

Abstract
The endogenous ligands for free fatty acid receptor 1 (FFA1) are medium and longer chain free fatty acids. However, a range of selective, small molecule ligands have recently been developed as tool compounds to explore the therapeutic potential of this receptor, whereas clinically employed thiazolidinedione “glitazone” drugs are also agonists at FFA1. Each of these classes of agonist was able to promote phosphorylation of the ERK1/2 mitogen-activated protein (MAP) kinases in cells able to express human FFA1 on demand. However, although both lauric acid and the synthetic agonist GW9508X produced rapid and transient ERK1/2 MAP kinase phosphorylation, the thiazolidinedione rosiglitazone produced responses that were sustained for a substantially longer period. Despite this difference, the effects of each ligand required FFA1 and were transduced in each case predominantly via G proteins of the Gαq/Gα11 family. Different glitazone drugs also displayed markedly different efficacy and kinetics of sustainability of ERK1/2 MAP kinase phosphorylation. A number of orthosteric binding site mutants of FFA1 were generated, and despite variations in the changes of potency and efficacy of the three ligand classes in different functional end point assays, these were consistent with rosiglitazone also binding at the orthosteric site. Four distinct polymorphic variants of human FFA1 have been described. Despite previous indications that these display differences in function and pharmacology, they all responded in entirely equivalent ways to lauric acid, rosiglitazone, and GW9508X in measures of ERK1/2 MAP kinase phosphorylation, enhancement of binding of [35S]GTPγS (guanosine 5′-O-(3-[35S]thio)triphosphate) to Gαq, and elevation of intracellular [Ca2+], suggesting that individuals expressing each variant are likely to respond equivalently to orthosteric agonists of FFA1.
Introduction
Fatty acids have long been known to produce a variety of effects in the body. However, until recently, these actions were thought to be mediated exclusively via regulation of cellular metabolism. Thus, the recent identification and deorphanization (1–5) of the free fatty acid (FFA)3 family (reviewed in Refs. 6 and 7) of G protein-coupled receptors (GPCRs) has prompted re-evaluation of the mechanism of action of FFAs in health and disease. The initial deorphanization studies of free fatty acid receptor 1 (FFA14), which at that time was designated GPR40 (1–3), also demonstrated expression of receptor mRNA in the pancreas, and further analysis showed levels to be enriched in islets and, in particular, the insulin-producing β-cells (1, 2). Coupled with the long appreciated effects of fatty acids to elevate glucose-dependent insulin secretion, this suggested FFA1/GPR40 as a potential target for the treatment of diabetes (7–10). This has resulted in efforts to identify small molecule ligands able to act as selective agonists or antagonists of FFA1 (11–16) to act as tool compounds and the generation of knock-out lines of mice (15, 17–19) to explore the physiological function of FFA1. FFA1 expression has also been detected in various pancreatic-derived cell lines, including MIN6, β-TC-3, HIT-T15, and INS-1E (1–3, 20), and such lines have also, therefore, been used widely to explore the function of this receptor. The action of the thiazolidinedione “glitazone” drugs as agonists at FFA1 (3, 21) is of particular interest because although a number have been employed clinically, they are well established as regulators of the peroxisome proliferator-activated receptor γ (PPARγ) group of nuclear receptors (22), and this is assumed to be their key site of action. However, it is also well established that glitazones can rapidly generate a series of intracellular signals, including activation of the ERK1/2 MAP kinases (23) that, kinetically, appear unlikely to reflect actions toward PPARγ. Combinations of mutagenesis and molecular modeling of FFA1 (16, 24) have implicated key residues involved in the binding and function of both fatty acids and structurally related, synthetic small agonist ligands, but the relevance of these residues to the binding and function at glitazones at FFA1 remains unknown.
Recently, a series of polymorphic variants, D175N (25), G180S (26), and R211H (27), of human (h)FFA1 have been described and, in each case, some element of the response of the variant receptor or physiological function of individuals expressing the variant has been described to be different from the predominant form. Linkage of polymorphisms of GPCRs to altered physiological response or drug treatment may provide validation of the encoded proteins as therapeutic drug targets and are an underpinning driver of expectations for the concepts of personalized medicine (28). Here, therefore, as well as exploring potential functional differences between the different classes of FFA1 agonists and comparing the response of glitazone drugs with both the fatty acid lauric acid and the best studied small molecule agonist GW9508X (11, 12) at a number of orthosteric binding pocket mutations, we have also explored potential variation in the signaling characteristics of the three ligand classes at each of the currently reported open reading frame polymorphisms of hFFA1.
EXPERIMENTAL PROCEDURES
Materials
Tissue culture reagents were from Invitrogen (Paisley, Strathclyde, UK). All ligands and experimental reagents were from Sigma-Aldrich (Dorset, UK) with the following exceptions: rosiglitazone, troglitazone, ciglitazone, and pioglitazone were from Axxora (Nottingham, UK); T0070907 and GW9662 were from Calbiochem (Nottingham, UK); GW1100 and GW9508X were a generous gift from Dr. Andrew J. Brown (GlaxoSmithKline, Stevenage, UK); YM-254890 (29) was a gift from Astellas Pharmaceuticals (Tsukuba, Japan); and the radiochemical [35S]GTPγS and ERK1/2 SureFire AlphaScreen kits were from PerkinElmer Life Sciences (Buckinghamshire, UK). Phospho-specific and total anti-ERK1/2 antibodies were from Cell Signaling (Boston, MA). Antiserum directed against green fluorescent protein and cross-reactive with enhanced yellow fluorescent protein (eYFP) was produced in-house. The Proteome Profiler human phospho-kinase array kit was from R&D Systems (Minneapolis, MN).
Site-directed Mutagenesis
Human FFA1 was fused via the C terminus to eYFP or Gαq and, in the case of hFFA1-eYFP, subcloned into pcDNA5/FRT/TO (Invitrogen), as described previously (30). Individual binding site mutations and polymorphisms were introduced into pcDNA5/FRT/TO-hFFA1-eYFP or pcDNA3/hFFA1-Gαq using the QuikChange® II site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions.
Cell Culture and Generation of Stable Flp-In T-REx HEK293 Cells
Cells were maintained in Dulbecco's modified Eagle's medium without sodium pyruvate (Invitrogen, catalog number 41965) supplemented with 10% (v/v) dialyzed fetal bovine serum, 1% penicillin/streptomycin mixture, and 10 μg/ml blasticidin at 37 °C in a humidified atmosphere of air/CO2 (19:1). Inducible Flp-In T-REx HEK293 cells were generated for hFFA1-eYFP and the various binding mutants and human polymorphisms. Briefly, cells were transfected with a mixture containing the desired receptor cDNA in pcDNA5/FRT/TO vector and the pOG44 vector (1:9) using Effectene® transfection reagent (Qiagen, West Sussex, UK) according to the manufacturer's instructions. Cell maintenance and selection were as described elsewhere (30). Antibiotic-resistant clones were screened for receptor expression by both fluorescence and Western blotting. To induce expression of receptors cloned into the Flp-In locus, cells were treated with 0.5 μg/ml doxycycline as indicated.
Cell Stimulations and Protein Assays
Cell stimulations and lysates were prepared for Western blotting, AlphaScreen, and Proteome Profiler experiments in a similar manner. Briefly, cells were plated on poly-d-lysine-coated plates (6- and 12-well tissue culture grade plates except AlphaScreen, which required 40,000 cells/well of a 96-well plate) and simultaneously induced with 0.5 μg/ml doxycycline as required. Before stimulation, cells were serum-starved either overnight or for 4 h (AlphaScreen) in cell culture medium lacking dialyzed fetal bovine serum (FBS).
