski R., Malakian K., Lin L., Stahl M. L., Joseph-McCarthy D., Benander C., Fitz L., Greco R., Somers W. S., Mosyak L. ( 2004) J. Biol. Chem. 279, 50401– 50409 [DOI] [PubMed] [Google Scholar]
- 48.Batkin M., Schvartz I., Shaltiel S. ( 2000) Biochemistry 39, 5366– 5373 [DOI] [PubMed] [Google Scholar]
- 49.Sterne-Marr R., Leahey P. A., Bresee J. E., Dickson H. M., Ho W., Ragusa M. J., Donnelly R. M., Amie S. M., Krywy J. A., Brookins-Danz E. D., Orakwue S. C., Carr M. J., Yoshino-Koh K., Li Q., Tesmer J. J. ( 2009) Biochemistry, in press [DOI] [PMC free article] [PubMed] [Google Scholar]