

Abstract
Aims
Periventricular white matter injury in premature infants occurs following hypoxia/ischaemia and systemic infection, and results in hypomyelination, as well as neuromotor and cognitive deficits later in life. Inflammatory infiltrates are seen within human cerebral white matter from periventricular leucomalacia (PVL) cases.
Methods
In this study, we examine the time course of CD-68+ microglial cell responses relative to cell death within white matter following hypoxia/ischaemia in a rat model of PVL. We also tested the efficacy of the minocycline, an agent that suppresses microglial activation, in this model when administered as a post-insult treatment.
Results
We show that preoligodendrocyte injury in the post-natal day 6 begins within 24 h and continues for 48–96 h after hypoxia/ischaemia, and that microglial responses occur primarily over the first 96 h following hypoxia/ischaemia. Minocycline treatment over this 96 h time window following the insult resulted in significant protection against white matter injury, and this effect was concomitant with a reduction in CD-68+ microglial cell numbers.
Conclusions
These results suggest that anti-inflammatory treatments may represent a useful strategy in the treatment of PVL, where clinical conditions would favour a post-insult treatment strategy.
Keywords: hypoxia ischaemia, microglia, minocycline, oligodendrocyte, periventricular leucomalacia
Introduction
Periventricular leucomalacia (PVL) is the major pathological substrate of cerebral palsy in preterm infants suffering cerebral hypoxia/ischaemia (HI) or maternal-fetal infection, resulting in oligodendrocyte (OL) injury in developing white matter (WM) [1]. PVL describes focal WM necrosis and diffuse gliosis, resulting in hypomyelination and associated motor and cognitive disabilities [1,2]. Candidate mechanisms for this WM injury include oxidative stress, glutamate-mediated excitotoxicity and inflammation; immature premyelinating OLs (pre-OLs) have been shown to be uniquely susceptible to these forms of injury compared with mature OLs [3–5]. While the cause of PVL is likely to be multifactorial, the present study addresses the role of the microglial responses in an in vivo model of PVL.
The age window of greatest susceptibility to PVL in the human brain is between 23 and 32 weeks' gestation, when subcortical WM is populated predominantly by pre-OLs, including both OL precursors and immature OLs [6]. In vitro studies have demonstrated that pre-OLs are more vulnerable to injury than mature OLs under conditions of oxidative stress, oxygen-glucose deprivation and glutamate receptor (GluR)-mediated excitotoxicity [3,4,7,8]. We have previously shown that pre-OLs in developing rat and human WM express GluRs [1,4,8–10]. We developed a rat model of PVL in the post-natal day 6 (P6) Long-Evans rat, in which unilateral carotid ligation and hypoxia lead to predominantly WM injury and hypomyelination [4,8]. Using this model, we have previously demonstrated that WM injury after HI can be attenuated by systemic post-treatment with 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione and topiramate [2,3:4,5-bis-O-(1-methyl ethylidene)-β-D-fructopyranose sulfamate], which are antagonists of the alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) and kainate GluR subtypes [4,8]. Additional studies have implicated a central role for microglia in a synergistic effect of HI and infection in WM injury [11–13]. Given that the therapeutic effect of AMPA receptor antagonists is subtotal, we aimed to investigate the role of microglial involvement in pre-OL injury.
Post mortem studies of human PVL tissue reveal that activated microglia are abundant in the diffuse component of PVL, along with macrophages in the periventricular necrotic foci, but there is minimal microglial activation in the overlying cortex [2]. In addition, oxidative and nitrosative stress markers are increased in the diffuse component of PVL in reactive astrocytes and pre-OLs [2]. Both human and rodent developmental studies demonstrate that the density of developing microglia in WM is higher during early development than in later life. In the human, WM is preferentially populated with amoeboid microglia during preterm [23–35 post-conceptional weeks (PCW)] [14,15]. Similarly in the rodent, microglia are increased in density in WM and deep cortex during the first post-natal week [16,17]. These time points correspond in both species to periods of increased susceptibility to WM injury, and hence targeting microglial activation may be especially effective in the immature brain.
When stimulated, microglia secrete reactive oxygen and nitrogen species and express a variety of cytokines, which have been implicated in inflammatory and hypoxic/ischaemic brain injury [2,14,18–20]. In the rodent, microglia are activated following hypoxic/ischaemic cortical injury [21,22] as well as after intracerebral administration of the endotoxin lipopolysaccharide [11,23].
Several studies have targeted microglial activation as a therapeutic strategy in models of inflammation and hypoxic/ischaemic injury in the adult brain [24,25]. The tetracycline derivatives, minocycline and doxycycline, have been widely used and shown to have protective efficacy in cellular injury via microglial inactivation [22,26– 29]. Recently, minocycline has been reported to variably inhibit microglial activation and to reduce WM damage in focal cerebral ischaemia in the immature rat [20]. Combination before and after treatment around a monophasic hypoxic/ischaemic injury in the immature rat brain suppressed the inflammatory markers nitric oxide (NO), IL-1β and TNFα in WM [20,26,30,31].
In the present study, we aimed to determine the protective efficacy of minocycline against microglial inactivation when administered solely after hypoxic/ischaemic injury. We show that pre-OL injury begins within 24 h, but continues for 48–96 h after HI, and a rise in microglial cell numbers within the WM occurs primarily over the first 96 h following HI. Minocycline treatment over this 96 h time window, following the insult, resulted in significant protection against WM injury, and this was concomitant with a reduction in microglial cell numbers.
Materials and methods
Unilateral carotid ligation with hypoxia
Selective WM injury was produced in P6 Long-Evans rat pups (Charles River Laboratories, Wilmington, MA) by unilateral carotid artery ligation (UCL) followed by severe hypoxia (6% O2 for 1 h), as previously described [4,8]. In brief, rats were anaesthetised with ether, and the proximal internal carotid artery was isolated from the sympathetic chain, clamped and cauterised. The neck wound was closed, and the animals were allowed to recover for 1 h on a thermal blanket, maintaining body temperature at 33–34°C (baseline temperature for P6–P10 rats). The rats were then placed in a sealed chamber infused with nitrogen to a level of 6% O2, also on a thermal blanket maintaining body temperature at 33–34°C throughout hypoxia. After 1–2 h of recovery, the rats were returned to their dam. Rats were sacrificed 96 h after the procedure (P10), and brains were perfused with 4% paraformaldehyde, post-fixed for 1 h, and then cryoprotected in 30% sucrose in phosphate buffered saline (PBS). All procedures were approved and in accordance with guidelines set up by the institutional animal care and use committee.