Stimulation of cells was arrested on ice, and cells were washed twice with ice-cold phosphate-buffered saline. For detection of hFFA1-eYFP or ERK1/2 by immunoblot, lysates were harvested in ice-cold radioimmunoprecipitation assay buffer (50 mm HEPES, 150 mm NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate supplemented with 10 mm NaF, 5 mm EDTA, 10 mm NaH2PO4, 5% ethylene glycol, 1 mm Na3VO4, and a protease inhibitor mixture (Complete; Roche Diagnostics, Herfordshire, UK, pH 7.4), rocked for 30 min at 4 °C, and transferred to microcentrifuge tubes for centrifugation at 4 °C and 14,000 × g for 15 min to pellet insoluble cell debris. Supernatant was diluted in Laemmli buffer (63 mm Tris, 50 mm dithiothreitol, 80 mm SDS, 10% glycerol, pH 6.8, with 0.004% bromphenol blue) and either boiled for 5 min (ERK1/2) or heated to 65 °C degrees (hFFA1 receptor detection). SDS-PAGE and Western blotting were performed as described recently (31). For SureFire pERK1/2 and Proteome Profiler experiments, cells were lysed in appropriate lysis buffers provided with the kits, and samples were processed according to the manufacturer's instructions.
Immunocytochemistry and Live Cell Imaging
For immunocytochemistry and live cell imaging, hFFA1-eYFP Flp-In T-REx HEK293 cells were plated on poly-d-lysine-coated coverslips in the presence or absence of 0.5 μg/ml doxycycline for incubation times as indicated. To determine localization and extent of ERK1/2 phosphorylation, cells were stimulated with ligand as indicated and then fixed with 4% (v/v) formaldehyde solution before immunostaining with the same pERK1/2 antibody as for Western blots and an Alexa Fluor-594 anti-mouse secondary antibody. Hoechst 33342 was used for nuclear staining. For live cell imaging of receptor expression, coverslips were placed into a microscope chamber containing physiological saline solution (130 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 20 mm HEPES, 10 mm d-glucose, pH 7.4), illuminated with an ultra high point intensity 75-watt xenon arc lamp (Optosource, Cairn Research, Faversham, Kent, UK) at 500 nm, and imaged using a Nikon Diaphot inverted microscope equipped with a Nikon ×40 oil immersion Fluor objective lens (NA = 1.3) and a monochromator (Optoscan, Cairn Research). Fluorescence emission at 535 nm was monitored using a high resolution interline transfer-cooled digital CCD camera (Cool Snap-HQ, Roper Scientific/Photometrics, Tucson, AZ). MetaMorph imaging software (Universal Imaging Corp., Downing, PA) was used for control of the monochromator and CCD camera and for processing of the cell image data. MetaMorph software was used to analyze the images.
Calcium Assays
Population cell calcium changes were assessed in Flp-In T-REx HEK293 cells harboring hFFA1-eYFP and polymorphisms/binding mutants that were treated with or without 0.5 μg/ml doxycycline. Cells were grown in poly-d-lysine-coated wells of a 96-well microtiter plate. 24 h after induction, cells were loaded with the calcium-sensitive dye Fura-2, as described previously (32), and the response to FFA1 ligands was assessed using a FLEXStation (Molecular Devices, Sunnydale, CA).
[35S]GTPγS Incorporation Assays
For hFFA1 activation experiments, HEK293 cells were transfected with 5 μg of wild type or mutant hFFA1-Gαq fusion receptor cDNA, and membranes were prepared after 24 h, as described elsewhere (21). [35S]GTPγS binding experiments were initiated by the addition of 5 μg of cell membranes to an assay buffer (20 mm HEPES, pH 7.4, 3 mm MgCl2, 100 mm NaCl, 1 μm GDP, 0.2 mm ascorbic acid, and 50 nCi of [35S]GTPγS) containing the given concentration of agonist. Each reaction was performed in the presence of 10 μm fatty acid-free bovine serum albumin as we have previously demonstrated that this step is required to prevent FFA1 binding of endogenous agonists released by membrane preparation (21). Nonspecific binding was determined in the above conditions with the addition of 100 μm GTPγS. Reactions were incubated for 30 min at 30 °C and were terminated by the addition of 500 μl of ice-cold buffer containing 20 mm HEPES, pH 7.4, 3 mm MgCl2, 100 mm NaCl, and 0.2 mm ascorbic acid. The samples were centrifuged at 14,000 × g for 10 min at 4 °C. The resulting pellets were resuspended in solubilization buffer (100 mm Tris, 200 mm NaCl, 1 mm EDTA, and 1.25% Nonidet P-40) plus 0.2% SDS. Samples were precleared with Pansorbin followed by immunoprecipitation with C-terminal Gq/G11 antiserum CQ (33). Finally, the immunocomplexes were washed once with solubilization buffer, and bound [35S]GTPγS was estimated by liquid scintillation spectrometry.
Data Analysis
Densitometry was performed on pERK1/2 Western blots and x-ray films from Proteome Profiler experiments using Quantity One software (Bio-Rad). All data were quantified and analyzed using GraphPad Prism 4.0 and are expressed as mean ± S.E. Differences were considered statistically significant when p < 0.05 according to Student's unpaired t test or one-way analysis of variance with Bonferroni's correction for multiple comparisons, as appropriate.
RESULTS
hFFA1 was C-terminally tagged with eYFP and cloned into the Flp-In locus of Flp-In T-REx HEK293 cells as described previously (21). Expression of hFFA1-eYFP could not be detected in the absence of inducer (supplemental Fig. 1), but the addition of doxycycline (0.5 μg/ml) for varying times resulted in expression of proteins with apparent mass close to 70 kDa in lysates of these cells that were detected by an in-house-generated anti-green fluorescent protein antiserum and that reached maximal levels within 36 h (supplemental Fig. 1A) and were sustained for at least 72 h (data not shown). Fluorescence imaging of these cells showed the expressed eYFP-tagged receptor to be located predominantly at the cell surface (supplemental Fig. 1B). We have previously used these cells (21) in intracellular [Ca2+] imaging studies to confirm that medium and longer chain free fatty acids (1–3), certain synthetic ligands including GW9508X (11), and a number of thiazolidinedione drugs (3), including rosiglitazone and troglitazone (21), act as agonists for hFFA1-eYFP.
Many GPCRs are able to cause phosphorylation and activation of the ERK1/2 MAP kinases (34, 35). hFFA1-eYFP also produced stimulation of ERK1/2 MAP kinase phosphorylation from a very low basal level in response to either lauric acid as a typical free fatty acid or GW9508X as a prototypic, selective small molecule agonist (Fig. 1A). In both cases, phosphorylation of these kinases was rapid, reaching a maximal level within 5–10 min of exposure to the ligand, and transient, returning to close to basal levels within 60 min (Fig. 1A). As reported by others using assays based on the elevation of intracellular [Ca2+], lauric acid, as with other medium chain free fatty acids (1), displayed relatively poor potency (pEC50 ∼ 4.7), whereas GW9508X was significantly more potent (pEC50 ∼ 6.5) (11, 24). Such extensive but transient activation of the ERK1/2 MAP kinases in response to lauric acid and GW9508X was confirmed via immunocytochemistry studies in which both ligands caused rapid activation and nuclear localization of the ERK1/2 MAP kinases, whereas the reduction in these signals at longer time points was again evident (supplemental Fig. 2A). By contrast, the addition of rosiglitazone (pEC50 ∼ 5.0), although producing a slightly less rapid elevation of ERK1/2 MAP kinase phosphorylation, resulted in a sustained response in which maximum signal was maintained throughout the 60-min period (Fig. 1B) and for up to 3 h (data not shown but see later). As a slower and more sustained profile of ERK1/2 activation has been linked in some situations with β-arrestin scaffolding and cytosolic retention of ERK1/2 (35, 36), we next examined the subcellular localization of ERK1/2 by immunocytochemistry. Staining of ERK1/2 MAP kinase phosphorylation in response to rosiglitazone was consistent with the time course observed in immunoblots (supplemental Fig. 2B); however, activated ERK1/2 was found to be nucleus-localized at both 5 min and 30 min, arguing against a substantial role for β-arrestin in producing the altered time course. As with the effects of lauric acid and GW9508X, the induced expression of hFFA1-eYFP was required for both rapid and more prolonged ERK1/2 MAP kinase activity in response to rosiglitazone in Flp-In T-REx HEK293 cells that harbored hFFA1-eYFP at the inducible locus (supplemental Fig. 2B).
FIGURE 1.

