

Abstract
Sphingolipid biosynthesis begins with the condensation of l-serine and palmitoyl-CoA catalyzed by the PLP-dependent enzyme serine palmitoyltransferase (SPT). Mutations in human SPT cause hereditary sensory autonomic neuropathy type 1, a disease characterized by loss of feeling in extremities and severe pain. The human enzyme is a membrane-bound hetereodimer, and the most common mutations are located in the enzymatically incompetent monomer, suggesting a “dominant” or regulatory effect. The molecular basis of how these mutations perturb SPT activity is subtle and is not simply loss of activity. To further explore the structure and mechanism of SPT, we have studied the homodimeric bacterial enzyme from Sphingomonas paucimobilis. We have analyzed two mutants (N100Y and N100W) engineered to mimic the mutations seen in hereditary sensory autonomic neuropathy type 1 as well as a third mutant N100C designed to mimic the wild-type human SPT. The N100C mutant appears fully active, whereas both N100Y and N100W are significantly compromised. The structures of the holoenzymes reveal differences around the active site and in neighboring secondary structure that transmit across the dimeric interface in both N100Y and N100W. Comparison of the l-Ser external aldimine structures of both native and N100Y reveals significant differences that hinder the movement of a catalytically important Arg378 residue into the active site. Spectroscopic analysis confirms that both N100Y and N100W mutants subtly affect the chemistry of the PLP. Furthermore, the N100Y and R378A mutants appear less able to stabilize a quinonoid intermediate. These data provide the first experimental insight into how the most common disease-associated mutations of human SPT may lead to perturbation of enzyme activity.
Sphingolipids are ubiquitous constituents of eukaryotic cells, where they play important roles in signaling, differentiation, and apoptosis (1–4). Defects in sphingolipid catabolism have been linked to several human diseases, such as hypertension, cancer, and disorders of the peripheral nervous system. The most common inherited peripheral neuropathy is hereditary sensory autonomic neuropathy type 1 (HSAN1).6 The disease leads to progressive loss of sensation in extremities and is often associated with searing pain (5–8). Genetic studies by two independent groups mapped the disease-associated mutations to the lcb1 (long chain base 1) gene on chromosome 9q22, which encodes the SPT1 subunit of serine palmitoyltransferase (SPT; EC 2.3.1.50) (9–11). SPT catalyzes the first and rate-limiting step of the sphingolipid biosynthetic pathway in all organisms studied to date (12). The reaction is a pyridoxal 5′-phosphate (PLP)-dependent, decarboxylative, Claisen condensation of the amino acid l-serine and the long chain (C16) fatty acid palmitoyl-CoA, which produces the sphingolipid precursor, 3-ketodihydrosphingosine (KDS).
SPT belongs to the α-oxoamine synthase subfamily of PLP-dependent enzymes, which contains three other well characterized members: 8-amino-7-oxononanoate synthase (AONS) (13, 14), 5-aminolevulinate synthase (ALAS) (15–17), and 2-amino-3-ketobutyrate-CoA ligase (KBL) (18). These enzymes catalyze the Claisen-like condensation between an amino acid and an acyl-coenzyme A thioester (19). A common mechanism has been proposed (12, 14, 20–23) comprising the following steps: formation of an external aldimine via displacement of the lysine-PLP internal aldimine (holo-SPT) by the incoming amino acid substrate; formation of a quinonoid intermediate by abstraction of the α-proton from the PLP-amino acid external aldimine; a Claisen condensation with the fatty acid-CoA thioester substrate, followed by displacement of the CoASH to form a β-ketoacid intermediate; decarboxylation of this species to form a product quinonoid; protonation of this quinonoid to form the product external aldimine; and finally release of the α-oxoamine product and regeneration of the enzyme PLP-internal aldimine (Fig. 1). Structural studies have shown AONS, ALAS, and KBL isoforms from various species to be homodimers (13, 17, 18). In marked contrast, eukaryotic SPTs are heterodimeric, membrane-bound proteins consisting of two subunits, SPT1 and SPT2 (encoded by the lcb1 and lcb2 gene, respectively) (24).
FIGURE 1.

Proposed catalytic mechanism of SPT. a, the internal aldimine (holo-SPT) is displaced by l-serine to form the external adlimine; b, binding of second substrate palmitoyl-CoA causes conformational change to give the Dunathan intermediate; c, formation of the quinonoid by deprotonation of Cα hydrogen; d, thioester bond hydrolysis and release of CoASH to give β-keto acid intermediate; e, decarboxylation to form KDS product quinonoid; f, reprotonation to form KDS product external aldimine; g, KDS product release and reformation of the holo-form. The l-serine external aldimine has been trapped in the current study. The dotted line shows that steps e and f may be bypassed by an alternative mechanism that does not proceed via a product quinonoid (see Ref. 21).
Analysis of a number of HSAN1 patients has revealed the four most common mutations to be C133W, C133Y, V144D, and G387A, with the cysteine mutations appearing to be the most prevalent in populations (10, 25). The impact that these mutations have on SPT activity and sphingolipid metabolism has been the focus of attention by a number of groups. Most surprisingly, these disease-associated mutations occur in the SPT1 monomer, a protein that must be inactive, since it lacks the key lysine, histidine, and aspartate residues necessary to bind and stabilize the PLP cofactor. It is the SPT2 protein that contains the conserved, active site lysine residue that forms a Schiff base internal aldimine with the PLP. However, both subunits are essential to produce functionally active SPT heterodimer (26–29), indicating that the inactive subunit has a crucial role in function. Dunn and co-workers (30) created several mutations in the yeast lcb1 and lcb2 genes, including those corresponding to the HSAN1 SPT1 mutations, and found that they dominantly inhibit SPT activity. A transgenic mouse model also revealed that the LCB1 C133W mutation led to mice with decreased tissue SPT activity and HSAN1 symptoms despite unaltered ceramide concentrations (31). A different study found that SPT activity was decreased in the tissues of HSAN1 patients, and in a Chinese hamster ovary model, hamster LCB1 C133Y and C133W mutations could not rescue cells lacking endogenous LCB1 (32). These combined studies revealed that the HSAN1 mutations act directly on the SPT enzyme but the pathological consequence on sphingolipid metabolism is unclear. Very recently, Dunn and colleagues discovered a small 80-amino acid protein (TSC3p) in yeast that stimulated yeast SPT activity, but its role is still unclear (33). Recently, a third eukaryotic subunit (SPT3) has been characterized and appears to be required for optimum SPT activity, although it is expressed most highly in placental tissue and human trophoblasts (34). It has been suggested that these three subunits could form a higher order SPT complex (35). The instability and hydrophobic nature of eukaryotic SPTs has made their isolation and therefore biophysical characterization particularly challenging (24, 36–38).
We have targeted homodimeric SPT from Sphingomonas paucimobilis EY2395 to provide molecular insight into the enzymatic properties of SPT (39, 40). Recently, we reported the first high resolution x-ray crystal structure of the S. paucimobilis holo-SPT (41) and showed that the active site containing the PLP cofactor is at the dimer interface. We used this structure to model the human enzyme and map the human cysteine residue (Cys133) of SPT1 onto Asn100 of the bacterial SPT (41). We found that Asn100 is proximal to the PLP binding site and lies at the dimer interface. To explore the effects of the HSAN1 mutations on human SPT activity and structure, we have studied mutants of the bacterial enzyme. We made the N100C mutant to better mimic native human SPT and N100W and N100Y to mimic the most common mutations in human SPT. We have characterized the mutations using kinetics, spectroscopy, and structural biology, including structures of the external l-Ser aldimine. These data provide molecular insights into the effects of HSAN1 mutations. Also, as part of these studies, we identify residues that play a role in the stabilization of intermediates in the SPT reaction and have been able to generate new insights into the substrate specificity and mechanism of the α-oxamine synthase family.