Minocycline treatment
The P6 rat pups were divided into minocycline (n = 16) and vehicle (PBS) treated (n = 15) groups and underwent UCL, followed by a hypoxia (6% O2 for 1 h). Minocycline (50 mg/kg i.p.) was initiated immediately following HI and continued every 12 h. Healthy controls consisted of six male P6 pups without treatment. All rats were sacrificed at P10, that is, 96 h after HI. Nine rats per group were used for myelin basic protein (MBP) staining, while all rats were used for CD-68 staining.
Histological analysis and immunofluorescence
For all experimental rats, serial 20 μm coronal sections were cut by cryostat from the anterior extent of the lateral ventricles through the posterior extent of the dorsal hippocampus. Brain tissue sections (20 μm) were stained with: (i) haematoxylin and eosin (HE), and in situ end labelling (ISEL) to measure cell death; (ii) double labelling with O4 antibodies followed by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling assay (TUNEL) toevaluate changes in the pre-OL population and in situ cell death; (iii) MBP to determine mature OLs; and (iv) CD-68 and CD-74 (major histocompatibility complex class II, MHC II) to identify activated microglia in the WM.
Assessment of cell death following unilateral carotid ligation and hypoxia
Representative sections were stained with HE for routine evaluation and with ISEL for detection of DNA fragmentation as a sensitive method for evidence of apoptotic and necrotic cell death [32]. Mounted sections were treated with pronase (1 g/ml; Boehringer Mannheim, Indianapolis, IN), rinsed in 2% glycine and then H2O, and incubated for 1 h at room temperature with 50 μg/ml DNA polymerase I (Promega, Madison, WI, USA) and 10 μM each biotin-21-dUTP (Clontech, Cambridge, UK), dCTP, dATP, and dGTP dissolved in buffer (50 mM Tris-HCl, 5 mM MgCl2, 10 mM β-mercaptoethanol and 0.005% BSA). Biotin end-labelled DNA fragments were detected using avidin-biotin-peroxidase complex amplification (Vectastain Elite; Vector Laboratories, Burlinghame, CA, USA) with diaminobenzidine tetrahydrochloride detection. Cell counts were made based on a total area measuring 63 μm2 that was assessed by two independent observers blind to treatment condition, as per our previously described methods [8]: 0 ≤ 2 positive cells/field, 1 = 3–10 positive cells/field, 2 = 11–20 positive cells/field, 3 = 21–30 positive cells/field, 4 ≥ 31 positive cells/field.
Assessment of O4 premyelinating oligodendrocyte population and evaluation of in situ cell death by deoxynucleotidyl transferase-mediated dUTP nick end-labelling assay
Double labelling was done sequentially with O4 labelling always performed first as these antigens are destroyed by detergent used in other labelling protocols. Serial 20 μm coronal sections of all brains were stained using mouse anti-rat O4 antibody (Gift from Dr S. Pfeiffer, Farmington, CT, USA). Adjacent mounted sections were first incubated in 5% normal goat serum for 1 h to block non-specific binding. Slides were incubated with O4 antibody at a dilution of 1:500 in PBS with 1% normal goat serum overnight at 4°C, followed by 1 h incubation with Alexa Fluor anti-mouse IgM antibody (1:1000; Molecular Probes, Invitrogen, Eugene, OR). To determine the specificity of immunostaining with the antibodies, control sections were processed without primary and/or secondary antibodies, which resulted in no labelling. Three adjacent pairs of coronal sections were evaluated for each rat, and cells were counted (number per 63 μm2) in pericallosal WM ipsilateral and contralateral to the ligation, and in the WM of healthy controls by two observers blind to experimental group. In situ cell death was evaluated by TUNEL assay (TMR red kit, Roche Applied Science) according to the supplied protocol. The sections were briefly fixed 2 min in 4% paraformaldehyde after completing the O4 staining and prior to TUNEL assay. Slides were viewed via epifluorescence microscopy on a Zeiss Axioscope. Images were captured with a Spot digital camera and the Spot Advanced software 4.5 (Diagnostic Instruments, Sterling Heights, MI, USA).
Assessment of microglia numbers
Serial 20 μm coronal sections of all brains were stained with mouse anti-rat CD-68 (Serotec-MCA341A, Raleigh, NC, USA). Adjacent mounted sections were first incubated in 5–10% normal goat serum for 1 h to block nonspecific binding and permeabilised in 0.1% Triton X-100. Slides were incubated with CD-68 antibody at a dilution of 1:100 in PBS with 1% normal goat serum with addition of 0.1% Triton X-100 overnight at 4°C, followed by 1 h incubation with Alexa Fluor 568 anti-mouse IgG antibody (1:1000; Molecular Probes, Invitrogen). A limited number of 20 μm coronal sections were stained with mouse anti-rat CD-74 (OX6; Santa Cruz Biotechnology, CA, USA). Adjacent mounted sections were first incubated in 5% normal goat serum for 1 h. Slides were incubated with CD-74 antibody at a dilution of 1:50 in PBS with 1% normal goat serum overnight at 4°C, followed by 1 h incubation with Oregon Green 488 anti-mouse IgG antibody (1:100; Molecular Probes, Invitrogen). To determine the specificity of immunostaining with the antibodies, control sections were processed without primary and/or secondary antibodies, which resulted in no labelling. In stereotactically identical sections, cells were counted (number per 63 μm2) in pericallosal WM ipsilateral and contralateral to the ligation, and in the WM of healthy controls by two observers blind to treatment group.
Assessment of myelin basic protein staining in the rodent periventricular leucomalacia model
Sections from nine minocycline and nine vehicle-treated animals were available for examination with MBP staining (SMA-99; 1:800, Sternberger Monoclonals, Baltimore, MD, USA), and were compared for WM injury. Adjacent mounted sections from stereotactically identical sections were incubated in 5–10% normal goat serum for 1 h to block non-specific binding and concurrently permeabilised in 0.1% Triton X-100. Slides were incubated with MBP antibody (SMA-99; Sternberger Monoclonals) at a dilution of 1:800 in PBS with 1% normal goat serum plus 0.1% Triton X-100 overnight at 4°C, followed by 1 h incubation with Oregon Green 488 anti-mouse IgG antibody (1:1000; Molecular Probes, Invitrogen). To determine the specificity of immunostaining with the antibodies, control sections were processed without primary and/or secondary antibodies, which resulted in no labelling. Lesion severity was assessed as per our previously published methods [4,8]. In brief, the WM staining for MBP was compared ipsilateral and contralateral to the ligation, and lesion severity was quantified on a scale of 0–3, as follows: 0 = the ipsilateral and contralateral hemispheres are similar; 1 = change ipsilateral to the ligation is limited to a loss of staining in the cortical processes; 2 = loss of staining includes thinning of the periventricular WM; and 3 = thinning of the WM tracts includes a full thickness loss of staining in the capsule. A mean score was obtained, and the lesion severity was compared between the group treated with minocycline and the vehicle-treated controls.