FFA1 receptor agonists stimulate phosphorylation of the ERK1/2 MAP kinases with different kinetics. Cells treated with doxycycline for 48 h to induce expression of hFFA1-eYFP were processed to detect both phosphorylated (upper panels) and total levels (lower panels) of the ERK1/2 MAP kinases. FBS was employed as a positive control to cause phosphorylation of ERK1/2. A, cells were treated with either lauric acid (100 μm) (panel i) or GW9508X (10 μm) (panel ii) for varying times or with varying concentrations of lauric acid (panel iii) or GW9508X (panel iv) for 10 min (n = 3–4). IB, immunoblot. B, cells were treated with rosiglitazone (100 μm) for varying times (panel i) or with varying concentrations of rosiglitazone for 30 min (panel ii) and were processed as in A. WT, wild type.
Although FFA1 is considered to couple predominantly to members of the Gαq family of G proteins (1, 6), there are many avenues and pathways by which activation of a GPCR can result in ERK1/2 MAP kinase activation (34, 35, 37). To explore this, the mechanism(s) of rapid activation of the ERK1/2 MAP kinases in response to lauric acid, GW9508X, and rosiglitazone was determined. In each case, activation was almost eliminated by treatment of the cells with the selective Gαq/Gα11 inhibitor YM-254890 (20 nm) (29) (Fig. 2) but maintained, although somewhat reduced, following pretreatment of the cells with pertussis toxin (25 ng/ml, 16 h) (Fig. 2), which causes ADP-ribosylation of members of the Gi/Go G protein group and prevents their potential activation by GPCRs. Furthermore, although thiazolidinediones are known as direct activators of the PPARγ group of nuclear receptors (22, 38, 39), the PPARγ antagonists T0070907 and GW9662 (40) were both without effect on rosiglitazone-mediated ERK1/2 MAP kinase phosphorylation (Fig. 2). As anticipated, however, the selective FFA1 receptor antagonist GW1100 (11, 21) blocked the effect of each of the three classes of agonist ligand (Fig. 2). Interestingly, although pretreatment with either GW1100 or YM-254890 fully blocked both initial and sustained ERK1/2 MAP kinase phosphorylation in response to rosiglitazone (Fig. 3), pretreatment with pertussis toxin resulted in inhibition of the sustained and maintained elevation of ERK1/2 MAP kinase phosphorylation and may suggest a role of Gi-family proteins in this element of the function of rosiglitazone (Fig. 3). Immunocytochemistry studies (supplemental Fig. 3 and data not shown) also confirmed the lack of effect of the PPARγ antagonist T0070907 in preventing short or longer term ERK1/2 phosphorylation in response to rosiglitazone. In no case did inhibitor treatment alter the subcellular localization of activated ERK1/2 MAP kinases (supplemental Fig. 3 and data not shown).
FIGURE 2.

hFFA agonists acutely promote phosphorylation of the ERK1/2 MAP kinases via activation of Gαq/Gα11. Cells were induced with doxycycline to induce expression of hFFA1-eYFP, as in Fig. 1, and then treated with or without inhibitors to block activation of Gαq/Gα11 (20 nm YM-254890 for 40 min, YM), Gαi/Gαo (25 ng/ml for 16 h; pertussis toxin, PTox), FFA1 (10 μm GW1100 (GW) for 40 min), or PPARγ (1 μm T0070907 or GW9662 for 40 min). Cells were then stimulated with lauric acid (100 μm, 10 min), rosiglitazone (100 μm, 30 min), or GW9508X (10 μm, 10 min). Samples were processed to detect phosphorylated (upper panels) and total (lower panels) levels of the ERK1/2 MAP kinases (n = 3). IB, immunoblot.
FIGURE 3.

Sustained phosphorylation of the ERK1/2 MAP kinases via activation of FFA1 by rosiglitazone does not reflect activation of PPARγ. Cells were untreated (−dox) or induced with doxycycline (+dox and all other sets) to induce expression of hFFA1-eYFP. They were then treated with inhibitors to potentially block activation of FFA1 (10 μm GW1100), Gαq/Gα11 (20 nm YM-254890), Gαi/Gαo (PTox), or PPARγ (1 μm T0070907) as in Fig. 2. Cells were then stimulated with rosiglitazone (100 μm) for varying times and processed to detect phosphorylated ERK1/2 MAP kinases. In each case, the effect of FBS was also assessed following stimulation for 5 min and in the presence of relevant inhibitors. IB, immunoblot.
A series of thiazolidinedione drugs was then tested for their capacity to promote and maintain ERK1/2 MAP kinase phosphorylation status in hFFA1-eYFP-expressing cells. Troglitazone, ciglitazone, and pioglitazone all produced relatively rapid activation of the ERK1/2 MAP kinases, but restoration to basal levels of phosphorylation followed a clear profile in which troglitazone and ciglitazone were more rapid than pioglitazone (Fig. 4A). Furthermore, although maintaining the signal for the most extended period, rosiglitazone appeared to be a partial agonist for ERK1/2 MAP kinase activation at early time points when compared with ciglitazone, whereas both rosiglitazone and ciglitazone were markedly more efficacious than troglitazone and pioglitazone (Fig. 4A). A major limitation of immunoblotting studies to identify phosphorylated forms of ERK1/2 MAP kinases is the very limited dynamic range of the signal. As such, to further explore the apparent partial agonism of pioglitazone and troglitazone, we repeated such studies using the bead-based SureFire ERK AlphaScreen assay. These studies confirmed the relatively weak partial agonism of both troglitazone and pioglitazone (Fig. 4B) and the markedly prolonged duration of signal induced by rosiglitazone when compared with ciglitazone (Fig. 4B). Concentration-response curves were then constructed for each of the glitazones at a single time point of 15 min using the SureFire ERK AlphaScreen assay. In agreement with the previously reported potencies of rosiglitazone and troglitazone for [35S]GTPγS binding (21), troglitazone displayed greater potency (although as noted above, substantially reduced efficacy) than rosiglitazone for pERK1/2 production, and both were more potent than ciglitazone and pioglitazone (Fig. 4C). The rank order of potencies is: troglitazone (5.72 ± 0.07) > rosiglitazone (4.94 ± 0.07) > ciglitazone (4.37 ± 0.06) > pioglitazone (3.09 ± 0.93).
FIGURE 4.

Different thiazolidinedione drugs provide distinct kinetics and efficacy of ERK1/2 MAP kinase phosphorylation but produce similar overall regulation of a series of phospho-proteins. Cells as in Fig. 1 were treated with doxycycline for 48 h to induce expression of hFFA1-eYFP. A, the cells were exposed to a range of thiazolidinedione drugs, rosiglitazone (100 μm), ciglitazone (100 μm), troglitazone (10 μm), and pioglitazone (100 μm) for varying times and then processed to detect phosphorylated ERK1/2 MAP kinases (n = 3). Stimulation of the cells with FBS for 5 min provided a positive control. B, the pERK1/2 SureFire AlphaScreen assay was used to quantify ERK1/2 activation over a 3-h period. (n = 3). RLU, relative light units. C, concentration-response curves for pERK1/2 in response to various thiazolidinediones measured at 15 min of stimulation (n = 3). In B and C, the error bars indicate S.E. D, quantification of phosphorylation of various kinases and kinase targets by lauric acid (100 μm), rosiglitazone (100 μm), or GW9508X (10 μm) in the absence (light bars, −dox) or presence (dark bars, +dox) of doxycycline. Shown are results from selected targets; data reflect an individual experiment representative of two performed.