EXPERIMENTAL PROCEDURES
Chemicals
Plasmids and Escherichia coli competent cells were purchased from Novagen, and all chromatography columns were from GE Healthcare. All buffers and reagents were from Sigma. Palmitoyl-CoA was from Fluka and Avanti Lipids, and [U-14C]l-serine (specific activity 1.85 MBq/ml) was from GE Healthcare. The QuikChange site-directed mutagenesis kit was purchased from Stratagene. The decanoyl-CoA (10:0), lauroyl-CoA (12:0), myristoyl-CoA (14:0), stearoyl-CoA (18:0), and arachidoyl-CoA (20:0) were from Sigma. Oligonucleotide primers were sourced from SigmaGenosys. PEG 3350 was from Fluka. S-(2-oxoheptadecyl)-CoA was a kind gift from Dr. Hiroko Ikushiro (Department of Biochemistry, Osaka Medical College, Japan).
Cloning and Expression of S. paucimobilis SPT N100W, N100Y, N100C, R378A, and R378N Mutants in E. coli
Plasmid pET-28a/SPT WT was used as the template for mutations (N100W, N100Y, N100C, R378A, and R378N) that were introduced into the SPT gene by site-directed mutagenesis using the following mutagenic primers (the mutagenic bases are shown in boldface type) and confirmed by big Dye sequencing: 5′-GGGTCGGGCACCTGGGGCAGCCGGATG-3′ (SPT N100W forward), 5′-CATCCGGCTGCCCCAGGTGCCCGACCC-3′ (SPT N100W reverse), 5′-GGGTCGGGCACCTATGGCAGCCGGATG-3′ (SPT N100Y forward), 5′-CATCCGGCTGCCATAGGTGCCCGACCC-3′ (SPT N100Y reverse), 5′-GGGTCGGGCACCTGTGGCAGCCGGATG-3′ (SPT N100C forward), 5′-CATCCGGCTGCCACAGGTGCCCGACCC-3′ (SPT N100C reverse), 5′-CTCTACGTCAACATGGCGGCCCCGCCC-3′ (SPT R378A forward), 5′-GGGCGGGGCCGCCATGTTGACGTAGAG-3′ (SPT R378A reverse), 5′-CTCTACGTCAACATGGCGAACCCGCCC-3′ (SPT R378N forward), and 5′-GGGCGGGTTCGCCATGTTGACGTAGAG-3′ (SPT R378N reverse).
The recombinant plasmids were used to transform E. coli BL21 (DE3) competent cells, and selection was carried out on LB agar containing 30 μg/ml kanamycin. For each mutant (and native), a single colony was used to inoculate 500 ml of 2YT broth (16 g/liter Bacto-tryptone, 10 g/liter Bacto-yeast extract, 5 g/liter sodium chloride (pH 7.5)), which was shaken at 250 rpm overnight at 37 °C. The overnight culture was added to 4 liters of 2YT broth supplemented with kanamycin and grown at 37 °C to an A600 of 0.6. Protein expression was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside to give a final concentration of 0.1 mm, and growth was continued for 5 h at 30 °C. Cells were harvested (Sorvall RC5B centrifuge) by centrifugation at 3500 rpm for 20 min at 4 °C.
Purification of SPT WT, N100W, N100Y, N100C, R378A, and R378N
The proteins were purified essentially according to the previously described method, which is based polyhistidine-tagged protein binding to Ni2+-nitrilotriacetic acid-agarose (Qiagen) (41). After elution, fractions were pooled and desalted by dialysis against 20 mm potassium phosphate (pH 7.5), 150 mm NaCl, and 25 μm PLP. A gel filtration step (Sephacryl S200 26/60 HR; GE Healthcare) was carried out in the same buffer. For enzymatic assays and UV-visible spectroscopy, this buffer is also used. For x-ray crystal trials, the protein was exchanged into 10 mm Tris (pH 7.5), 150 mm NaCl, and 25 μm or 250 μm PLP. For storage, the enzymes were transferred to the same buffer containing 20% glycerol (v/v) and stored at −80 °C until use. Protein identity and integrity were confirmed by high pressure liquid chromatography electrospray mass spectrometry on a MicroMass Platform II quadrupole mass spectrometer equipped with an electrospray ion source. The experimentally determined masses of recombinant S. paucimobilis SPT WT and each of the mutants were within 0.1% of the theoretical mass, which includes the C-terminal LEHHHHHH fusion affinity tag with the enzyme lacking the N-terminal methionine (41).
Spectroscopic Measurements
All UV-visible spectra were recorded on a Cary 50 UV-visible spectrophotometer (Varian) and analyzed using Cary WinUV software (Varian). Prior to enzymatic assays, SPT was converted to the holo-form by dialysis against freshly prepared 20 mm potassium phosphate (pH 7.5) containing 150 mm NaCl and 25 μm PLP for 1 h at 4 °C. Excess PLP was removed by passing the protein through a PD-10 (Sephadex G-25M) desalting column (GE Healthcare) before concentration to 10–20 mg/ml using a VivaSpin 30 kDa cut-off concentration filter. For UV-visible assays, the concentration of recombinant SPT was 10 μm, and the spectrophotometer was blanked with 20 mm potassium phosphate (pH 7.5) containing 150 mm NaCl.
Determination of Dissociation Constants
Assays were carried out in 0.5-ml quartz cuvettes (1-cm path length) and typically contained 10 μm SPT in 20 mm potassium phosphate (pH 7.5). Varying amounts of l-serine (0–80 mm) were used in each assay. After the addition of substrate, the reactants were mixed and allowed to equilibrate for 15 min at 25 °C. Small base-line changes were corrected using Sigma Plot software. Changes in absorbance at 425 nm (SPT WT, N100C, R378A, and R378N) and 420 nm (SPT N100W and N100Y) were plotted against l-serine concentrations, and data points were fitted to a hyperbolic saturation curve (Equation 1) using Sigma Plot software,
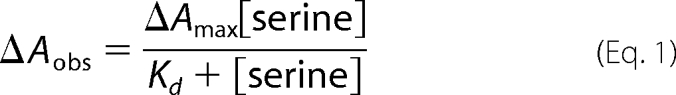 |
where ΔAobs represents the observed change in absorbance at 422 nm, and ΔAmax is the maximal absorbance change, [serine] is the l-serine concentration, and Kd is the dissociation constant (Table 1). The errors are asymptotic S.E. values of each parameter derived from the curve fitting procedure within Sigma Plot, and values were reproducible across a number of enzyme preparations.
TABLE 1.
Kinetic parameters for the purified SPT wild-type and mutant enzymes
| Enzyme | kcat × 103 | KmSer | KmPCoA | kcat/KmSer | KdSer |
|---|---|---|---|---|---|
| s−1 | mm | μm | m−1 s−1 | mm | |
| SPT WT | 1150.0 ± 30.0 | 1.4 ± 0.1 | 35.4 ± 2.0 | 821.4 | 1.1 ± 0.1 |
| SPT N100C | 250.0 ± 4.0 | 7.0 ± 0.4 | 60.0 ± 9.8 | 35.7 | 2.7 ± 0.2 |
| SPT N100W | 9.0 ± 0.4 | 1.6 ± 0.4 | 19.3 ± 2.2 | 5.6 | 16.2 ± 3.1 |
| SPT N100Y | 5.0 ± 0.1 | 2.5 ± 0.4 | 31.4 ± 3.3 | 2.0 | 7.5 ± 0.5 |
| SPT R378A | 78.3 ± 3.2 | 3.8 ± 1.0 | 39.2 ± 3.1 | 20.5 | 1.1 ± 0.1 |
| SPT R378N | 33.0 ± 1.0 | 2.4 ± 0.2 | 31.0 ± 6.0 | 13.8 | 3.1 ± 0.2 |
SPT WT, N100Y, and R378A Quinonoid Formation
Assays were carried out in 0.5-ml quartz cuvettes and typically contained 5 μm SPT in 20 mm potassium phosphate (pH 7.5). To this 45 mm l-serine and 1.45 mm S-(2-oxoheptadecyl)-CoA were added in the assay. After the addition of the substrate, the reactants were mixed and allowed to equilibrate for 20 min at 25 °C. The spectroscopic data were recorded between 200 and 800 nm. Small base-line changes were corrected using Sigma Plot software.