Statistical analysis
All values are expressed as mean ± SEM. Data distribution normality was evaluated with the Kolmogorov-Smirnov test. Groups were compared using the one-way ANOVA test with the Bonferroni multiple comparison post hoc tests. A two-way ANOVA was used for comparison of CD-68 in the normal development vs. HI. Myelin basic protein staining results were evaluated using the Mann-Whitney U-test. P-values ≤ 0.05 were considered statistically significant.
Results
Temporal profile of cell death following unilateral hypoxic/ischaemic injury at post-natal day 6/7
Following UCL and hypoxia at P6/7, pups were sacrificed at 24, 48, 72, 96 h and 7 days (n = 6/group), with litter mate control rats for comparison (n= 3/age). Consistent with our previously published reports [4], ISEL-positive cells within pericallosal WM were apparent by 24 h after hypoxic/ischaemic injury and peaked at 24–48 h, decreasing by 96 h (Figures 1A,B). These data indicate that cell death occurs early, and additional staining using the pre-OL marker O4 showed a commensurate decrease in O4+ cell number ipsilateral to the ligation between 24 and 96 h from 52.9 ± 14.6 to 34.5 ± 1.3 cells/field compared with the average density in control pups of 57.2 ± 12.8 cells/field (P<0.01). In addition, double staining demonstrated TUNEL-positive O4-positive pre-OLs (Figure 1C). Several studies show that the marker O4 stains the major pre-OL population present in WM in the preterm human (23–35 PCW) and in the first post-natal week in the rat, when WM is most susceptible to hypoxic/ischaemic injury [4,6].
Figure 1.
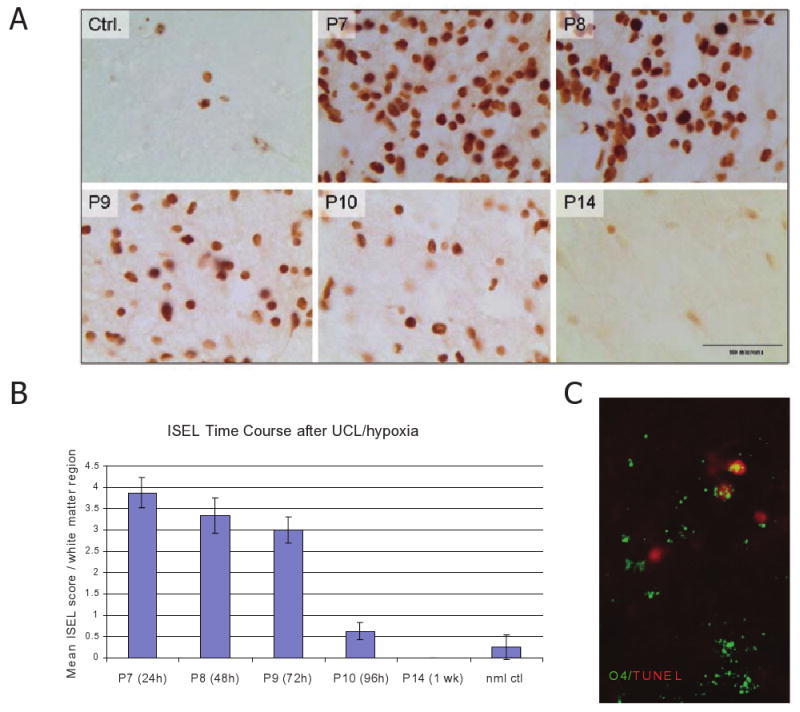
Temporal profile of cell death following unilateral hypoxic-ischaemic injury at post-natal day 6. (A) Highest levels of cells detected by in situ end labelling (ISEL) are seen at 24 and 48 h (upper middle and right: P7 and P8), decreasing by 96 h (lower middle: P10) following injury. One week (lower right: P14) after injury a rare ISEL+ cells were detected, a finding comparable to normal cell levels (upper left: Ctrl, ×1000 magnification). (B) Quantification of the time course of cell death following UCL/hypoxia at P6 (score: 0 ≤ 2 cells/field, 1 = 3–10 cells/field; 2 = 11–20 cells/field; 3 = 21–30 cells/field; 4 ≥ 31 cells/field). Normal control values at P6. (C) White matter pre-OL cell death 24 h post UCL/hypoxia at P6. TUNEL (red)/O4 (green) double staining. Scale bar = 20 μm. P, post-natal day; pre-OL, premyelinating oligodendrocyte; TUNEL, transferase-mediated dUTP nick end-labelling assay; UCL, unilateral carotid artery ligation.
Developmental profile of CD-68+ microglia in white matter during the second postnatal week
Because the second post-natal week exhibits the greatest susceptibility to WM injury in the Long-Evans rat, we evaluated the developmental regulation of the microglial population in WM in this specific rat strain. CD-68-positive cell counts were measured in developing WM in normal rat pups at P7 through P13. The density of CD-68-positive cells steadily increased from 19.6 ± 1.8 at P7 to 47.2 ± 5.2 at P10, and returned to a lower density of 12.7 ± 2.3 per unit area by P13 (Figure 2A).
Figure 2.

Microglial infiltration in the white matter under (A) normal oxygenation and (B) after P6 UCL/hypoxia. Blinded cell counts CD-68+ cells in pericallosal white matter are shown as means ± SEM. CD-68+ cells are significantly increased ipsilateral to the lesion by 24 h and remain significantly increased for at least 96 h. CONTRA, contralateral; IPSI, ipsilateral; NML, normal; SEM, standard error of the mean; UCL, unilateral carotid artery ligation.