Although ERK1/2 MAP kinase phosphorylation is often assessed as a convenient end point of signal generation via GPCR activation, a series of complex kinase cascades and inter-related pathways may also be potentially regulated. To explore this for ligands at hFFA1-eYFP, analysis of the phosphorylation status of a wide range of kinases and kinase targets was assessed in response to lauric acid, GW9508X, and rosiglitazone following treatment of cells for 5 or 45 min using a human phospho-kinase array that allows simultaneous assessment of the phosphorylation status of 38 kinases and other cellular proteins based on a dot blot array of 46 phospho-specific antibodies (Proteome Profiler human phospho-kinase array, R&D Systems). All three ligands induced FFA1-specific enhanced phosphorylation of a number of other potential targets including p38MAPKα (Fig. 4D) and N-terminal c-Jun kinase (JNK) (Fig. 4D), although in the case of JNK, this was not observed at 5 min but only at the later time point. As expected, not all targets on the kinase array were phosphorylated in response to ligands (for example, Akt) (Fig. 4D and data not shown) or in a hFFA1-specific manner (for example, RSK1/2 and AMPKα2) (Fig. 4D), where equal signals above basal were produced both with and without induction of expression of the receptor and were therefore considered to reflect off-target mechanisms. Enhanced p38MAPKα was also detected in response to each ligand without induction of FFA1-eYFP expression (Fig. 4D), but in this case, substantially greater levels of phosphorylation were produced following receptor induction (Fig. 4D). Despite the different kinetic profile of ERK1/2 activation seen in the previous experiments for rosiglitazone, the unbiased screen of kinase targets failed to identify differential signal pathway activation. Thus, taken together with the results of the inhibitor studies in Figs. 2 and 3, it seems that rosiglitazone activates ERK1/2 and related kinase targets through the same pathway as GW9508X and lauric acid but with a different time course of stimulation.
Key residues of the binding pocket of FFA1 for GW9508X identified by Sum et al. (24) include Arg183 (position 5.39 in the nomenclature of (41)), Asn244 (6.55), Arg258 (7.35), and His137 (4.56), whereas for linoleic acid, the same amino acids, except His137, were also predicted to be of major importance (24). We mutated each of these residues individually to Ala, as well as generating a double R183A,R258A mutant, initially in the context of hFFA1-Gαq fusion proteins (21). Each of these was expressed transiently in HEK293 cells, and following membrane preparation, [35S]GTPγS binding studies were performed with an associated Gαq immunoprecipitation step (21). Both GW9508X (pEC50 = 6.01 ± 0.16) and rosiglitazone (pEC50 = 5.17 ± 0.16) enhanced binding of [35S]GTPγS in a concentration-dependent manner in samples containing wild type hFFA1-Gαq. By contrast, although H137A hFFA1-Gαq responded to GW9508X with similar potency (pEC50 = 6.31 ± 0.29) as the wild type hFFA1 construct, the efficacy of GW9508X in this assay was greatly reduced (Fig. 5A). N244A hFFA1-Gαq displayed a significant response to GW9508X only at concentrations at and above 1 μm, and this prevented accurate assessment of EC50 (Fig. 5A). However, best estimates suggested that the N244A mutation reduced the potency of GW9508X by ∼100-fold. No significant response to GW9508X could be recorded in such [35S]GTPγS binding studies employing membranes transfected to express R183A hFFA1-Gαq, R258A hFFA1-Gαq, or R183A,R258A hFFA1-Gαq (Fig. 5A). Apart from a weak response via H137A hFFA1-Gαq, of the constructs generated, only wild type hFFA1-Gαq was able to respond significantly to rosiglitazone in this assay at concentrations that were practical to employ (Fig. 5A). To explore the functionality of the hFFA1 receptor more fully, each of the above mutants was also introduced into hFFA1 in the context of C-terminally eYFP-tagged forms. These were employed to produce further stable and inducible Flp-In T-REx HEK293 cell lines, able to express the construct only upon the addition of doxycycline (Fig. 5B). As with wild type hFFA1-eYFP, when induced, each of these mutants was present predominantly at the cell surface (Fig. 5B). In contrast to the [35S]GTPγS binding studies, H137A hFFA1-eYFP produced a robust, concentration-dependent elevation of [Ca2+] in response to both lauric acid (pEC50 = 4.91 ± 0.08) and rosiglitazone with little difference in the potency of either ligand when compared with wild type (lauric acid pEC50 = 5.08 ± 0.1, rosiglitazone = 4.26 ± 0.20) (Fig. 5C). However, at concentrations below 30 μm, lauric acid was unable to cause a substantial elevation of intracellular [Ca2+] via any of the other mutants (Fig. 5C), whereas the apparent potency of rosiglitazone at N244A hFFA1 was not different from that at H137A hFFA1 (Fig. 5C). Again responses to rosiglitazone were lacking in cells expressing alanine mutations at either or both of Arg183 and Arg258 (Fig. 5C).
FIGURE 5.

Selective effects of hFFA1 binding pocket mutants on the function of lauric acid, GW9508X and rosiglitazone. A, receptor activation by rosiglitazone or GW9508X was measured in [35S]GTPγS incorporation assays performed on membranes prepared from HEK293 cells transfected with Gαq fusion proteins of wild type (WT) hFFA1 and H137A, R183A, N244A, R258A, and R183A,R258A receptor mutants (n = 2–7). B, wild type, H137A, R183A, N244A, R258A, and R183A,R258A hFFA1-eYFP were produced and used to generate Flp-In T-REx HEK293 cells. Each receptor construct was detected only following treatment of the cells with doxycycline. C, the effect of the above mutations was then tested in cell population calcium assays in response to lauric acid and rosiglitazone (n = 2–6). In A and C, the error bars indicate S.E.
A similar general pattern was noted in SureFire ERK AlphaScreen assays (Fig. 6). Responses to GW9508X were lacking at Ala mutants of either or both Arg183 and Arg258, were of reduced efficacy but similar potency at H137A hFFA1 (pEC50 = 6.97 ± 0.16), and were of markedly reduced potency, although as the data were fitted poorly, this was impossible to assess accurately, at N244A hFFA1 when compared with wild type (pEC50 = 6.32 ± 0.13) (Fig. 6). These observations were essentially replicated when using lauric acid as ligand (Fig. 6). However, rosiglitazone provided a different pattern. Now both at H137A hFFA1 (pEC50 = 5.81 ± 0.09) and, more surprisingly, at R258A hFFA1 (pEC50 = 5.72 ± 0.08), rosiglitazone acted as a partial agonist and, indeed, in both situations displayed enhanced potency when compared with wild type hFFA1 (pEC50 = 4.76 ± 0.12) (Fig. 6).
FIGURE 6.
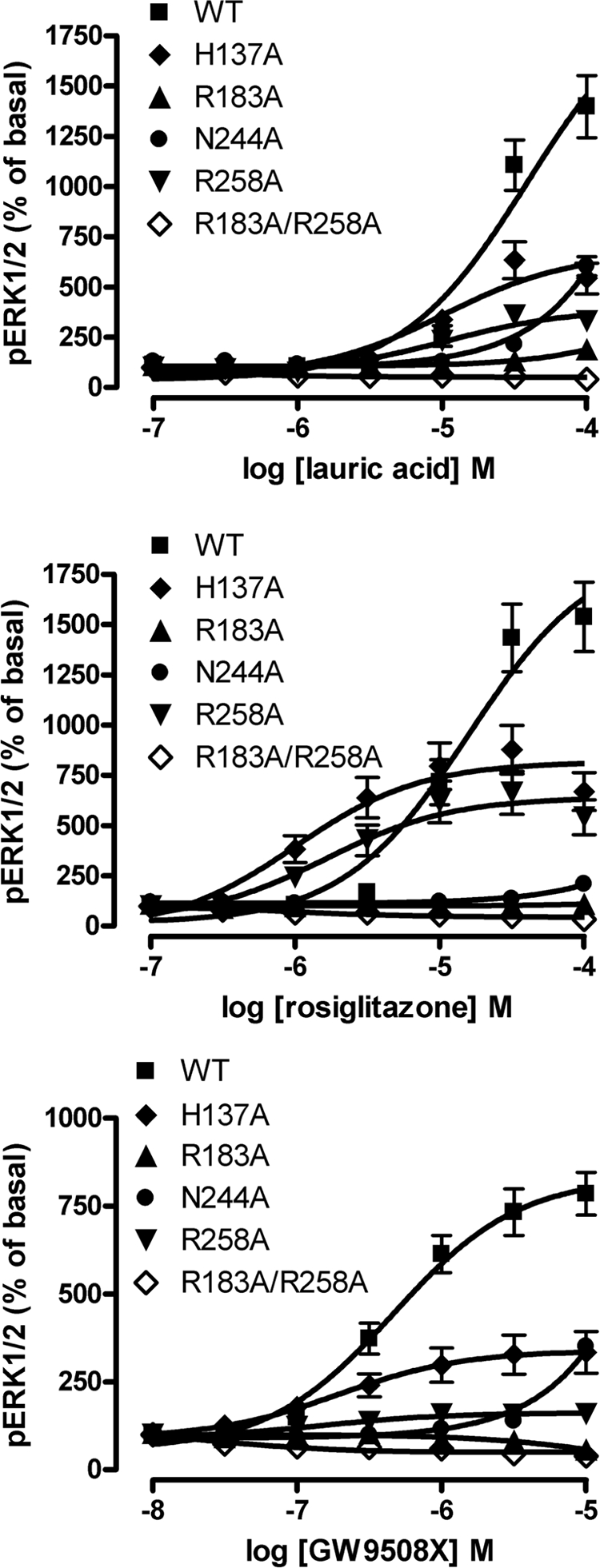
Orthosteric binding site mutants of FFA1 display different responses to rosiglitazone when compared with lauric acid or GW9508X. Cells induced to express wild type (WT) hFFA1-eYFP or each of the individual mutants were then used to monitor potential activation of the ERK1/2 MAP kinases using the SureFire AlphaScreen assay. Response to the ligands was measured at 15 min of stimulation (n = 3). The error bars indicate S.E.