Assay of Recombinant SPT Activity
The activity of SPT was measured by monitoring the formation of [14C]KDS. A final enzyme concentration of 10 μm SPT in 20 mm potassium phosphate buffer, pH 7.5, 150 mm NaCl) was incubated with 20 mm [U-14C]l-serine (9250 Bq, 0.250 μCi; GE Healthcare) and 1.6 mm palmitoyl-CoA in a final volume of 125 μl. The reaction was incubated at 37 °C for 20 min, and then the reaction was quenched by the addition of 30 μl of 2.0 m NH4OH (final concentration 0.4 m). This was then extracted with an equal volume of CHCl3/CH3OH (2:1, v/v). The sample was centrifuged at 13,000 rpm for 5 min, and the aqueous phase was discarded before the organic phase was allowed to evaporate overnight. The resulting lipid residue was resuspended in 15 μl of CHCl3/CH3OH (2:1, v/v) and spotted onto a silica gel 60 TLC plate. Separation was carried out with a mobile phase of CHCl3/CH3OH/NH4OH (40:10:1, v/v/v), using a KDS reference standard (Matreya). The TLC was developed with a Storage PhosphorImager (GE Healthcare) for 4 days at room temperature, and the phosphor screen was visualized using ImageJ software.
SPT activity was also measured using a continuous spectrophotometric assay by monitoring the release of CoASH from acyl-CoA substrates and reaction with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) (42). Assays were carried out in 0.5-ml cuvettes in a Varian Cary-50 UV-visible spectrophotometer. The enzyme was incubated with l-serine in a buffered solution containing DTNB, and the assay was started by the addition of the second substrate, palmitoyl-CoA. The CoASH thiol product was monitored by observation of the TNB− anion at 412 nm ϵmax = 14,150 m−1 cm−1 (43). Initial rates were measured at increasing concentrations of l-serine (0.12–50 mm) while maintaining palmitoyl-CoA in excess. A typical experiment to determine the Km value for l-serine contained 0.16 μm SPT, 0.1–50 mm l-serine, 250 μm palmitoyl-CoA, and 0.2 mm DTNB in 50 mm potassium phosphate buffer, pH 7.5. Kinetic constants (kcat and Km values; Table 1) were calculated from Michaelis-Menten plots using Sigma Plot. Kinetic constant values were calculated in a similar way for CoA substrates (decanoyl, lauroyl, myristoyl, palmitoyl, stearoyl, and arachidoyl) but by maintaining l-serine (100 mm) in excess (Table 2). As a check, the kcat/Km parameters for CoA substrates were also determined by the method of complete condensation when [CoA] was ≪Km. In this case, kcat/Km is calculated at a single substrate concentration as an observed rate constant (kobs) by monitoring disappearance of the substrate. The data were fitted to the equation, A = Alim × (1 − exp(−kobs × t)), where A is the absorbance at 412 nm, Alim is the limiting absorbance value when the substrate has completely disappeared, kobs is the observed rate constant, and t is the time in seconds. The errors are asymptotic S.E. values of each parameter derived from the curve fitting procedure within Sigma Plot. The values obtained were reproducible over a number of enzyme preparations, and the errors can be used as a gauge of the fitted curve's accuracy and were generally less than 5% of the value of the parameter.
TABLE 2.
Chain length specificity of wild-type SPT
| Acyl-CoA | kcat | KmCoA | kcat/KmCoA |
|---|---|---|---|
| s−1 | μm | m−1 s−1 | |
| Decanoyl (10:0) | 0.045 ± 0.002 | 2324.9 ± 175.1 | 19.4 |
| Lauroyl (12:0) | 0.262 ± 0.006 | 822.2 ± 32.6 | 318.7 |
| Myristoyl (14:0) | 0.601 ± 0.012 | 97.1 ± 5.1 | 6189.5 |
| Palmitoyl (16:0) | 1.150 ± 0.030 | 35.4 ± 2.0 | 32,485.9 |
| Stearoyl (18:0) | 0.898 ± 0.012 | 13.7 ± 0.8 | 65,547.4 |
| Arachidoyl (20:0) | 0.327 ± 0.014 | <10 | >32,700 |
Structural Biology
The three mutant proteins were screened for suitable crystallization conditions at the Scottish Structural Proteomics Facility. The proteins were dialyzed in the presence of excess PLP to ensure complete reloading prior to crystallization. Conditions used to obtain crystals of the wild-type/PLP protein (41) were used as a starting point to formulate stochastic optimization screens using software developed in house. The screens were built on a Hamilton Microstar liquid-handling robot controlled by Rhombix system software (Thermo). Crystallization trials were set up in hanging drop plates (EasyXtal DG-CrystalSupport; Qiagen) using 2–3 μl of the protein solution and 1 μl of well solution in the hanging drop.
Crystals of the mutant SPTs were obtained in the form of yellow plates corresponding to the high resolution form of the holo wild-type SPT. The SPT N100W mutant was crystallized at 20 mg/ml in 10 mm Tris (pH 7.5), 150 mm NaCl, 25 μm PLP using a well solution of 0.1 m MgCl2, 22% (w/v) PEG 3350, and 0.10 m Hepes (pH 6.5). The N100Y mutant was crystallized at 20 mg/ml in 10 mm Tris (pH 7.5), 150 mm NaCl, 250 μm PLP, using a well solution of 0.15 m MgCl2, 22% (w/v) PEG 3350, and 0.10 m Hepes (pH 6.5). Finally, the N100C mutant was crystallized at 20 mg/ml in 10 mm Tris (pH 7.5), 150 mm NaCl, 250 μm PLP, using a well solution of 0.1 m MgCl2, 27% (w/v) PEG 3350, and 0.10 m Hepes (pH 6.5).
The solutions used for equilibrating harvested crystals varied slightly according to the crystallization conditions and consisted of 22–29% PEG 3350 (w/v), 0.1 m HEPES (pH 6.5), 0.08–0.12 m MgCl2. All crystals used for data collection were soaked for a few minutes in the respective equilibrating solution plus 1–2 mm PLP. Additionally, the external aldimine form of the SPT wild-type was produced by soaking the crystal in the equilibrating solution plus 5 mm l-serine for 10 min, and that of the N100Y mutant was produced by soaking the crystal in the equilibrating solution plus 50 mm l-serine for 15 min at room temperature. The crystals were mounted in a cryo-loop (Molecular Dimensions) and cryo-protected in solutions varying slightly for each crystal and containing the equilibrating solutions plus 15–20% PEG 400 (v/v). The crystals were then frozen by plunging them into liquid nitrogen and carried in a dry cryogenic Dewar to the European Synchrotron Radiation Facility (Grenoble, France) for data collection. The data sets were collected at 100 K to varying resolutions using three different beam lines (supplemental Table 1). The data for the wild-type serine external aldimine were processed with Mosflm and scaled with Scala from the CCP4 suite of programs (44). The data for the N100C, N100Y, N100W, and N100Y serine external aldimine structures were processed using XDS and scaled using XSCALE (available on the World Wide Web) (45).
Analysis of the density revealed that the PLP was covalently bound to Lys265 in every mutant, confirming that we had obtained the holo-form of the proteins. All mutant models were refined using Refmac5 (46) and manually adjusted, including the addition of water molecules with WinCoot (47). Data and structures have been deposited in the Protein Data Bank. Density was clearly visible to the N100W mutation, but we could not satisfactorily locate the tryptophan side chain; there is disordered electron density, which indicates multiple conformations.