Increased microglial density in white matter following hypoxic/ischaemic injury
Immunostaining revealed a significant increase in CD-68-positive cells in pericallosal WM ipsilateral to the carotid ligation compared with the same region in normal litter mates. CD-68-positive cell density increased between 24 and 96 h, but peak density was apparent between 48 and 96 h (P < 0.001), with a gradual decline over 7 days post-insult (Figure 2B). CD-68-positive microglia were not significantly increased in the hemisphere contralateral to the lesion compared with controls, and did not differ from the developmentally regulated increase seen in normal controls in this age window.
Systemic minocycline attenuates hypomyelination following unilateral carotid artery ligation and hypoxia/ischaemia at post-natal day 10
Myelin basic protein-positive staining for OL processes was used to assess WM injury ipsilateral and contralateral to the ligation in the minocycline and vehicle (PBS) treatment groups (n = 9/group). Using a previously published scoring system of MBP loss (see Materials and methods), vehicle-treated rats showed a significant decrease in MBP staining in the ipsilateral hemisphere (P = 0.04; Figure 3A,B), as previously reported [4,8]. At 96 h post ligation, ipsilateral pericallosal WM staining with MBP shows disruption of horizontal OL processes, as well as dorsally radiating fibers into overlying cortex (Figure 3A).
Figure 3.
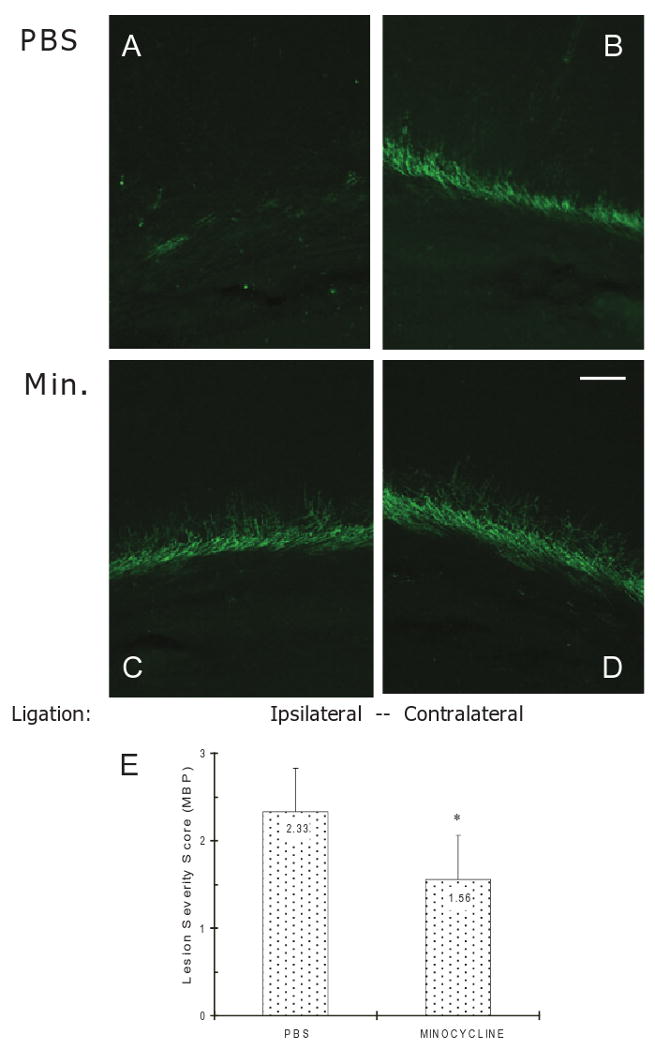
Effect of minocycline on the pericallosal white matter (WM) damage in P6 rats following UCL/hypoxia at 96 h. Myelin basic protein (MBP) staining shows less damage in minocycline-treated rats in comparison with vehicle-treated controls ipsilateral to UCL. (A) WM of vehicle-treated animal ipsilateral to UCL. (B) WM of vehicle-treated animal contralateral to UCL. (C) WM of minocycline-treated animal ipsilateral to UCL. (D) WM of minocycline-treated animal contralateral to UCL. (E) Quantification of WM injury using the lesion severity score (means ± SEM) based on MBP staining in minocycline-treated (n = 9) and vehicle (PBS)-treated animals (n = 9). Scale bar = 100 μm. Min, Minocycline; PBS, phosphate buffered saline; UCL, unilateral carotid artery ligation.
In contrast, minocycline post treatment every 12 h for 4 days significantly attenuated MBP loss ipsilateral to the ligation compared with vehicle (Figure 3C,D). A semiquantitative analysis of lesion severity by MBP staining confirmed significant protection against WM injury in rats treated with minocycline compared with vehicle-treated controls (lesion severity score 1.56 ± 0.5 vs. 2.33 ± 0.7, respectively, p = 0.04, Figure 3E).
Systemic minocycline reduces CD-68 positive microglial cell numbers following hypoxia/ischaemia at post-natal day 9
As minocycline has been reported to inhibit microglial activation following injury, we next determined the effect of minocycline treatment on the number of CD-68-positive microglia compared with vehicle treatment (PBS). Counts of CD-68-positive cells were performed in minocycline-treated P6 pups (n =16) and compared with vehicle-treated controls (n = 15). At 96 h, increased density of CD-68-positive cells was observed throughout the pericallosal WM ipsilateral to the ligation compared with the contralateral hemisphere (Figure 4A,B). CD-68-positive cell density was significantly higher on the ipsilateral side compared with contralateral (85.33 ± 3.4 vs. 47.06 ± 5.5, P < 0.001), and ipsilateral side was significantly higher than WM from control littermates (28.33 ± 4.3, P < 0.001, Figure 5). Minocycline treatment significantly attenuated this ipsilateral elevation in CD-68-positive cells compared with the vehicle ipsilaterally (61.75 ± 5.5 vs. 85.33 ± 3.4, P < 0.001), but had no effect on microglial density on the contralateral side (42.62 ± 3.6 vs. 47.06 ± 5.5, Figure 4C,D and Figure 5).
Figure 4.