Recently, three distinct polymorphic variants of hFFA1, D175N (5.31) (25), G180S (5.36) (26), and R211H (5.67) (27) have been reported. The G180S polymorphism is located close to Arg183 (5.39), D175N is within the second extracellular loop and is close to Glu172 (5.28), which, in concert with Arg183 (5.39), is reported to generate an ionic lock that may restrain FFA1 in an inactive state (42), whereas R211H (5.67) is within the proximal section of the third intracellular loop and may, therefore, contribute to G protein coupling and selectivity. As such, we also generated each of these variants, initially again within the context of hFFA1-Gαq fusion proteins. Following transient transfection into HEK293 cells and membrane preparation, rosiglitazone was equipotent and displayed equal efficacy in promoting binding of [35S]GTPγS to the G protein element of each of these variants (Fig. 7A and Table 1). Following production of eYFP-tagged forms of each polymorphic variant and the generation of Flp-In T-REx HEK293 cells to allow for their inducible expression, each variant was again undetectable in the absence of doxycycline and turned on rapidly and to similar levels in response to doxycycline (Fig. 7B). Equally, each variant displayed a similar, predominantly plasma membrane-delineated, pattern of expression (Fig. 7C). In both single cell imaging (data not shown) and cell population (Fig. 7D) [Ca2+] measurements, lauric acid and rosiglitazone produced equivalent responses at each variant (Table 2). When examining ERK1/2 MAP kinase phosphorylation, each variant responded to both lauric acid and GW9508X without detectable differences in concentration dependence (data not shown) or the time scale of activation or maintenance of ERK1/2 phosphorylation (Fig. 8), and this was also true for responses to rosiglitazone (Fig. 8). Finally, we examined the G protein-coupling profile of each polymorphic variant because Asn175 hFFA1 was previously reported to respond less efficaciously via the Gi/o pathway (25). Similarly, there were no detected differences in the mechanism of ERK1/2 MAP kinase phosphorylation between the variants (Fig. 9) in that the effect of each ligand was blocked by both the FFA1 antagonist GW1100 and the Gαq/Gα11 inhibitor YM-254890 and only impaired to a limited extent by pertussis toxin pretreatment. Thus, despite the clinical and pharmacological studies suggesting functional differences for each of the polymorphic variants of FFA1, thorough pharmacological characterization failed to unveil differential pharmacology.
FIGURE 7.

Polymorphic variants of hFFA1 regulate intracellular Ca2+ and stimulate binding of [35S]GTPγS equally in response to both lauric acid and rosiglitazone. Three distinct polymorphic variants of hFFA1, D175N, G180S, and R211H, have been described. A, each of these variants was generated in the context of the hFFA1-Gαq fusion proteins. Receptor activation in response to rosiglitazone was measured by [35S]GTPγS incorporation in membranes prepared from HEK293 transfected with each Gαq fusion protein (n = 3–5). hFFA1-eYFP forms of these variants were generated and cloned into the Flp-In locus of Flp-In T-REx HEK293 cells. Cells were exposed to doxycycline (0.5 μg) for varying periods of time. WT, wild type. B and C, lysates of these cells were resolved by SDS-PAGE and immunoblotted (IB) with an anti-green fluorescent protein (anti-GFP) antiserum (B), or cells were grown on glass coverslips and then imaged (C). D, the effect of the above polymorphisms was then tested in cell population calcium assays in response to varying concentrations of lauric acid and rosiglitazone (n = 3–6). In A and D, the error bars indicate S.E.
TABLE 1.
Potency and efficacy of rosiglitazone at hFFA1 polymorphic variants fused to Gαq in [35S]GTPγS binding assays
Membranes of HEK293 cells transfected to express variants of hFFA1 fused to Gαq were employed in [35S]GTPγS binding studies as described under “Experimental Procedures.” Data are means ± S.E. from between 3 and 5 individual experiments. WT, wild type.
| hFFA1 variant | pEC50 | Efficacy (% of basal) |
|---|---|---|
| FFA1 WT-Gαq | 5.20 ± 0.12 | 162.8 ± 4.0 |
| FFA1 D175N-Gαq | 5.69 ± 0.35 | 150 ± 7.6 |
| FFA1 G180S-Gαq | 5.36 ± 0.26 | 140 ± 4.3 |
| FFA1 R211H-Gαq | 5.46 ± 0.39 | 146 ± 8.5 |
TABLE 2.
Potency and efficacy of lauric acid and rosiglitazone at inducible hFFA1 polymorphic variants in population calcium assays
Variants of hFFA1-eYFP were expressed from the Flp-In locus of Flp-In T-REx HEK293 cells by treatment with doxycycline (0.5 mg/ml, 48 h). Cells were then used to measure ligand regulation of intracellular [Ca2+] levels. Data are means ± S.E. from between 3 and 5 individual experiments. WT, wild type.
| hFFA1variant | Lauric acid |
Rosiglitazone |
||
|---|---|---|---|---|
| pEC50 | Efficacy(% of wild type) | pEC50 | Efficacy(% of wild type) | |
| FFA1 WT | 5.09 ± 0.11 | 100 | 4.51 ± 0.4 | 100 |
| FFA1 D175N | 5.37 ± 0.23 | 95 ± 9 | 4.77 ± 0.28 | 121 ± 23 |
| FFA1 G180S | 5.01 ± 0.16 | 91 ± 10 | 4.00 ± 0.57 | 126 ± 87 |
| FFA1 R211H | 5.38 ± 0.24 | 78 ± 9 | 4.77 ± 0.36 | 97 ± 23 |
FIGURE 8.

Lack of variation in pERK1/2 responsiveness of polymorphic variants of hFFA1. Expression of D175N hFFA1-eYFP, G180S hFFA1-eYFP, or R211H hFFA1-eYFP was induced in appropriate Flp-In T-REx HEK293 cells by exposure to doxycycline (0.5 μg/ml, 48 h). Each variant responded to lauric acid (100 μm), to GW9508X (10 μm), and to rosiglitazone (100 μm) with no detectable differences in the time scale of activation or maintenance of ERK1/2 MAP kinase phosphorylation (upper panels). Total levels of ERK1/2 MAP kinases (lower panels) were assessed as loading controls (n = 3). IB, immunoblots.
FIGURE 9.

Polymorphic variants of hFFA1 all regulate ERK1/2 MAP kinase phosphorylation predominantly via activation of Gαq/Gα11. Expression of D175N hFFA1-eYFP, G180S hFFA1-eYFP, or R211H hFFA1-eYFP was induced in appropriate Flp-In T-REx HEK293 cells by exposure to doxycycline (0.5 μg/ml, 48 h). The ability of each variant to promote phosphorylation of the ERK1/2 MAP kinases was assessed following exposure to vehicle (1% dimethyl sulfoxide (DMSO)) or lauric acid (100 μm, 10 min) following treatment of the cells with (+) or without (−) the inhibitors YM-254890 (YM, 20 nm, 40 min), pertussis toxin (PTox, 25 ng/ml, 16 h), or GW1100 (GW, 10 μm, 40 min). Total levels of ERK1/2 MAP kinases (lower panels) were assessed as loading controls. Immunoblots (IB) are representative of experiments performed on three separate occasions. WT, wild type.