RESULTS
Spectroscopic Properties of SPT N100 Mutants
The three mutants were obtained in milligram quantities as the internal aldmine, holo-form, confirming their capacity to bind the PLP cofactor (Fig. 2). The SPT N100C mutant has absorbance maxima at 335 and 425 nm, akin to the wild-type SPT values of 340 and 425 nm (Fig. 2, A and B). In both native and N100C, the enolimine peak at 335 nm is dominant, indicating that the PLP cofactor is in a very similar chemical environment in both proteins. In contrast, the UV-visible spectra of the SPT N100W and N100Y mutants have maximum absorbance values at 340 and 415 nm (Fig. 2, C and D). We noted that the ketoenamine peak (415 nm) had not only blue-shifted; it had also become the dominant form in the spectrum in these enzymes. These differences suggest a common perturbation in the PLP binding site of the mutants compared with the wild type (Fig. 2, C and D, solid lines). The ability of the mutants to form the external aldimine form by binding l-serine was measured by the addition of increasing concentrations of the amino acid (ranging between 0 and 100 mm). This addition resulted in an increase of the absorbance at 425 nm for the wild-type and the N100C enzyme. Titration of l-serine into the SPT N100W and N100Y mutants led to a small shift of a few nm to a longer wavelength. Although all mutants form the external aldimine, the N100W and N100Y mutants have clear differences (Fig. 2, A–D). By measuring the change in absorbance at ketoenamine-specific wavelengths that are specific to the external aldimine, the apparent serine dissociation constants (KdSer) have been determined for the wild type SPT and each of the mutants (Table 1). The wild-type SPT KdSer is 1.1 mm, in good agreement with the Kdapp found by Ikushiro et al. (40) (1.4 mm). A similar KdSer was obtained for the N100C mutant (2.7 mm; ∼2.5-fold higher). Higher KdSer values were obtained for two SPT mutants: N100W, 16.2 mm, a ∼15-fold increase, and N100Y, 7.5 mm, a ∼7-fold increase, compared with the wild-type SPT.
FIGURE 2.

UV-visible analysis of SPT wild-type and mutants. Absorbance spectra of S. paucimobilis SPT wild-type (A) and mutants N100C (B), N100W (C), and N100Y (D). In spectrum A the enolimine (335 nm) and ketoenamine (420 nm) forms of the external aldimine are drawn. The solid line in each spectrum is the holo-form of the enzyme (10 μm SPT, 20 mm potassium phosphate buffer (pH 7.5), 150 mm NaCl, 25 °C). Increasing concentrations of l-serine were added (0, 0.1, 0.5, 1, 2, 5, 10, 20, and 40 mm; dotted and dashed lines), and the spectrum was recorded after 15 min. For the SPT N100C, N100W, and N100Y spectra the final concentrations of l-serine were 0, 0.1, 0.5, 1, 2, 5, 10, 20, 40, 60, and 80 mm. AU, absorbance units.
KDS Formation by SPT WT and Mutants
The ability of each enzyme to convert [14C]l-serine and palmitoyl-CoA to the product [14C]KDS was examined (Fig. 3A), using the method previously described (48). The N100C mutant showed an activity similar to the wild-type SPT; however, both the N100W and N100Y mutants showed a large decrease in activity (∼84%) compared with the wild-type enzyme (with saturating substrate concentrations) (41). The R378N and R378A produced 50 and 40% KDS, respectively, in comparison with the wild-type enzyme (Fig. 3B).
FIGURE 3.

Assay of SPT activity. A and B, TLC of radiolabeled products obtained by SPT-catalyzed conversion of l-serine and palmitoyl-CoA to [14C]KDS. Recombinant holo-SPT was incubated with U-14C-labeled l-serine and palmitoyl-CoA for 20 min at 37 °C. The assay was stopped by the addition of NH4OH and extracted with an equal volume of chloroform/methanol (2:1, v/v). Organic phases were removed, dried, separated by TLC, and analyzed by a PhosphorImager. The arrow marks the position of the standard KDS. A, products observed after reaction of the SPT N100C (lane 1), SPT N100W (lane 2), SPT N100Y (lane 3), and SPT WT (lane 4). B, products observed after reaction of the SPT WT (lane 1), SPT R378A (lane 2), and SPT R378N (lane 3). C, kinetic analysis of wild-type SPT using a spectrophotometric assay. i, initial rates of CoASH release measured spectrophotometrically at 412 nm and at several final concentrations of l-serine: 6.25 mm (a), 1.56 mm (b), 0.78 mm (c), 0.39 mm (d), and 0.12 mm (e). Each assay contained 0.16 μm enzyme, 250 μm palmitoyl-CoA, and 0.2 mm DTNB. ii, the initial rates were converted to kcatapp values and plotted against l-serine concentration to determine the Km and kcat parameters. iii, complete condensation of 5 μm palmitoyl-CoA by 0.57 μm wild-type SPT in the presence of 100 mm l-serine. kcat/Km for palmitoyl-CoA was calculated by dividing kobs by the enzyme concentration.
Kinetic Analyses of Wild-type, HSAN1 Mimics, and Arg378 Mutants
The raw data for wild-type SPT are shown in Fig. 3C. The SPT WT bound l-serine and palmitoyl-CoA with Km values of 1.4 mm and 35.4 μm, respectively (Table 1). The enzyme turned over with a kcat of 1.150 s−1 and an efficiency (kcat/Km) of 821.4 m−1 s−1 for l-serine. We observed kcat/Km values of 35.7, 5.6, and 2.0 m−1 s−1 for the mutant SPTs N100C, N100W, and N100Y, respectively. The lower values observed for the HSAN1 mimics N100W and N100Y (147- and 410-fold respectively) confirm that the mutations have had an impact on catalysis. We attribute the lower value for the N100C mutant (23-fold lower) to reactivity of the Cys100 thiol with the DTNB reagent (data not shown), since this mutant showed comparable KDS formation in the radioactive assay. The coupled assay was used to investigate the chain length specificity of wild-type SPT with various acyl-CoA substrates. We found palmitoyl-CoA (C16:0) to have the fastest turnover (kcat = 1.150 s−1) of all of the acyl-thioesters, with a Km of 35.4 μm (Table 2). When comparing the kcat/Km values, stearoyl-CoA (C18:0) was the most efficient substrate (65,547 m−1 s−1). The thioester with the longest chain, arachidoyl-CoA (C20:0) was a good substrate, with a Km of <10 μm, but below this concentration, accurate rates could not be determined due to the limit of detection with the DTNB reagent. In contrast, the shortest substrate, decanoyl-CoA (C10:0), was the poorest substrate in terms of binding and turnover. In the first study of S. paucimobilis SPT carried out by Ikushiro et al. (39), they found a similar trend, with palmitoyl-CoA (C16) being the best substrate matching the human SPT. The recently discovered, unusual fused, viral SPT displayed highest activity with myristoyl-CoA (C14) (12, 49).
Similar analysis was also carried out on the SPT R378A and R378N mutants. These bound PLP in a manner similar to wild-type with absorbance maxima at 335 and 425 nm (data not shown). The values obtained were similar to that of the wild-type SPT (KdSer values of 1.1 and 3.1 mm for the R378A and R378N, respectively). Using the coupled spectrophotometric assay, we measured 60- and 40-fold decreases in kcat/KmSer for each mutant (R378A and R378N, respectively; Table 1).
SPT WT, N100Y, and R378A Quinonoid Formation
The ability of the wild-type enzyme and both the N100Y and R378A mutants to form a quinonoid intermediate was tested by the addition of a thioether analogue of palmitoyl-CoA reported by Ikushiro et al. (50). The addition of the analogue to the l-serine external aldimine form of the wild-type enzyme led to the appearance of a clear peak at 495 nm due to the quinonoid (increase in absorbance ∼0.06), in agreement with that found previously (Fig. 4A). In contrast, the SPT N100Y mutant, under the same conditions, produced a small, broad shoulder 490–500 nm (increase in absorbance ∼0.01 absorbance units; Fig. 4B). We could not observe the formation of a quinonoid species at all in the R378A mutant (Fig. 4C).
FIGURE 4.

Quinonoid formation in the wild-type SPT, N100Y, and R378A mutants. UV-visible spectra of the SPT wild type (A), N100Y (B), and R378A (C) external aldimine forms after titration of the thioether analog (S-(2-oxoheptadecyl)-CoA) of palmitoyl-CoA. Incubations were carried out with 5 μm enzyme, 45 mm l-serine, 1.4 mm analog, 20 mm potassium phosphate buffer (pH 7.5), 150 mm NaCl for 15 min at 25 °C. The solid line in each spectrum represents the holo-form; the dotted line is the external aldimine form after the addition of l-ser; and the dashed line is after the addition of the analogue. An absorbance maximum at 495 nm (marked with an arrow), attributed to a quinonoid species (50), is clearly observed in the wild-type enzyme, but only a broad shoulder is produced in the N100Y mutant, and no peak is observed for the R378A mutant. AU, absorbance units.