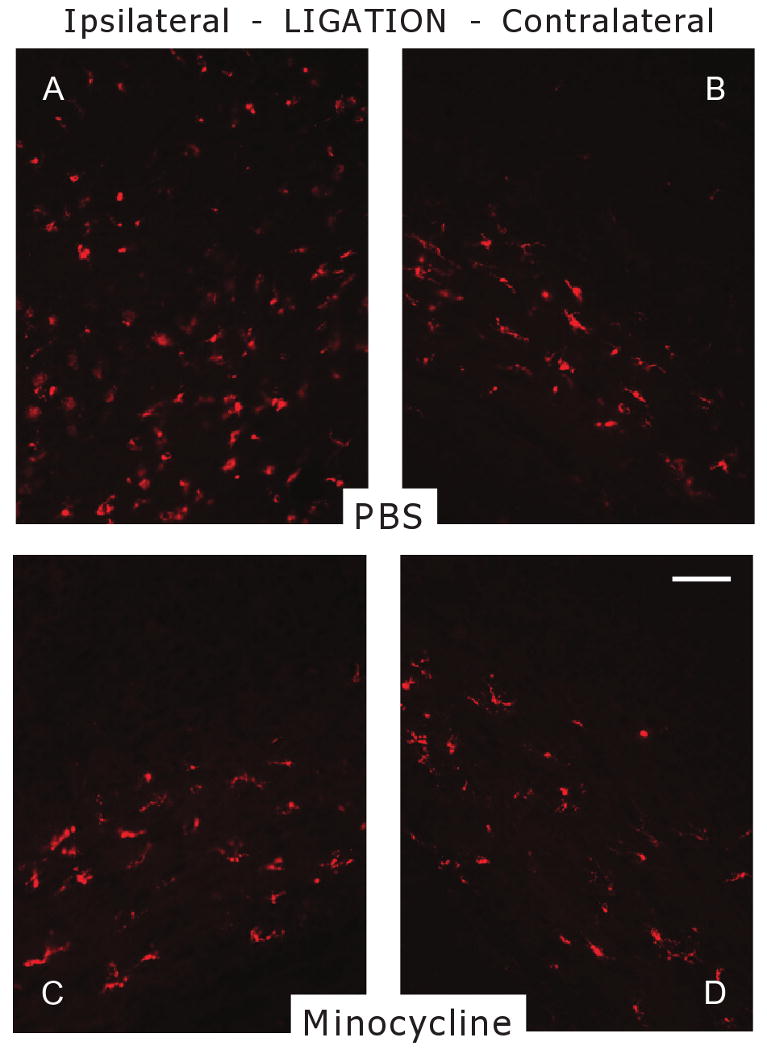
Effect of minocycline on the number of microglia in the pericallosal white matter (WM) in P6 rats following UCL/hypoxia at 96 h. CD-68 staining shows decreased number of CD-68+ cells in minocycline-treated rats in comparison with vehicle-treated controls ipsilateral to UCL. (A) WM of vehicle-treated animal ipsilateral to UCL. (B) WM of vehicle-treated animal contralateral to UCL. (C) WM of minocycline-treated animal ipsilateral to UCL. (D) WM of minocycline-treated animal contralateral to UCL. Scale bar, 100 μm. P6, post-natal day 6; PBS, phosphate buffered saline; UCL, unilateral carotid artery ligation.
Figure 5.
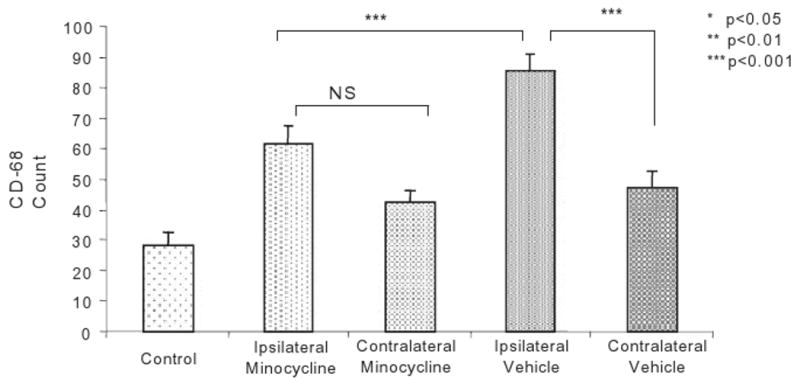
Effect of minocycline on the number of CD-68-positive cells in the pericallosal white matter (WM) of P6 rats following UCL/hypoxia at 96 h. Results are shown as means ± SEM of CD-68+ count in brains of: minocycline-treated (n = 16) rats ipsilateral (IL-Minocycline) and contralateral (CL-Minocycline) to UCL; vehicle (PBS)-treated (n = 15) rats ipsilateral (IL-PBS) and contralateral (CL-PBS) to UCL; and healthy controls (n= 6). NS, not significant; P6, post-natal day 6; PBS, phosphate buffered saline; SEM, standard error of the mean; UCL, unilateral carotid artery ligation.
CD-68-positive microglia are present in non-ischaemic, normal human cortex during development [14], and here we report that in control rats there is a transient population of CD-68 microglia. CD-68 has been reported to binds to activated microglia and stain phagocytic macrophages in inflamed and/or ischaemic tissue [33,34]. To further characterise the increased microglial population seen after hypoxic/ischaemic injury in this study, we used the CD-74 (OX-6) marker for MHC II expression, often taken as a marker of activated microglia and macrophages [34,35]. A limited number of slides from the hypoxic/ischaemic vehicle-treated group that were stained with CD-74 revealed an increase in CD-74-positive cells in pericallosal WM ipsilateral to the carotid ligation compared with the same region on the contralateral side (Figure 6A,B), similar to the pattern observed with CD-68. Like CD-68, staining was highest on the side ipsilateral to the HI, but there were a limited number of cells on the contralateral hemisphere that received hypoxia alone (Figure 6B). However, in normoxic control brains, only rare CD-74-positive cells were present in the control while scattered cells were seen with CD-68 (data not shown). Finally, CD-74 staining was also applied to slides adjacent to those stained with CD-68 from the minocyline-treated group, and showed that treatment attenuated the number of CD-74-positive cells in cortical WM ipsilateral to the hypoxic/ischaemic insult (Figure 6C), and little to no CD-74-stained cells was observed in the same region contralaterally. Taken together, the CD-74 staining shows similar patterns to that of CD-68, suggesting that at least a subset of the microglia ipsilateral to the hypoxic/ischaemic insult are involved in an immune reaction, and that this is diminished by minocycline treatment.
Figure 6.