DISCUSSION
hFFA1 (also known as GPR40) was originally identified as an orphan seven-transmembrane-element protein and likely GPCR based on sequence similarity with other rhodopsin family or class A GPCRs (43). Specific attention to its potential as a therapeutic target for the treatment of type 2 diabetes was driven by a combination of the known effect of fatty acids to elevate glucose-dependent insulin secretion and expression of FFA1 receptor mRNA in the insulin-producing β-cells of the pancreas (1, 2). This has resulted in the search for, and identification of, novel and selective ligands for FFA1 via both traditional function-based screening of chemical libraries (11–15) and virtual screening of the receptor based on the generation of receptor homology and pharmacophore models (16). Studies using both knockdown of FFA1 levels in pancreatic cell lines (2, 20, 44, 45) and FFA1 knock-out lines of mice (15, 17, 46, 47) have provided further support for this receptor as an important regulator of insulin secretion and in the effects of fatty acids. However, as there are both positive effects of fatty acids in promoting insulin release acutely but also potentially negative effects of long term elevation of fatty acids on the function and viability of pancreatic β-cells (48, 49), there are differing views as to whether agonism or antagonism of FFA1 might prove to be the most effective treatment (6, 47). As well as the FFA1-selective, synthetic small molecule ligands identified recently, it has been known for some time that thiazolidinedione glitazone drugs that are used clinically in the treatment of diabetes can also act as FFA1 agonists (3, 21), although their clinical effectiveness is generally considered to reflect activation of PPARγ.
Recently, we have made considerable use of Flp-In T-REx HEK293 cells to explore the pharmacology and regulation of a range of GPCRs (21, 31, 32, 50, 51). As these allow tight regulation of expression of proteins located at the Flp-In locus, they have particular value in circumstances in which ligands might be pleiotropic and regulate cellular function by a range of mechanisms, as is the case for both free fatty acids and thiazolidinediones (7). Indeed, initial generation of Flp-In T-REx HEK293 cells that allowed inducible expression of hFFA1-eYFP allowed us to demonstrate that rapid elevation of intracellular [Ca2+] in response to either troglitazone or rosiglitazone required expression of the receptor construct (21). This was equally true when activation of the ERK1/2 MAP kinases was measured in response to these ligands but, importantly, also demonstrated that phosphorylation of certain kinases, e.g. RSK1/2, by FFA1 ligands did not reflect activation of the receptor.
Intriguingly, in these studies, the kinetics of phosphorylation of the ERK1/2 MAP kinases was markedly different when employing rosiglitazone when compared with prototypic examples of free fatty acids and synthetic small molecules. Two distinct differences were noted. Firstly, in response to rosiglitazone, the onset of activation was somewhat delayed. This could be interpreted to reflect a slow on-rate of rosiglitazone, but as elevation of intracellular [Ca2+] in response to rosiglitazone was just as rapid as in response to lauric acid and these two ligands display similar (although modest) potency at FFA1, this seems unlikely. Secondly, and more obviously, phosphorylation of the ERK1/2 MAP kinases was sustained for a substantially longer period in response to rosiglitazone than to either lauric acid or GW9508X. Although phosphorylation of the ERK1/2 MAP kinases was more prolonged in response to rosiglitazone, the mechanism responsible for activation appears to be the same. In each case, pretreatment of the cells with the selective Gαq/Gα11 inhibitor YM-254890 (29) almost fully blocked the FFA1-mediated effect of the ligand. In a number of situations, sustained ERK1/2 MAP kinase phosphorylation has been shown to reflect interactions of the receptor with a β-arrestin (35). Such examples have been important in understanding the contribution of β-arrestins to the generation of novel signals (36) as opposed to their initially established role in the termination of GPCR-and G protein-mediated signaling (52). The basis for the prolonged effect of rosiglitazone remains to be clearly established, but as the activated ERK1/2 was not sequestered in the cytosol and activation was blocked in the presence of YM-254890, it does appear to be G protein-mediated rather than β-arrestin-mediated. Furthermore, it was interesting to note that not all the glitazone ligands were able to maintain ERK1/2 MAP kinase phosphorylation as effectively. These results indicate clearly that not all FFA1 agonists should be considered as functionally equivalent, although ligands from each of the three classes were also able to promote increases in both intracellular [Ca2+] and stimulated binding of [35S]GTPγS to Gαq, whereas for free fatty acids and the best studied synthetic agonist, the mode of binding is well established (24).
Although activation of the ERK1/2 MAP kinases is routinely monitored via immunoblotting studies employing antisera that specifically identify an adjacent pair of Thr and Tyr residues that become phosphorylated to promote activation, the dynamic range of such assays, as with other immunoblots, is limited. We also, therefore, employed the SureFire ERK AlphaScreen assay. This approach provided a much greater dynamic range and demonstrated much more clearly the differences in both the efficacy and the different kinetics of ERK1/2 MAP kinase phosphorylation produced by different thiazolidinediones, including the two, rosiglitazone (AvandiaTM) and pioglitazone (ActosTM in the United States, GlustinTM in Europe), that have been used in clinical settings. The basis for these differences in regulation of the ERK1/2 MAP kinases by individual glitazone drugs will be explored more fully in future studies.
The binding pocket for the endogenous ligand linoleic acid has previously been explored by Sum et al. (24), but no relevant information was available for the glitazones. We, therefore, took advantage of the efforts of Sum and colleagues (16, 24) by generating Flp-In T-REx HEK293 cell lines able to express a range of binding pocket mutants of hFFA1-eYFP on demand. Furthermore, because previous work from Sum and colleagues (16) was restricted to measurement of the elevation of [Ca2+]i and we have recently demonstrated differential pharmacological outcomes by measuring a series of distinct end points when studying orthosteric binding site mutants of the related receptors FFA2 and FFA3 (32), in these studies, we measured [Ca2+]i, ERK1/2 MAP kinase phosphorylation, and stimulation of binding of [35S]GTPγS. As anticipated from the studies of Sum et al. (24), the double R183A,R258A hFFA1 mutant was entirely unable to respond to any of the FFA1 ligand classes in any assay format, consistent with the importance of the carboxylate function of the free fatty acids and small molecule agonists such as GW9508X. We presume that the 2,4-dione function of the thiazolidinediones is coordinated to Arg183 and Arg258 in a similar manner and that this provides orientation of these ligands. Thiazolidinediones can exist in a number of distinct tautomeric states, and an amide-iminol tautomerization to generate form 5 in Ref. 53 is likely to be promoted in polar solvents. The charge provided by Arg183 and/or Arg258 would further stabilize and drive tautomerism toward this state. Such effects also reinforce the view that thiazolidinediones act as orthosteric FFA1 ligands. Interestingly, although the effect of the H137A mutation was shown by Sum et al. (24) to have only a marginal effect on the potency of linoleic acid but a large effect on the potency of GW9508X, in our [35S]GTPγS binding studies, the potency of GW9508X was unaffected, whereas efficacy was greatly reduced. Similarly, in ERK1/2 MAP kinase assays, although efficacy of GW9508X was reduced, potency was actually increased slightly. The basis for these apparent discrepancies is not clear. Furthermore, in various assays, N244A hFFA1 displayed substantially reduced potency for all the ligand classes in each assay conducted and, in the case of lauric acid and rosiglitazone that have relatively low potency at wild type FFA1, this made function difficult to observe at any ligand concentration that could be employed. This should also be contrasted with the results of Sum et al. (24) in which the N244A mutant also displayed reduced potency but, at least for linoleic acid, this was reported to be only some 4-fold. Clearly, measures of efficacy are difficult to compare between assays and between studies. It is not surprising that an amplified, downstream end point, such as [Ca2+]i, should display higher relative efficacy for mutant forms of the receptor than an early step in signal transduction such as the binding of [35S]GTPγS and, thus, we have avoided attempting to make potentially confusing and misleading cross-assay and cross-study comparisons of this parameter.