The Structure of the Wild-type SPT·l-Ser External Aldimine Complex
The dimeric form of the l-Ser external aldimine SPT complex is shown in Fig. 5A. Of course, the major difference between the internal (holo-) and external aldimine forms is that in the internal adimine, the Schiff base is between Lys265 of the protein and the PLP co-factor, whereas in the l-Ser external aldimine, this C–N bond has broken. The PLP·l-ser external aldmine is clearly observed in the electron density (Fig. 5B). An overlay of the structures of the internal (41) and external aldimine shows an r.m.s. deviation of 0.3 Å and reveals that most of the interactions between the enzyme and the PLP cofactor are unchanged. In both structures, the PLP hydrogen-bonds to the side chains of Asp231, His234, Thr262, and Ser264, the main chain of Gly134 and Thr135, and π-stacks with His159. His159 is conserved throughout all of the members of the α-oxoamine synthase family (41). As a result of external aldimine formation, the PLP has shifted, essentially rotating around the phosphate, with the Schiff base nitrogen moving over 1.5 Å. Lys265 adopts a different conformation as a result of breaking its link to PLP and now makes a hydrogen bond with the hydroxyl of the l-ser component of the external aldimine (Fig. 6A). Accompanying the change in Lys265 conformation, Tyr73 substantially alters its position. The carboxylate group of l-Ser hydrogen-bonds to the conserved His159 and makes a salt link with Arg378, which has “swung” into the active site. In the native structure, this residue is at the end of a β-strand and hydrogen-bonded to the side chain of Gln357. In order to make the salt contact, the β-strand has been disrupted, and the Cα of Arg378 has been moved over 5 Å. The loop following Arg378 (the “PPATP” loop) has significantly altered its conformation and position (e.g. the Cγ atom of Pro379 has moved over 10 Å). The reordering of the structure around Arg378 seems to be linked to other movements at Phe47 (1.2-Å Cα shift). These residues are contained within the one monomer. The phosphate of PLP binds to the side chain of Thr294 from one subunit and to the main chain of Ala294 from the other subunit. The hydroxyl of l-Ser makes a polar contact to the main chain of Ala294. In the other monomer, the side chain Met104 adjusts its conformation, and there is a slight rigid body shift of Leu105 to Asn106. The side chain of Asn100 from the other subunit has altered its position, but the hydrogen bond to Lys265 is maintained.
FIGURE 5.

Structure of the SPT·l-serine external aldimine complex. A, overall structure of the SPT dimer showing one monomer in light blue and the other in purple. The residues Lys265 and Asn100 from the light blue monomer are shown in stick form and are colored yellow. The equivalent residues from the purple monomer are colored green. This clearly shows that the Asn100 from one monomer interacts with the active site of the other monomer. The PLP·l-ser external aldmine is also shown in stick form. B, a stereo image of an Fo − Fc electron density map contoured at 3 σ around the PLP·l-ser external aldimine found in the native SPT. The phases were calculated from a model that had never included the PLP or aldimine.
FIGURE 6.

Structure of the SPT·l-serine external aldimine complex and impact of the N100Y HSAN1 mutation. A, a stereo image showing residues involved in binding the PLP·l-ser external aldimine complex are shown. His159, Asp231, His234, Thr262, Lys265, and Arg378 are from monomer A (light blue) in yellow stick format. Residues Thr294 and Asn100 are from monomer B (purple), are colored green, and are annotated with an asterisk. Residues His159 and Arg378 interact with the carboxylate group of the l-serine; Lys265 (denoted by dashed lines) interacts with the hydroxyl group of the l-serine; and His234 interacts with the hydroxyl group of the PLP. Residue Asp231 binds to the nitrogen atom of the PLP cofactor, and the side chain of Asn100 (from monomer B) interacts with the backbone of Lys265 of the opposite monomer A. B, a stereo image of an overlay of the external aldimine forms of the wild-type and N100Y. The wild-type structure has been rendered with monomer A colored with a blue backbone and yellow side chains, and monomer B is shown with a purple backbone and green side chains. In the N100Y mutant, the monomer A backbone and side chains are colored gold, and monomer B is colored with a red backbone and salmon pink side chains. The N100 side chain from monomer B of the wild-type SPT (green stick, asterisk) interacts with residues from monomer A. However, in the SPT N100Y mutant, the Tyr100 side chain has flipped into a pocket formed by displacement of Phe109 of the same monomer B, residue Asn106 from monomer B has also undergone a large shift, and residue Asn375 from monomer A has also changed. The backbone containing residue Tyr100 has also undergone a large conformational change. C, a stereo image of an overlay of the external aldimine forms of the wild-type and N100Y showing the impact of mutation on Arg378 and the PPATP loop. The coloring is the same as in B. In wild-type SPT, residue Arg378 (side chain yellow on the blue backbone) is in the swung in position and interacts with the l-Ser carboxylic acid. In the SPT N100Y mutant, the “PPATP loop” and the Arg378 residue (backbone and side chain colored gold) are in the “swung out” conformation observed in the wild-type internal aldimine form. The interaction between Arg378 and Gln357 (both in gold) and the different positions of the Asn100 and Tyr100 residues are shown.
Structural Biology of the Holo-forms of the HSAN1 Mutant Mimics
The wild-type SPT and N100C structures superimpose with an r.m.s. deviation of 0.2 Å along the 396 Cα residues and are largely identical. Similarly, the N100W and N100Y mutant structures superimpose with 0.1 Å r.m.s. deviation for their Cα positions, mirroring their similar spectroscopic properties. However, both N100W and N100Y superimpose with the native structure (and N100C) with an r.m.s. deviation of 0.6 Å. For ease of discussion, only the differences between N100Y and the native enzyme are reported. The N100W structure is essentially identical to N100Y, whereas N100C is essentially identical to native. Reflecting the large deviation in Cα positions, there are a number of small shifts in secondary structure elements throughout the structure when comparing the N100Y mutant with the native SPT. These shifts are largely conserved, regardless of which structures one compares (N100W versus native, N100W versus N100C, N100Y versus N100C, and N100Y versus native), suggesting that they are real and not simply a crystallographic artifact. The most obviously visible change is the N-terminal helix (Asp23–Gly42), which has undergone an approximately 1.7-Å shift. This helix is an important part of the dimeric interface of SPT (41). At the site of mutation itself, the tyrosine side chain has “flipped” out the pocket occupied by Asn100 (Fig. 6B). At the Cα level, this is a displacement of 3.4 Å and is accomplished by an almost 180° rotation of the Ramachandran angle at Thr99. As a result, the hydrogen bonds between the side chain of Asn100 with the key Lys265 from the other monomer have been broken. Instead, the Tyr100 now occupies a new pocket at the dimer interface formed by hydrophobic residues from both monomers (Phe109 from the same monomer; Met51, Thr72, and Ile69 from the other monomer) (Fig. 6B). These hydrophobic residues have adjusted their position to accommodate the bulky tyrosine side chain. The most pronounced of these changes appear to accompany the movement of Phe109, which connects to an α-helix. Residues of the first turn of the helix, His110, Asp111, His112, Met113, and Glu114, all show disturbed side chain and main chain positions relative to the native structure. The main chain at Leu105, Asn106, and Gly107 sits opposite and close to Asn100 in the native structure. The movement of the Cα atom at position 100 by 3.4 Å causes Leu105–Gly107 to undergo large positional movements (over 4 Å for the Cα positions of Gly107) and changes in their Ramachandran angles.
Structural Biology of Wild Type and N100Y l-Ser External Aldimines
The r.m.s. deviation between the two external aldimines, native SPT, and N100Y mutant at 0.4 Å is less than the value for the internal aldimine structures (0.6 Å). Thus, it appears that formation of the external aldimine drives N100Y toward a more “native” conformation. This can be seen most clearly in the loop between Tyr100 and Gly107, which is more similar to the native structure in the external aldimine complex. The most striking difference between the two external aldimine structures is that in the N100Y structure, the “PPATP loop” remains in its internal aldimine conformation, and consequently Arg378 does not enter the active site and does not make salt links with the carboxylate of l-ser (Fig. 6C). Rather, in the external aldimine of N100Y, Arg378 is hydrogen-bonded to Gln357, an interaction that is absent in the N100Y internal aldimine due to a small shift in structure.
