

Abstract
The serine/threonine protein kinase Akt is a major signal transducer of the phosphoinositide 3-kinase (PI 3-K) pathway in all cells and tissues and plays a pivotal role in the maintenance of cellular processes including cell growth, proliferation, survival and metabolism. The frequent aberrant activation of the PI 3-K/Akt pathway in human cancer has made it an attractive therapeutic target. Numerous studies have provided a comprehensive understanding of the specific functions of Akt signaling in cancer cells as well as the surrounding tumor microenvironment and this has informed and enabled the development of therapeutic drugs to target both PI 3-K and Akt. However, recent studies have provided evidence for distinct functions of the three mammalian Akt isoforms, particularly with respect to the regulation of cell motility and metastasis of breast cancer. Here we discus the mechanisms by which Akt signaling contributes to invasive migration and tumor metastasis, and highlight recent advances in our understanding of the contribution of the Akt pathway in the tumor-associated stroma.
Keywords: Akt, PI 3-K, breast cancer, cell motility, metastasis, signal transduction
1. Introduction
Since the discovery of Akt/PKB (protein kinase B) as the human homolog of the viral oncogene v-Akt [1, 2] and the seminal finding that Akt is a major effector of PI 3-K (phosphoinositide 3-kinase) signaling [3], the PI 3-K/Akt pathway has emerged as a key regulator of numerous cellular phenotypes associated with cancer including cell survival, proliferation, growth, metabolism, angiogenesis and malignant transformation. Indeed, recent studies have revealed that the PI 3-K/Akt signaling cascade harbors some of the most prevalent genetic lesions in cells and tissues derived from virtually all human solid tumors [4, 5]. In this context, increased Akt protein expression and activity have been detected in aggressive human gastric cancers as well as breast, prostate, ovarian and brain tumors [2, 6]. The detection of activated Akt in breast cancer patients has also been associated with poor prognosis with a higher probability of relapse accompanied by distant metastases [7]. Recent human cancer genome sequencing studies have identified a number of somatic mutations in Akt isoforms. One such mutation has received particular attention because it was identified in human breast, colorectal and ovarian cancers. The Glu17Lys somatic mutation comprises a gain-of-function genetic lesion in Akt1 which renders the kinase constitutively active by localizing it to the plasma membrane [8]. Similar mutations in Akt2 or Akt3 have not yet been reported. Over the past two decades, a multitude of studies have revealed the unequivocal importance of signaling through the PI 3-K and Akt pathway leading to tumorigenesis. As but one example, Akt was shown to promote tumorigenesis and drug resistance via the translational regulators mammalian target of rapamycin (mTOR) and eukaryotic initiation factor 4E (eIF4E) in a murine lymphoma model [9]. Consistent with the notion that signaling through Akt promotes cell growth, expression of constitutively active Akt1 alleles transforms fibroblast cells in vitro [6]. Although most studies have demonstrated the importance of Akt in modulating epithelial cell phenotypes leading to dysplasia, it is increasingly recognized that in addition to the genetic background of carcinoma cells, the surrounding environment plays an equally important role. Mesenchymal cells such as fibroblasts, and also endothelial cells and immune cells comprise the stromal component of human solid tumors and regulate the growth, progression and metastasis of carcinoma, ultimately determining clinical outcome [10]. Because Akt isoforms are ubiquitously expressed in all cells and tissues, including all the cellular compartments of the stroma, investigation into the role by which Akt isoforms control phenotypes in cancer cells as well as the associated stroma has provided insights into the pathophysiological mechanisms of tumor development and metastasis. Several recent reviews have focused on the mechanisms by which PI 3-K and Akt control tumorigenesis and cell growth (for example refs. [5, 11],). Here, we focus on the role of Akt signaling in tumor invasive migration and metastatic dissemination, highlighting the differential effects of Akt isoforms on breast cancer cell motility. Recent advances in our understanding of the importance of Akt signaling in the tumor-associated stroma will also be discussed.