A further area of interest in current hFFA1 receptor studies is the potential for variation in function between polymorphic variants. D175N (25), G180S (26), and R211H (25, 27) variants have been reported. The R211H polymorphism has been suggested to contribute to the variation of insulin secretory capacity in Japanese men (25), although this clinical effect was not found in later studies (26), nor was alteration in allelic frequency observed in type 2 diabetics (25) or subjects with abnormalities in glucose metabolism (26). In the case of the D175N variant, which was very infrequent in the population studied by Hamid et al. (25), 5,8,11-eicosatriynoic acid displayed lower efficacy to stimulate inositol phosphate production when FFA1 was co-expressed with a chimeric G protein that trafficked Gi/o-mediated signaling through phospholipase Cβ (54). Meanwhile, the most recently described polymorphism, G180S FFA1, has been reported as a reduction in function variant in response to oleic acid and linked to reduced insulin secretion in a small number of Gly/Ser heterozygotes (26). Despite these observations, we were unable to note any substantial variation in Flp-In T-REx HEK293 cells and membranes of HEK293 cells that expressed any of these variants when compared with the wild type hFFA1 receptor sequence in response to the range of agonists examined in any of the assays we employed. Clearly, although factors other than basic pharmacological responses to the sequence alterations may be important for the reported variation in function in individuals, the current studies do not provide support for previous indications of variations in the response of these polymorphisms in biochemical studies using transfected cells. Although numerous polymorphic variants within the open reading frame sequences of a wide range of GPCRs have been reported (55), their relevance to function is often unclear or hotly debated (28, 56).
These studies provide detailed insight into the mode of action of three distinct classes of ligands that act as agonists of FFA1. Despite the distinctiveness of the kinetics and, indeed, the efficacy of different glitazone ligands as regulators of ERK MAP kinase phosphorylation status, there is no indication that these should be considered as anything other than orthosteric agonists with respect to free fatty acids. The current studies also question the basis of reported variation in pharmacology and function of the currently described polymorphic variants of this GPCR.
Supplementary Material
Acknowledgment
We thank Dr. Sabine Schlyer (Structural Research, Boehringer Ingelheim Pharmaceutical, Inc., Ridgefield, CT) for helpful discussions on the tautomeric states of thiazolidinedione ligands.
This work was supported in part by Biotechnology and Biosciences Research Council Grant BB/E019455/1.
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
4 FFA1 is the provisional International Union of Pharmacology designation for the receptor previously called GPR40.
- FFA
- free fatty acid
- FFA1
- free fatty acid receptor 1
- hFFA1
- human FFA1
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- GPCR
- G protein-coupled receptor
- T0070907
- 2-chloro-5-nitro-N-(4-pyridyl)benzamide
- GW9662
- 2-chloro-5-nitrobenzanilide
- GW9508X
- 3-[4-({3-(phenyloxy)phenyl]methyl}amino)phenyl]propanoic acid
- GW1100
- (ethyl 4-[5-{[2-(ethyloxy)-5-pyrimidinyl]methyl}-2-{[4-fluorophenyl)methyl]thio}-4-oxo1(4H)-pyrimidinyl]-benzoate
- eYFP
- enhanced yellow fluorescent protein
- MAP
- mitogen-activated protein
- ERK
- extracellular signal-regulated kinase
- pERK1/2
- phosphorylated ERK1/2
- PPARγ
- peroxisome proliferator-activated receptor γ
- FBS
- fetal bovine serum.
REFERENCES
- 1.Briscoe C. P., Tadayyon M., Andrews J. L., Benson W. G., Chambers J. K., Eilert M. M., Ellis C., Elshourbagy N. A., Goetz A. S., Minnick D. T., Murdock P. R., Sauls H. R., Jr., Shabon U., Spinage L. D., Strum J. C., Szekeres P. G., Tan K. B., Way J. M., Ignar D. M., Wilson S., Muir A. I. ( 2003) J. Biol. Chem. 278, 11303– 11311 [DOI] [PubMed] [Google Scholar]
- 2.Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M. ( 2003) Nature 422, 173– 176 [DOI] [PubMed] [Google Scholar]
- 3.Kotarsky K., Nilsson N. E., Flodgren E., Owman C., Olde B. ( 2003) Biochem. Biophys. Res. Commun. 301, 406– 410 [DOI] [PubMed] [Google Scholar]
- 4.Brown A. J., Goldsworthy S. M., Barnes A. A., Eilert M. M., Tcheang L., Daniels D., Muir A. I., Wigglesworth M. J., Kinghorn I., Fraser N. J., Pike N. B., Strum J. C., Steplewski K. M., Murdock P. R., Holder J. C., Marshall F. H., Szekeres P. G., Wilson S., Ignar D. M., Foord S. M., Wise A., Dowell S. J. ( 2003) J. Biol. Chem. 278, 11312– 11329 [DOI] [PubMed] [Google Scholar]
- 5.Le Poul E., Loison C., Struyf S., Springael J. Y., Lannoy V., Decobecq M. E., Brezillon S., Dupriez V., Vassart G., Van Damme J., Parmentier M., Detheux M. ( 2003) J. Biol. Chem. 278, 25481– 25489 [DOI] [PubMed] [Google Scholar]
- 6.Stoddart L. A., Smith N. J., Milligan G. ( 2008) Pharmacol. Rev. 60, 405– 417 [DOI] [PubMed] [Google Scholar]
- 7.Milligan G., Stoddart L. A., Brown A. J. ( 2006) Cell. Signal. 18, 1360– 1365 [DOI] [PubMed] [Google Scholar]
- 8.Costanzi S., Neumann S., Gershengorn M. C. ( 2008) J. Biol. Chem. 283, 16269– 16273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swaminath G. ( 2008) Arch. Pharm. (Weinheim) 341, 753– 761 [DOI] [PubMed] [Google Scholar]
- 10.Telvekar V. N., Kundaikar H. S. ( 2008) Curr. Drug Targets 9, 899– 910 [DOI] [PubMed] [Google Scholar]
- 11.Briscoe C. P., Peat A. J., McKeown S. C., Corbett D. F., Goetz A. S., Littleton T. R., McCoy D. C., Kenakin T. P., Andrews J. L., Ammala C., Fornwald J. A., Ignar D. M., Jenkinson S. ( 2006) Br. J. Pharmacol. 148, 619– 628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrido D. M., Corbett D. F., Dwornik K. A., Goetz A. S., Littleton T. R., McKeown S. C., Mills W. Y., Smalley T. L., Jr., Briscoe C. P., Peat A. J. ( 2006) Bioorg. Med. Chem. Lett. 16, 1840– 1845 [DOI] [PubMed] [Google Scholar]
- 13.Bharate S. B., Rodge A., Joshi R. K., Kaur J., Srinivasan S., Kumar S. S., Kulkarni-Almeida A., Balachandran S., Balakrishnan A., Vishwakarma R. A. ( 2008) Bioorg. Med. Chem. Lett. 18, 6357– 6361 [DOI] [PubMed] [Google Scholar]