DISCUSSION
Mechanistic Implications
The seminal hypothesis, forwarded by Dunathan (51) over 40 years ago, to explain how PLP-dependent enzymes can catalyze numerous chemical reactions is still valid. The key steps involve the deprotonation of the amino acid·PLP external aldmine to form the quinonoid species, which condenses with the incoming acyl-CoA thioester (Fig. 1). This is thought to generate a β-keto acid intermediate that then decarboxylates to give a product aldimine (23). The first intermediate in the catalytic cycle of SPT is the external aldimine formed between l-ser and PLP. Deprotonation of the Cα of this external aldimine intermediate is the crucial next step. In studies with ALAS, this deprotonation has been observed to be accelerated by over 250,000-fold by the binding of the incoming thioester substrate (14, 52). Studies on AONS, which uses l-alanine and pimeloyl-CoA, suggest that this enzyme undergoes conformational change during intermediate formation. Incubation of the enzyme with palmitoyl-CoA preequilibrated with l-alanine gave a 30-ms lag before quinonoid formation at 486 nm was observed with a rate of 45 s−1 (14). Recently, a thioether CoA substrate analogue (S-(2-oxoheptadecyl)-CoA) was shown to increase the rate of proton abstraction from the serine external aldimine of SPT by around 100-fold (50).
Our new structure provides insight into the residues that control the l-Ser external aldimine conformation (Fig. 7, A–C). How was this intermediate trapped in the crystal? The high resolution data confirm that the l-serine·PLP external aldimine is sp3-hybridized at the Cα (the angles are 111.5, 106.4, and 109.6°, which are close to the theoretical values for a tetrahedral carbon). In the external aldimine structure, the Nz atom of Lys265 contacts (3.4 Å) the Cα proton of the l-Ser. This double role for the Schiff base forming lysine was suggested by studies of members of the α-oxoamine synthase family (14, 17, 18, 20), and SPT has now been shown to use the same feature. The angles between each functional group and the N-imine (which is co-planar with the PLP ring) were measured as follows: H-Cα-N, 107.7°; CO2-Cα-N, 108.4°; CH2-Cα-N, 113.3°. None of these have the desired perpendicular orientation to the plane, indicating that in the crystal structure, the external aldimine is not in the optimal “Dunathan conformation” suitable for deprotonation.
FIGURE 7.

Schematic representation of the residues involved in SPT catalysis, conformations of the external aldimines, and roles of active site arginine residues in controlling. A, key residues involved in forming the active site are highlighted. The Arg378 side chain is in the swung out conformation and interacts with Gln357. B, binding of l-Ser leads to PLP·l-Ser external aldimine formation. The residues observed in the wild-type crystal structure are shown. The carboxylate of l-Ser interacts with the side chains of His159 and Arg378, which is now in the swung in conformation. Rotation around the Cα–Nα bond (arrow) brings the intermediate into the desired Dunathan conformation shown in C. The carboxylate is shown to possibly interact with Arg390, and the Cα-H is now perpendicular to the PLP ring. This is then deprotonated by Lys265 to form the quinonoid. D, stereo image of a model of the external aldimine Dunathan conformation using the external aldimine structure.
If, as seems likely, palmitoyl-CoA binding leads to rapid deprotonation, we suggest that it does so by triggering the rotation of the external aldimine about the imine N–Cα bond. A small turn of ∼15–20° would bring the Cα hydrogen into the required perpendicular geometry (Fig. 7B). Of course, this would cause the simultaneous movement of the -CO2− group and the -CH2OH side chain. Ikushiro proposed a role for the conserved residue Arg390 in the SPT mechanism (50). The structural data show that Arg390 is over 6.5 Å distant from the carboxylate; rotating around the Cα–N bond to reach the likely quinonoid conformation shortens this distance to 4.6 Å (Fig. 7, C and D). An additional rearrangement, promoted by palmitoyl-CoA binding, could allow Arg390 to interact with the carboxylate, as seen in the KBL external aldimine. We note that Arg390 plays an important role in stabilizing the conformation of Tyr73, which forms the active site. The rotation to form the quinonoid would disrupt the salt link to Arg378 in the absence of any additional structural adjustment. The interplay of Arg390 and Arg378 in interacting with the carboxylate group may be an important component in controlling the formation and deprotonation of the external aldimine. Together, they could act to pick up, ferry, then anchor the carboxylic acid group. This functional group is ultimately lost from the β-keto acid as CO2, and future work will be required to understand how this important step is controlled. Ikushiro et al. (50) also proposed a role for His159 acting as a hydrogen donor to accommodate the developing negative charge on the C O of the thioester. The structure shows that the Nϵ atom of His159 hydrogen-bonds to the l-serine CO2− group as well as maintaining a π-stacking interaction with the PLP ring. This is consistent with the reported SPT H159F mutant, which bound l-serine weakly (Kd = 11.1 mm) and was incapable of KDS formation. Whether the residue also plays a role in the chemistry of the thioester activation remains an open question.
O of the thioester. The structure shows that the Nϵ atom of His159 hydrogen-bonds to the l-serine CO2− group as well as maintaining a π-stacking interaction with the PLP ring. This is consistent with the reported SPT H159F mutant, which bound l-serine weakly (Kd = 11.1 mm) and was incapable of KDS formation. Whether the residue also plays a role in the chemistry of the thioester activation remains an open question.
That Arg378 binds to the external aldimine was unsuspected, since it required a very large conformational change from the holoenzyme structure. Mutation of this residue (R378A and R378N) reduces enzyme efficiency by over a factor of 10 (Table 1). Thus, we suggest that this residue plays an important, but not essential, role possibly influencing the stabilization of an intermediate after both substrates have bound. Weight is given to this hypothesis by the fact that R378A cannot form a quinonoid in the presence of the thioether analogue (Fig. 4C). The nonessential nature of the residue is consistent with its lack of sequence conservation in all SPT isoforms, and an equivalent residue in the human SPT1/SPT2 heterodimer awaits identification. That there may be such a residue comes from analysis of enzymes in the α-oxoamine synthase family. In the two glycine-specific members, KBL and ALAS, residues Asn50 and Asn54, respectively, make direct contact with the -CO2− group (17, 18) of the amino acid. In AONS, Webster et al. (14) used the AONS-AON product external aldimine structure to predict that the l-alanine carboxylate would coordinate with residue Asn47. Sequence homology lines up residue Asn74 from S. paucimobilis SPT with the three Asn residues from AONS, ALAS, and KBL; however, analysis of the structure reveals that this residue in not involved in the active site. Thus, there is reason to suspect that some residue is required to coordinate to the carboxylic group of the external aldimine; however, given that the function is not essential, the conservation of the residue is not absolute.
The organization of the active sites of AONS, KBL, and ALAS is different compared with the bacterial SPT; the constellation of residues around the PLP cofactor is conserved, but there are subtle differences. SPT favors l-Ser as the substrate, and in the SPT external aldimine structure, the side chain hydroxyl makes a number of hydrogen bonds, which could explain this preference. Interestingly, although SPT can form an external aldimine with l-Thr (Kd = 3.8 mm) (40), it cannot be deprotonated to the quinonoid in our hands (data not shown). Looking at the structure of the aldimine, the additional methyl group of l-Thr would appear to clash with Ser102 and Arg378 in the proposed quinonoid model. In the ALAS structure, Thr83 would seem to select against any substituent at the Cα position, explaining the preference for Gly of this enzyme.