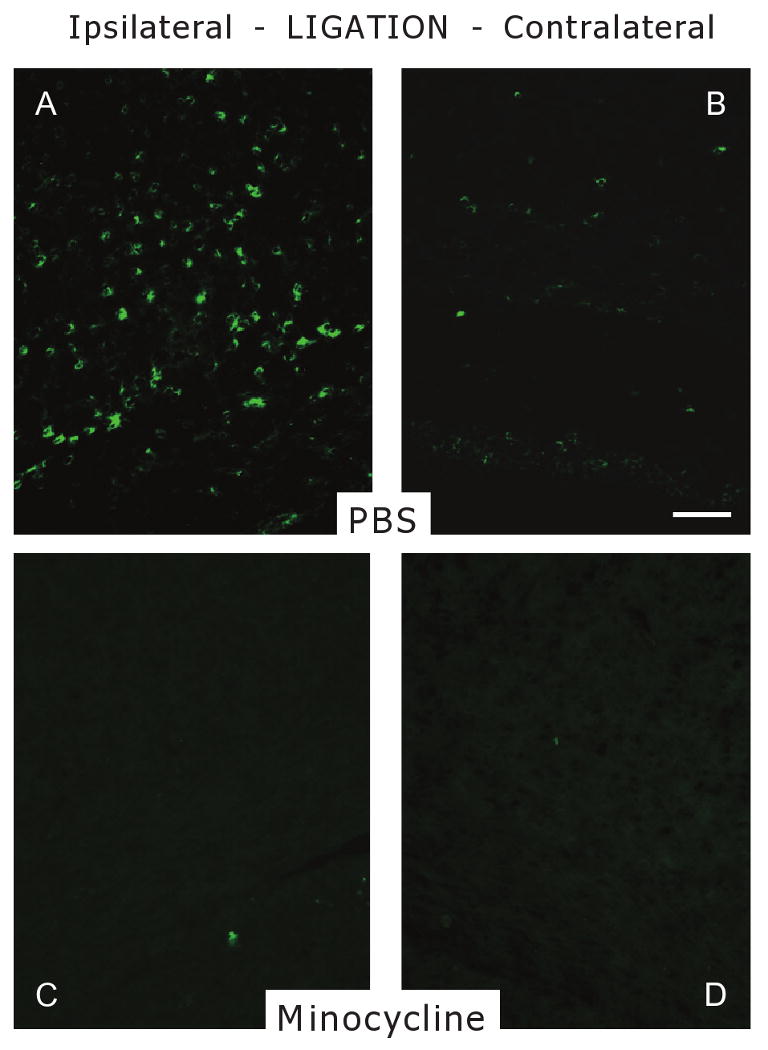
Effect of minocycline on the number of CD-74 (MHC II)-positive cells in the pericallosal white matter (WM) in P6 rats following UCL/hypoxia at 96 h. CD-74 staining shows decreased number of CD-74+ cells in minocycline-treated rats in comparison with vehicle-treated controls ipsilateral to UCL. (A) WM of vehicle-treated animal ipsilateral to UCL. (B) WM of vehicle-treated animal contralateral to UCL. (C) WM of minocycline-treated animal ipsilateral to UCL. (D) WM of minocycline-treated animal contralateral to UCL. Scale bar, 100 μm. MBP, myelin basic protein; P6, post-natal day 6; PBS, phosphate buffered saline; UCL, unilateral carotid artery ligation.
Discussion
The present study demonstrates the temporal relationship between HI-induced pre-OL death and microglial activity in WM in an immature rat model of PVL. Most importantly, we provide evidence for a protective effect of treatment with the microglial inactivator minocycline when administered following the hypoxic/ischaemic insult.
Hypoxic/ischaemic pre-OL injury in developing white matter
We have previously shown that WM is selectively vulnerable to HI in the first post-natal week [4,8], while similar insults in the second post-natal week preferentially cause neocortical strokes and seizures [36,37]. Pre-OLs are selectively vulnerable to injury and HI results in subsequent hypomyelination [4], similar to that observed in human WM in PVL cases [38]. We have demonstrated that one factor responsible for pre-OL injury in the immature rat is their relative overexpression of GluRs [9,10]. Subsequent studies have demonstrated enhanced AMPA receptor expression on pre-OLs in premature human brain, relative to younger and older ages [10]. Ca++-permeable AMPA receptors are activated in pre-OLs and subsequently trigger excitotoxic pre-OL death [5,7,8]. Indeed, treatment with the AMPA receptor antagonists following hypoxia and carotid ligation in P6/7 pups results in significant protection against ipsilateral MBP loss [4,8]. Given the known interaction between inflammation and excitotoxicity [22,30], we examined: (i) whether there were developmental differences in resident microglia in developing WM; and (ii) whether inflammatory mechanisms of pre-OL cell injury were operating in addition to those known to be excitotoxic.
Developmental regulation of microglial infiltration
Because the first post-natal week exhibits the greatest susceptibility of WM to hypoxic/ischaemic injury, we have evaluated whether the microglial population in the WM was developmentally regulated. An increase in microglial cell density occurs normally in the cerebral WM of the human fetus during the peak window of vulnerability for PVL [14,15]. Similarly, studies have shown that there is a developmental gradient of microglial migration into WM and cortex in rodents [17,19]. Here we show that the density of CD-68-positive microglia is significantly increased in WM at baseline during the age window that we have previously shown to exhibit regional WM susceptibility to hypoxic/ischaemic injury. This suggests, as has been reported in the developing human brain, that amoeboid migrating microglia appear to share antigenic characteristics with activated microglia in injured tissue. The mechanistic significance of this antigenicity is unknown. Interestingly, an alternate marker that has been previously used to quantify activated microglia, CD-74 (MHC II), did not show such increases in baseline expression during development. Similarly, Nikodemova et al. also reported that microglia do not express MHC II under resting conditions in primary microglial cultures [35]. Nevertheless, the increase inbasal levels of microglia that we observed during early post-natal development may enhance the susceptibility of developing WM to inflammatory-mediated injury. The functional significance of this transient overpopulation of microglia on WM development is not completely understood. However, microglia are known to mediate neuronal and glial differentiation [39,40], which are actively occurring in the first post-natal week.