- 14.Christiansen E., Urban C., Merten N., Liebscher K., Karlsen K. K., Hamacher A., Spinrath A., Bond A. D., Drewke C., Ullrich S., Kassack M. U., Kostenis E., Ulven T. ( 2008) J. Med. Chem. 51, 7061– 7064 [DOI] [PubMed] [Google Scholar]
- 15.Tan C. P., Feng Y., Zhou Y. P., Eiermann G. J., Petrov A., Zhou C., Lin S., Salituro G., Meinke P., Mosley R., Akiyama T. E., Einstein M., Kumar S., Berger J. P., Mills S. G., Thornberry N. A., Yang L., Howard A. D. ( 2008) Diabetes 57, 2211– 2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tikhonova I. G., Sum C. S., Neumann S., Engel S., Raaka B. M., Costanzi S., Gershengorn M. C. ( 2008) J. Med. Chem. 51, 625– 633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steneberg P., Rubins N., Bartoov-Shifman R., Walker M. D., Edlund H. ( 2005) Cell Metab. 1, 245– 258 [DOI] [PubMed] [Google Scholar]
- 18.Brownlie R., Mayers R. M., Pierce J. A., Marley A. E., Smith D. M. ( 2008) Biochem. Soc. Trans. 36, 950– 954 [DOI] [PubMed] [Google Scholar]
- 19.Lan H., Hoos L. M., Liu L., Tetzloff G., Hu W., Abbondanzo S. J., Vassileva G., Gustafson E. L., Hedrick J. A., Davis H. R. ( 2008) Diabetes 57, 2999– 3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapiro H., Shachar S., Sekler I., Hershfinkel M., Walker M. D. ( 2005) Biochem. Biophys. Res. Commun. 335, 97– 104 [DOI] [PubMed] [Google Scholar]
- 21.Stoddart L. A., Brown A. J., Milligan G. ( 2007) Mol. Pharmacol. 71, 994– 1005 [DOI] [PubMed] [Google Scholar]
- 22.Saltiel A. R., Olefsky J. M. ( 1996) Diabetes 45, 1661– 1669 [DOI] [PubMed] [Google Scholar]
- 23.Gardner O. S., Dewar B. J., Graves L. M. ( 2005) Mol. Pharmacol. 68, 933– 941 [DOI] [PubMed] [Google Scholar]
- 24.Sum C. S., Tikhonova I. G., Neumann S., Engel S., Raaka B. M., Costanzi S., Gershengorn M. C. ( 2007) J. Biol. Chem. 282, 29248– 29255 [DOI] [PubMed] [Google Scholar]
- 25.Hamid Y. H., Vissing H., Holst B., Urhammer S. A., Pyke C., Hansen S. K., Glümer C., Borch-Johnsen K., Jørgensen T., Schwartz T. W., Pedersen O., Hansen T. ( 2005) Diabet. Med. 22, 74– 80 [DOI] [PubMed] [Google Scholar]
- 26.Vettor R., Granzotto M., De Stefani D., Trevellin E., Rossato M., Farina M. G., Milan G., Pilon C., Nigro A., Federspil G., Vigneri R., Vitiello L., Rizzuto R., Baratta R., Frittitta L. ( 2008) J. Clin. Endocrinol. Metab. 93, 3541– 3550 [DOI] [PubMed] [Google Scholar]
- 27.Ogawa T., Hirose H., Miyashita K., Saito I., Saruta T. ( 2005) Metabolism 54, 296– 299 [DOI] [PubMed] [Google Scholar]
- 28.Insel P. A., Tang C. M., Hahntow I., Michel M. C. ( 2007) Biochim. Biophys. Acta 1768, 994– 1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takasaki J., Saito T., Taniguchi M., Kawasaki T., Moritani Y., Hayashi K., Kobori M. ( 2004) J. Biol. Chem. 279, 47438– 47445 [DOI] [PubMed] [Google Scholar]
- 30.Canals M., Jenkins L., Kellett E., Milligan G. ( 2006) J. Biol. Chem. 281, 16757– 16767 [DOI] [PubMed] [Google Scholar]
- 31.Canals M., Milligan G. ( 2008) J. Biol. Chem. 283, 11424– 11434 [DOI] [PubMed] [Google Scholar]
- 32.Stoddart L. A., Smith N. J., Jenkins L., Brown A. J., Milligan G. ( 2008) J. Biol. Chem. 283, 32913– 32924 [DOI] [PubMed] [Google Scholar]
- 33.Mitchell F. M., Buckley N. J., Milligan G. ( 1993) Biochem. J. 293, 495– 499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crespo P., Gutkind J. S. ( 2004) Methods Mol. Biol. 250, 203– 210 [DOI] [PubMed] [Google Scholar]
- 35.Smith N. J., Luttrell L. M. ( 2006) Hypertension 48, 173– 179 [DOI] [PubMed] [Google Scholar]
- 36.Lefkowitz R. J., Rajagopal K., Whalen E. J. ( 2006) Mol. Cell 24, 643– 652 [DOI] [PubMed] [Google Scholar]
- 37.Luttrell L. M. ( 2008) Mol. Biotechnol. 39, 239– 264 [DOI] [PubMed] [Google Scholar]
- 38.Jha R. J. ( 1999) Clin. Exp. Hypertens. 21, 157– 166 [DOI] [PubMed] [Google Scholar]
- 39.Tontonoz P., Spiegelman B. M. ( 2008) Annu. Rev. Biochem. 77, 289– 312 [DOI] [PubMed] [Google Scholar]
- 40.Leesnitzer L. M., Parks D. J., Bledsoe R. K., Cobb J. E., Collins J. L., Consler T. G., Davis R. G., Hull-Ryde E. A., Lenhard J. M., Patel L., Plunket K. D., Shenk J. L., Stimmel J. B., Therapontos C., Willson T. M., Blanchard S. G. ( 2002) Biochemistry 41, 6640– 6650 [DOI] [PubMed] [Google Scholar]
- 41.Ballesteros J. A., Weinstein H. ( 1995) in Methods in Neurosciences. Receptor Molecular Biology ( Conn M. P., Sealfon S. C. eds), pp. 366– 428, Academic Press, Orlando, FL [Google Scholar]
- 42.Sum C. S., Tikhonova I. G., Costanzi S., Gershengorn M. C. ( 2009) J. Biol. Chem. 284, 3529– 3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sawzdargo M., George S. R., Nguyen T., Xu S., Kolakowski L. F., O'Dowd B. F. ( 1997) Biochem. Biophys. Res. Commun. 239, 543– 547 [DOI] [PubMed] [Google Scholar]
- 44.Salehi A., Flodgren E., Nilsson N. E., Jimenez-Feltstrom J., Miyazaki J., Owman C., Olde B. ( 2005) Cell Tissue Res. 322, 207– 215 [DOI] [PubMed] [Google Scholar]
- 45.Schnell S., Schaefer M., Schöfl C. ( 2007) Mol. Cell. Endocrinol. 263, 173– 180 [DOI] [PubMed] [Google Scholar]
- 46.Kebede M., Alquier T., Latour M. G., Semache M., Tremblay C., Poitout V. ( 2008) Diabetes 57, 2432– 2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latour M. G., Alquier T., Oseid E., Tremblay C., Jetton T. L., Luo J., Lin D. C., Poitout V. ( 2007) Diabetes 56, 1087– 1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGarry J. D. ( 2002) Diabetes 51, 7– 18 [DOI] [PubMed] [Google Scholar]
- 49.Nolan C. J., Madiraju M. S., Delghingaro-Augusto V., Peyot M. L., Prentki M. ( 2006) Diabetes 55, Suppl. 2, S16– 23 [DOI] [PubMed] [Google Scholar]
- 50.Ellis J., Pediani J. D., Canals M., Milasta S., Milligan G. ( 2006) J. Biol. Chem. 281, 38812– 38824 [DOI] [PubMed] [Google Scholar]
- 51.Milasta S., Pediani J., Appelbe S., Trim S., Wyatt M., Cox P., Fidock M., Milligan G. ( 2006) Mol. Pharmacol. 69, 479– 491 [DOI] [PubMed] [Google Scholar]
- 52.Moore C. A., Milano S. K., Benovic J. L. ( 2007) Annu. Rev. Physiol. 69, 451– 482 [DOI] [PubMed] [Google Scholar]
- 53.Sundriyal S., Khana S., Saha R., Bharatam P. V. ( 2008) J. Phys. Org. Chem. 21, 30– 33 [Google Scholar]
- 54.Milligan G., Kostenis E. ( 2006) Br. J. Pharmacol. 147, Suppl. 1, S46– 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kazius J., Wurdinger K., van Iterson M., Kok J., Bäck T., Ijzerman A. P. ( 2008) Hum. Mutat. 29, 39– 44 [DOI] [PubMed] [Google Scholar]
- 56.Thompson M. D., Siminovitch K. A., Cole D. E. ( 2008) Methods Mol. Biol. 448, 139– 185 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.