Mimics of the HSAN1 Disease Causing Mutations
The two most common mutations (C133W and C133Y) associated with the human disease HSAN1 have intrigued a number of researchers as to how they impact on sphingolipid metabolism and regulation. The fact that these mutations are in the inactive subunit, LCB1 (SPT1), has added to their scientific interest. Tissue from HSAN1 patients contains relatively normal levels of total sphingolipids (10, 31, 32). However, there are differing reports as to the intrinsic SPT activity of cells from HSAN1 individuals, and questions remain as to the precise consequence of the HSAN1 mutants on SPT turnover, stability, and regulation. Gable et al. (30) produced mutants of the yeast lcb1 gene (C180Y and C180W) that mimic the HSAN1 substitutions (C133W and C133Y). Interestingly, these mutations dominantly inactivated the SPT activity of the yeast LCB1·LCB2 heterodimeric complex. This study proposed that the LCB1 HSAN1 mutations perturbed the active site of the LCB1·LCB2 dimer. McCampbell et al. (31) expressed the human LCB1 C133W mutant in a transgenic mouse and found that the SPT activity was decreased in various tissues and mice developed a number HSAN1 symptoms. However, the levels of total ceramide in these mice were unchanged, as was also observed in human HSAN1 individuals. This indicates that to some extent, SPT activity is preserved, and the mutants have a more subtle effect than simple loss of activity. It would also indicate that there is some regulation of sphingolipid biosynthesis after KDS formation by SPT.
We had previously mapped the human Cys133 residue to Asn100 on the bacterial SPT (41). We first engineered an N100C mutation as a better mimic for the human enzyme. This mutant enzyme behaves within the sensitivity of our UV-visible spectroscopic, kinetic, and high resolution structural analyses as native. This gave us confidence that the other mutations at Asn100 in the bacterial enzyme should provide a meaningful insight into human SPT.
N100W and N100Y mutants displayed marked spectroscopic, enzymatic, and structural differences when compared with wild-type SPT. Although both mutants are severely compromised in their activity, importantly, they do retain the ability to make the desired KDS product. It was obvious from the distinctive UV-visible properties of PLP-containing holo-forms of the enzymes that the mutants had altered the cofactor's chemical environment (Fig. 2, A–D). This change in the chemical environment apparently disturbs catalysis. After formation of an external aldimine, deprotonation gives a quinonoid intermediate (Fig. 1), which has been detected for both AONS and ALAS enzymes (14, 52). In the native SPT enzyme, the quinonoid form can be detected by adding the palmitoyl-CoA thioether analog. However, in the N100Y mutant, the signal for the quinonoid is essentially absent, indicating that the stabilization of this intermediate is significantly decreased (Fig. 4, A and B). In the wild-type holo-SPT (and N100C), the side chain of one monomer makes a direct contact with the amide backbone of the conserved PLP Schiff base Lys265 of the other monomer. This hydrogen bond is lost by the disruption introduced by the mutation. We suggest that this would increase the mobility of the internal aldimine. The most striking observation is that the N100Y and N100W mutations cause, because of the key location of Asn100 at the dimer interface, complex structural changes to ripple across the dimer interface (Fig. 6B). A striking illustration of this can be seen in the external aldimine of N100Y, particularly the role of Arg378. In the native enzyme, this residue plays an important role in binding to the carboxylate of the external aldimine, but in the N100Y mutant, Arg378 remains uninvolved with the intermediate (Fig. 6C). Although it is difficult to identify a clear cut interaction that disfavors Arg378 from entering the active site, this residue is in contact with Leu105 from the other subunit. Leu105 is one of the residues directly affected by the mutation at Asn100. The change in Leu105 position introduced by the mutation would increase the van der Waals contact between Arg378 (in the “swung in” state) and Leu105 in the mutant. It is this ability for structural changes to “reach across the interface” that we propose underlies the C133W and C133Y mutation effects in HSAN1. The perturbations in the neighboring structure give rise to the changed chemical environment of both the internal and external aldimine, altering the enzyme function.
Combined with previous studies on AONS, ALAS, and KBL, the SPT data presented here shed more light on the catalytic mechanism employed by members of the α-oxoamine synthase family. The eukaryotic SPT heterodimeric isozymes appear to be more complex than their bacterial homologs, since a third, tissue-specific SPT subunit has been identified recently (34). Furthermore, an abnormal, “dead end” sphingolipid product, 1-deoxysphinganine (also known as the natural product “spisulosine”), formed by SPT using l-Ala as a substrate, has been identified in mammalian cells (53, 54). Whether the SPT HSAN1 mutations impact directly on substrate and product specificity requires further detailed investigation. In the absence of large amounts of homogenous eukaryotic complexes, the bacterial enzymes provide some insight into how the wild-type and mutant forms behave and aid in the design of isolation strategies for the more complex SPTs (55). Studies aimed at the capture of complexes of various SPTs with a range of substrates, products, and inhibitors are under way.
Supplementary Material
Acknowledgments
We thank Dr. Thorsten Hornemann (Zürich) for help regarding the CoASH assay. We also extend thanks to Dr. Hiroko Ikushiro (Osaka) for the palmitoyl-CoA thioether analogue. The SSPF is supported by the Scottish Funding Council (grant reference SULSA).
This work was supported by Biotechnology and Biological Sciences Research Council (BBSRC) Grants BB/F009739/1 and BBS/B/14434 (to D. J. C. and J. H. N.) and the Scottish Structural Proteomics Facility (SSPF).
This paper is dedicated to the memory of Dr. Joe Spencer (University of Cambridge).
The atomic coordinates and structure factors (codes 2W8J, 2W8W, 2W8U, 2W8V, and 2W8T) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- HSAN1
- hereditary sensory autonomic neuropathy type 1
- SPT
- serine palmitoyltransferase
- PLP
- pyridoxal phosphate
- KDS
- 3-ketodihydrosphingosine
- DTNB
- 5,5′-dithiobis-2-nitrobenzoic acid
- r.m.s.
- root mean square
- AONS
- 8-amino-7-oxononanoate synthase
- ALAS
- 5-aminolevulinate synthase
- KBL
- 2-amino-3-ketobutyrate-CoA ligase
- PEG
- polyethylene glycol.
REFERENCES
- 1.Merrill A. H., Jr. ( 2002) J. Biol. Chem. 277, 25843– 25846 [DOI] [PubMed] [Google Scholar]
- 2.Futerman A. H., Hannun Y. A. ( 2004) EMBO Rep. 5, 777– 782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi T., Takahashi M., Nagatsuka Y., Hirabayashi Y. ( 2006) Biol. Pharm. Bull. 29, 1526– 1531 [DOI] [PubMed] [Google Scholar]
- 4.Hirabayashi Y., Igarashi Y., Merrill A. H. J. ( 2006) Sphingolipid Biology, 1st Ed., Springer-Verlag, Tokyo [Google Scholar]