Temporal profile of microglial density in white matter following hypoxic/ischaemic injury
We demonstrate that microglia show a robust and transient response to WM injury in the immature rat. Microglial density increases rapidly within 24 h and continues to rise until 72 h, and then gradually declines. As CD-68 is expressed by a mixed population of mononuclear phagocytes (including monocytes and macrophages as well as microglia), it is possible that both microglial proliferation and infiltration of CD-68-positive cells may have contributed to this increase. CD-74 staining showed similar patterns, suggesting that at least a sub-population exhibited activation of the immune response. Given the profile of cell death in WM following HI, it is likely that microglial responses could contribute to the pre-OL death occurring over the first several days post injury. We thus used these cell death profile data to specifically determine the optimal timing for minocycline treatment, which was administered in our model immediately after HI, and was continued every 12 h. A post-treatment paradigm has significant clinical utility as many premature infants are identified at hour to days following hypoxic/ischaemic cerebral injury.
Protective efficacy of minocycline post treatment
In this study, post-insult administration of minocycline, without pre-treatment, not only attenuated WM injury, but also significantly reduced the microglial number following HI in P6 rats. These observations are consistent with a potential role for microglial-mediated cell injury during hours to days following the hypoxic/ischaemic insult [20]. Minocycline-induced suppression of microglial responses may be a primary mechanism for the protection observed. Importantly, minocycline treatment was found to reduce numbers of CD-68-positive microglia (approaching the numbers of amoeboid microglia in the contralateral hemisphere) in the pericallosal WM ipsilateral to the ligation. Again, CD-74 staining was also reduced, confirming that minocycline affected the immune response in at least a sub-population of these cells, as has been previously reported in other models of cerebral inflammation [35]. Minocycline did not alter the contralateral counts, which did not differ from normal age-matched controls, suggesting that resident microglia were not significantly altered. Hence, minocycline appears to selectively affect the sub-population of microglia that responds to the hypoxic/ischaemic insult, which may be a factor improving the safety profile of this treatment paradigm.
A clinically relevant aspect of the present study is the apparent efficacy of post-treatment alone, in the absence of pre-treatment. This protective efficacy isconsistent with that of Cai et al. [20], in which that a pre- and post-injury minocyline treatment protected from hypomyelination in a bilateral carotid artery ligation model. Other studies in other models with pre- and/or post-treatment combination have yielded conflicting results. First, it appears that the protective effect may be species-specific, as minocycline treatment in mice actually produced toxicity rather than protection [41]. The timing of the injury may also play a role in the efficacy of minocycline here. In contrast to models in utero [42], the timing of injury here (P6) is coincident with the developmental peak of microglial infiltration in WM in the Long-Evans rat strain. Notably, human data show that microglia are at their highest density in human WM in the premature period of greatest susceptibility to PVL [14]. Other models in which minocycline has yet to be tested include those resulting in primarily grey matter injury in the immature rat [43,44], whereas the present model produces predominantly WM injury with little to no grey matter lesions.
Microglia/macrophage release of NO is thought to mediate neurotoxicity in several neurodegenerative diseases, and there is good evidence that minocycline may be neuroprotective through regulation of microglial NO production [20,22,26]. Minocycline pre-treatment alone or combination pre- and post-treatment reduces activation of inducible nitric oxide synthase in microglia, as well as NO and peroxynitrite production in hypoxic/ischaemic tissue [22,31,38]. Other reports reveal that minocycline can directly inhibit cytochrome C release from mitochondria during oxidative stress [28,45,46], suggesting that minocycline may have an antioxidant effect on multiple cell types within WM. Minocycline is reported to inhibit caspases following hypoxic/ischaemic injury, to delay apoptosis [47], reduce glutamate excitotoxicity and increase cell survival directly [48] as well as inhibit proteases, offering short-term cytoprotection following acute injury in ischaemic stroke [49]. Another potential action of minocycline specific to WM involves its ability to inhibit matrix metalloproteinase-2 (MMP-2), which is induced by HI [18,50] and subsequently degrades MBP in OLs [51,52]. MMPs are a family of proteins which play a role in injury-induced breakdown of the blood brain barrier [53].
In summary, minocycline appears to have protective efficacy against WM injury in a rodent model of PVL when administered solely following the insult. The density of microglia is higher in the injured developing WM in rodents and in humans, and hence this cell type may represent an age-specific target for therapy. Furthermore, we show efficacy of a post-treatment paradigm based on our observations of the time course of increased microglial density in WM following HI. Post-treatment efficacy has important clinical potential, as brain injury in premature infants is often detected hours to days after the insult. Minocycline protection against MBP loss in WM was concomitant with a decrease in the density of CD-68-positive cells compared with vehicle, suggesting that minocycline-mediated reduction in microglial cell numbers is a likely mechanism for protection. However, minocycline may also be acting directly on pre-OLs via interruption of oxidative stress pathways, blood brain barrier breakdown and MBP degradation. Given the likely synergistic actions of inflammation and excitotoxicity in PVL, future studies could investigate synergistic or additive efficacy of combination therapy of GluR antagonists and minocycline.
Acknowledgments
This study was supported by grants from the NIH (NS31718, NS38475, HD18655) and the United Cerebral Palsy Foundation. ML was recipient of NIH T32 Grant (NS007473).