- 5.Auer-Grumbach M. ( 2004) Drugs Today 40, 385– 394 [DOI] [PubMed] [Google Scholar]
- 6.Dyck P. ( 1993) Neuronal Atrophy and Degeneration Predominantly Affecting Peripheral Sensory and Autonomic Neurons, W.B. Saunders Co., Philadelphia [Google Scholar]
- 7.Houlden H., Blake J., Reilly M. M. ( 2004) Curr. Opin. Neurol. 17, 569– 577 [DOI] [PubMed] [Google Scholar]
- 8.Houlden H., King R., Blake J., Groves M., Love S., Woodward C., Hammans S., Nicoll J., Lennox G., O'Donovan D. G., Gabriel C., Thomas P. K., Reilly M. M. ( 2006) Brain 129, 411– 425 [DOI] [PubMed] [Google Scholar]
- 9.Bejaoui K., Wu C., Scheffler M. D., Haan G., Ashby P., Wu L., de Jong P., Brown R. H., Jr. ( 2001) Nat. Genet. 27, 261– 262 [DOI] [PubMed] [Google Scholar]
- 10.Dawkins J. L., Hulme D. J., Brahmbhatt S. B., Auer-Grumbach M., Nicholson G. A. ( 2001) Nat. Genet. 27, 309– 312 [DOI] [PubMed] [Google Scholar]
- 11.Nicholson G. A., Dawkins J. L., Blair I. P., Kennerson M. L., Gordon M. J., Cherryson A. K., Nash J., Bananis T. ( 1996) Nat. Genet. 13, 101– 104 [DOI] [PubMed] [Google Scholar]
- 12.Hanada K. ( 2003) Biochim. Biophys. Acta 1632, 16– 30 [DOI] [PubMed] [Google Scholar]
- 13.Alexeev D., Alexeeva M., Baxter R. L., Campopiano D. J., Webster S. P., Sawyer L. ( 1998) J. Mol. Biol. 284, 401– 419 [DOI] [PubMed] [Google Scholar]
- 14.Webster S. P., Alexeev D., Campopiano D. J., Watt R. M., Alexeeva M., Sawyer L., Baxter R. L. ( 2000) Biochemistry 39, 516– 528 [DOI] [PubMed] [Google Scholar]
- 15.Jordan P. M., Shemin D. ( 1972) in The Enzymes ( Boyer P. D. ed) 3rd Ed., Academic Press, Inc., London [Google Scholar]
- 16.Ferreira G. C., Gong J. ( 1995) J. Bioenerg. Biomembr. 27, 151– 159 [DOI] [PubMed] [Google Scholar]
- 17.Astner I., Schulze J. O., van den Heuvel J., Jahn D., Schubert W. D., Heinz D. W. ( 2005) EMBO J. 24, 3166– 3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt A., Sivaraman J., Li Y., Larocque R., Barbosa J. A., Smith C., Matte A., Schrag J. D., Cygler M. ( 2001) Biochemistry 40, 5151– 5160 [DOI] [PubMed] [Google Scholar]
- 19.Eliot A. C., Kirsch J. F. ( 2004) Annu. Rev. Biochem. 73, 383– 415 [DOI] [PubMed] [Google Scholar]
- 20.Hunter G. A., Ferreira G. C. ( 1999) Biochemistry 38, 3711– 3718 [DOI] [PubMed] [Google Scholar]
- 21.Zaman Z., Jordan P. M., Akhtar M. ( 1973) Biochem. J. 135, 257– 263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexeev D., Baxter R. L., Campopiano D. J., Kerbarh O., Sawyer L., Tomczyk N., Watt R., Webster S. P. ( 2006) Org. Biomol. Chem. 4, 1209– 1212 [DOI] [PubMed] [Google Scholar]
- 23.Kerbarh O., Campopiano D. J., Baxter R. L. ( 2006) Chem. Commun. 60– 62 [DOI] [PubMed] [Google Scholar]
- 24.Hanada K., Hara T., Fukasawa M., Yamaji A., Umeda M., Nishijima M. ( 1998) J. Biol. Chem. 273, 33787– 33794 [DOI] [PubMed] [Google Scholar]
- 25.Verhoeven K., Coen K., De Vriendt E., Jacobs A., Van Gerwen V., Smouts I., Pou-Serradell A., Martin J. J., Timmerman V., De Jonghe P. ( 2004) Neurology 62, 1001– 1002 [DOI] [PubMed] [Google Scholar]
- 26.Buede R., Rinker-Schaffer C., Pinto W. J., Lester R. L., Dickson R. C. ( 1991) J. Bacteriol. 173, 4325– 4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada K., Hara T., Nishijima M., Kuge O., Dickson R. C., Nagiec M. M. ( 1997) J. Biol. Chem. 272, 32108– 32114 [DOI] [PubMed] [Google Scholar]
- 28.Nagiec M. M., Baltisberger J. A., Wells G. B., Lester R. L., Dickson R. C. ( 1994) Proc. Natl. Acad. Sci. U. S. A. 91, 7899– 7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagiec M. M., Lester R. L., Dickson R. C. ( 1996) Gene 177, 237– 241 [DOI] [PubMed] [Google Scholar]
- 30.Gable K., Han G., Monaghan E., Bacikova D., Natarajan M., Williams R., Dunn T. M. ( 2002) J. Biol. Chem. 277, 10194– 10200 [DOI] [PubMed] [Google Scholar]
- 31.McCampbell A., Truong D., Broom D. C., Allchorne A., Gable K., Cutler R. G., Mattson M. P., Woolf C. J., Frosch M. P., Harmon J. M., Dunn T. M., Brown R. H., Jr. ( 2005) Hum. Mol. Genet. 14, 3507– 3521 [DOI] [PubMed] [Google Scholar]
- 32.Bejaoui K., Uchida Y., Yasuda S., Ho M., Nishijima M., Brown R. H., Jr., Holleran W. M., Hanada K. ( 2002) J. Clin. Invest. 110, 1301– 1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gable K., Slife H., Bacikova D., Monaghan E., Dunn T. M. ( 2000) J. Biol. Chem. 275, 7597– 7603 [DOI] [PubMed] [Google Scholar]
- 34.Hornemann T., Richard S., Rütti M. F., Wei Y., von Eckardstein A. ( 2006) J. Biol. Chem. 281, 37275– 37281 [DOI] [PubMed] [Google Scholar]
- 35.Hornemann T., Wei Y., von Eckardstein A. ( 2007) Biochem. J. 405, 157– 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han G., Gable K., Yan L., Natarajan M., Krishnamurthy J., Gupta S. D., Borovitskaya A., Harmon J. M., Dunn T. M. ( 2004) J. Biol. Chem. 279, 53707– 53716 [DOI] [PubMed] [Google Scholar]
- 37.Hanada K., Hara T., Nishijima M. ( 2000) J. Biol. Chem. 275, 8409– 8415 [DOI] [PubMed] [Google Scholar]
- 38.Chen J. K., Lane W. S., Schreiber S. L. ( 1999) Chem. Biol. 6, 221– 235 [DOI] [PubMed] [Google Scholar]
- 39.Ikushiro H., Hayashi H., Kagamiyama H. ( 2001) J. Biol. Chem. 276, 18249– 18256 [DOI] [PubMed] [Google Scholar]
- 40.Ikushiro H., Hayashi H., Kagamiyama H. ( 2004) Biochemistry 43, 1082– 1092 [DOI] [PubMed] [Google Scholar]
- 41.Yard B. A., Carter L. G., Johnson K. A., Overton I. M., Dorward M., Liu H., McMahon S. A., Oke M., Puech D., Barton G. J., Naismith J. H., Campopiano D. J. ( 2007) J. Mol. Biol. 370, 870– 886 [DOI] [PubMed] [Google Scholar]
- 42.Ellman G. L. ( 1959) Arch. Biochem. Biophys. 82, 70– 77 [DOI] [PubMed] [Google Scholar]
- 43.Riddles P. W., Blakeley R. L., Zerner B. ( 1979) Anal. Biochem. 94, 75– 81 [DOI] [PubMed] [Google Scholar]
- 44.Collaborative Computational Project 4 ( 1994) Acta Crystallogr. D 50, 760– 76315299374 [Google Scholar]
- 45.Kabsch W. ( 1993) J. Appl. Crystallogr. 26, 795– 800 [Google Scholar]
- 46.Murshudov G. N., Vagin A. A., Dodson E. J. ( 1997) Acta Crystallogr. D 53, 240– 255 [DOI] [PubMed] [Google Scholar]
- 47.Emsley P., Cowtan K. ( 2004) Acta Crystallogr. D 60, 2126– 2132 [DOI] [PubMed] [Google Scholar]
- 48.Williams R. D., Wang E., Merrill A. H., Jr. ( 1984) Arch. Biochem. Biophys. 228, 282– 291 [DOI] [PubMed] [Google Scholar]
- 49.Han G., Gable K., Yan L., Allen M. J., Wilson W. H., Moitra P., Harmon J. M., Dunn T. M. ( 2006) J. Biol. Chem. 281, 39935– 39942 [DOI] [PubMed] [Google Scholar]
- 50.Ikushiro H., Fujii S., Shiraiwa Y., Hayashi H. ( 2008) J. Biol. Chem. 283, 7542– 7553 [DOI] [PubMed] [Google Scholar]
- 51.Dunathan H. C. ( 1966) Proc. Natl. Acad. Sci. U. S. A. 55, 712– 716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J., Ferreira G. C. ( 2002) J. Biol. Chem. 277, 44660– 44669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornemann T., Penno A., von Eckardstein A. ( 2008) Chem. Phys. Lipids 154S, S63, PO111 [Google Scholar]
- 54.Zitomer N. C., Mitchell T., Voss K. A., Bondy G. S., Pruett S. T., Garnier-Amblard E. C., Liebeskind L. S., Park H., Wang E., Sullards M. C., Merrill A. H., Jr., Riley R. T. ( 2009) J. Biol. Chem. 284, 4786– 4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ikushiro H., Islam M. M., Tojo H., Hayashi H. ( 2007) J. Bacteriol. 189, 5749– 5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.