References
- 1.Volpe JJ. Neurology of the Newborn. Philadelphia: Saunders; 2001. [Google Scholar]
- 2.Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–50. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 3.Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–53. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–41. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg PA, Dai W, Gan XD, Ali S, Fu J, Back SA, Sanches RM, Segal MM, Follett PL, Jensen FE, Volpe JJ. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res. 2003;71:237–45. doi: 10.1002/jnr.10472. [DOI] [PubMed] [Google Scholar]
- 6.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–12. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci USA. 2003;100:6801–6. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen F. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–20. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischaemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol. 2006;497:42–60. doi: 10.1002/cne.20972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischaemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–19. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda T, Mishima K, Aoo N, Egashira N, Iwasaki K, Fujiwara M, Ikenoue T. Combination treatment of neonatal rats with hypoxia-ischaemia and endotoxin induces long-lasting memory and learning impairment that is associated with extended cerebral damage. Am J Obstet Gynecol. 2004;191:2132–41. doi: 10.1016/j.ajog.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 13.Larouche A, Roy M, Kadhim H, Tsanaclis AM, Fortin D, Sebire G. Neuronal injuries induced by perinatal hypoxic-ischaemic insults are potentiated by prenatal exposure to lipopolysaccharide: animal model for perinatally acquired encephalopathy. Dev Neurosci. 2005;27:134–42. doi: 10.1159/000085985. [DOI] [PubMed] [Google Scholar]
- 14.Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, Kinney HC. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- 15.Monier A, Evrard P, Gressens P, Verney C. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J Comp Neurol. 2006;499:565–82. doi: 10.1002/cne.21123. [DOI] [PubMed] [Google Scholar]
- 16.Earle KL, Mitrofanis J. Identification of transient microglial cell colonies in the forebrain white matter of developing rats. J Comp Neurol. 1997;387:371–84. doi: 10.1002/(sici)1096-9861(19971027)387:3<371::aid-cne4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 17.Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: a review. Microsc Res Tech. 1999;45:359–82. doi: 10.1002/(SICI)1097-0029(19990615)45:6<359::AID-JEMT4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 18.Cho KO, La HO, Cho YJ, Sung KW, Kim SY. Minocycline attenuates white matter damage in a rat model of chronic cerebral hypoperfusion. J Neurosci Res. 2006;83:285–91. doi: 10.1002/jnr.20727. [DOI] [PubMed] [Google Scholar]
- 19.Rezaie P, Dean A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology. 2002;22:106–32. doi: 10.1046/j.1440-1789.2002.00438.x. [DOI] [PubMed] [Google Scholar]
- 20.Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischaemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–35. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 21.Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H. Chemokine and inflammatory cell response to hypoxia-ischaemia in immature rats. Pediatr Res. 1999;45:500–9. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- 22.Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121:619–28. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- 23.Cai Z, Pang Y, Lin S, Rhodes PG. Differential roles of tumor necrosis factor-alpha and interleukin-1 beta in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res. 2003;975:37–47. doi: 10.1016/s0006-8993(03)02545-9. [DOI] [PubMed] [Google Scholar]
- 24.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–34. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Liu L, Barger SW, Mrak RE, Griffin WS. Vitamin E suppression of microglial activation is neuroprotective. J Neurosci Res. 2001;66:163–70. doi: 10.1002/jnr.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai AY, Todd KG. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia. 2006;53:809–16. doi: 10.1002/glia.20335. [DOI] [PubMed] [Google Scholar]
- 27.Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 2004;27:293–8. doi: 10.1097/01.wnf.0000150867.98887.3e. [DOI] [PubMed] [Google Scholar]
- 28.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Fer-rante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–8. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 29.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6–tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, Gressens P. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. 2001;11:56–71. doi: 10.1111/j.1750-3639.2001.tb00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol. 2000;47:54–63. [PubMed] [Google Scholar]
- 32.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH. A new method to detect apoptosis in paraffin section: in situ end-labeling of fragmented DNA. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- 33.Bauer J, Sminia T, Wouterlood FG, Dijkstra CD. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res. 1994;38:365–75. doi: 10.1002/jnr.490380402. [DOI] [PubMed] [Google Scholar]
- 34.McKay SM, Brooks DJ, Hu P, McLachlan EM. Distinct types of microglial activation in white and grey matter of rat lumbosacral cord after mid-thoracic spinal transection. J Neuropathol Exp Neurol. 2007;66:698–710. doi: 10.1097/nen.0b013e3181256b32. [DOI] [PubMed] [Google Scholar]
- 35.Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC) alpha/betaII. J Biol Chem. 2007;282:15208–16. doi: 10.1074/jbc.M611907200. [DOI] [PubMed] [Google Scholar]
- 36.Jensen FE. Developmental factors regulating susceptibility to perinatal brain injury and seizures. Curr Opin Pediatr. 2006;18:628–33. doi: 10.1097/MOP.0b013e328010c536. [DOI] [PubMed] [Google Scholar]
- 37.Jensen FE. Role of glutamate receptors in periventricular leukomalacia. J Child Neurol. 2005;20:950–9. doi: 10.1177/08830738050200120401. [DOI] [PubMed] [Google Scholar]
- 38.Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–33. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang SC, Goetz BD, Duncan ID. Suppression of activated microglia promotes survival and function of transplanted oligodendroglial progenitors. Glia. 2003;41:191–8. doi: 10.1002/glia.10172. [DOI] [PubMed] [Google Scholar]
- 40.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–7. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 41.Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens hypoxic-ischaemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189:58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Baud O, Daire JL, Dalmaz Y, Fontaine RH, Krueger RC, Sebag G, Evrard P, Gressens P, Verney C. Gestational hypoxia induces white matter damage in neonatal rats: a new model of periventricular leukomalacia. Brain Pathol. 2004;14:1–10. doi: 10.1111/j.1750-3639.2004.tb00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischaemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev. 2002;8:30–8. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 44.Roberson R, Woodard JE, Toso L, Abebe D, Poggi SH, Spong CY. Postnatal inflammatory rat model for cerebral palsy: too different from humans. Am J Obstet Gynecol. 2006;195:1038–44. doi: 10.1016/j.ajog.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 45.Ferriero DM. Oxidant mechanisms in neonatal hypoxia-ischaemia. Dev Neurosci. 2001;23:198–202. doi: 10.1159/000046143. [DOI] [PubMed] [Google Scholar]
- 46.Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischaemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun. 2003;304:551–9. doi: 10.1016/s0006-291x(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 47.Levkovitch-Verbin H, Kalev-Landoy M, Habot-Wilner Z, Melamed S. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol. 2006;124:520–6. doi: 10.1001/archopht.124.4.520. [DOI] [PubMed] [Google Scholar]
- 48.Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, Neusch C, Bahr M, Diem R. Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis. 2007;25:514–25. doi: 10.1016/j.nbd.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 49.Elewa HF, Hilali H, Hess DC, Machado LS, Fagan SC. Minocycline for short-term neuroprotection. Pharmacotherapy. 2006;26:515–21. doi: 10.1592/phco.26.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ihara M, Tomimoto H, Kinoshita M, Oh J, Noda M, Wakita H, Akiguchi I, Shibasaki H. Chronic cerebral hypoperfusion induces MMP-2 but not MMP-9 expression in the microglia and vascular endothelium of white matter. J Cereb Blood Flow Metab. 2001;21:828–34. doi: 10.1097/00004647-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 51.Chandler S, Miller KM, Clements JM, Lury J, Corkill D, Anthony DC, Adams SE, Gearing AJ. Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: an overview. J Neuroimmunol. 1997;72:155–61. doi: 10.1016/s0165-5728(96)00179-8. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg GA, Sullivan N, Esiri MM. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–8. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- 53.Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. A new approach for the investigation of reperfusion-related brain injury. Biochem Soc Trans. 2006;34:1366–9. doi: 10.1042/BST0341366. [DOI] [PubMed] [Google Scholar]