2. PI 3-K Signaling to Akt
In mammals, three distinct genes encode for Akt1 (PKBα), Akt2 (PKBβ) and Akt3 (PKBγ) [12]. Whereas Akt1 and Akt2 are ubiquitously expressed, Akt3 displays a more restricted tissue distribution and is found abundantly in neuronal tissues. All three Akt isoforms are activated by similar mechanisms in PI 3-K signaling [13, 14]. Upon stimulation with growth factors such as insulin-like growth factor-1 (IGF-1), PI 3-K synthesizes the second messenger PtdIns-3,4,5-P3 from the precursor PtdIns-4,5-P2. PtdIns-3,4,5-P3 binds to the Pleckstrin Homology (PH) domain of Akt effectively recruiting it to the plasma membrane. Full activation is achieved by the phosphorylation of two critical and highly conserved residues in the catalytic domain. Thr308 in the activation loop is phosphorylated by PDK-1 (phosphoinositide-dependent kinase-1), which is also recruited to membranes through PH domain binding to PtdIns-3,4,5-P3 [15]. The very carboxyl-terminal residue Ser473 is phosphorylated primarily by the mTORC2 (mammalian target of rapamycin complex 2) complex [16], although other mechanisms of Ser473 phosphorylation have been proposed, including phosphorylation by DNA-PK (DNA-dependent protein kinase), PDK-1, ILK (integrin-linked kinase) and Akt autophosphorylation [17]. Regardless of the mechanism, once phosphorylated at these two key residues, Akt then loses the PtdIns-3,4,5-P3 binding requirement and translocates to distinct subcellular compartments, including the nucleus, mitochondria and other organelles. Akt then transduces the signal by phosphorylating numerous substrate proteins, the first discovered being Glycogen Synthase Kinase 3β (GSK3β) [18]. Other substrates include BAD, MDM2 and Forkhead transcription factors (FOXO), many of which are implicated in cancer-associated phenotypes [19]. To date, over 100 proteins have been identified as Akt substrates (see review by Manning and Cantley, [20]). Importantly, genetic lesions in this pathway such as highly prevalent oncogenic mutations in the PI 3-K catalytic subunit PIK3CA as well as deletions or mutations in the PtdIns-3,4,5-P3 phosphatase PTEN result in hyperactivation of Akt signaling, phosphorylation of substrates and in turn the induction of cellular transformation leading to tumorigenesis.
Despite numerous studies which have provided unequivocal evidence of the importance of Akt signaling in cancer and other pathologies, what has remained more elusive is the identification of isoform-specific functions for the three mammalian Akt isoforms. Indeed, up until recently it was assumed that Akt isoforms might act redundantly to one another in signal relay mechanisms. Evaluation of specific functions of Akt proteins has been hampered by the lack of isoform-specific chemical inhibitors and other tools, and up until recently studies generally relied on the use of overexpression of individual isoforms which likely result in mis-localization of the exogenous kinases and thus do not accurately reflect the physiological functions of Akt. The first glimpse into isoform-specific functions of Akt1, Ak2 and Akt3 was provided by genetic deletion studies, revealing that despite their high sequence similarity, individual Akt isoforms exert non-redundant functions. Whereas Akt1 null mice have growth retardation [21, 22], Akt2 null mice develop insulin-resistant diabetes [23, 24]. In contrast, Akt3 null mice reveal reduced brain size [25, 26]. Thereafter, the development of approaches such as siRNA and more specific inhibitors allowed a more specific investigation into distinct roles of individual Akt isoforms. In this context, the realization that Akt1–3 have non-overlapping phenotypes in signal relay was first provided by studies into the mechanisms by which this pathway controls cell migration and invasion.
3. Regulation of Cancer Cell Invasive Migration by Akt
Up until recently, the paradigm stated that all three Akt isoforms enhance tumor survival and proliferation, making it an attractive target for the development and use of targeted cancer therapy [19]. The basic premise has been that genetic lesions in PI 3-K or PTEN, or amplifications of Akt isoforms, promote tumorigenesis in carcinoma due to enhanced proliferation and survival. Thus, targeted therapy using PI 3-K/Akt/mTOR inhibitors to attenuate these phenotypes would be predicted to reverse enhanced proliferation and survival, and only in the tumor cells but not in normal tissues which do not harbor hyperactive Akt. However, recent studies have demonstrated that depending on the cell and tissue type, distinct Akt isoforms function in an opposing manner in the modulation of an equally important cancer cell phenotype, invasive migration and metastatic dissemination.
3.1 Akt as an enhancer of cell motility in distinct cell types
Early studies had pointed to an important role for Akt in the regulation of multiple processes that control invasive migration, including actin organization, cell-to-cell adhesion, cell motility and extracellular matrix degradation. For example, Akt1 has been shown to enhance fibroblast motility by phosphorylating Girdin, an actin-binding protein that promotes stress fiber formation and lamellapodia [27]. In addition, Akt1 signaling enhances matrix metalloproteinase-2 (MMP2) activity in mouse mammary epithelial cells, thereby augmenting invasion [28]. Similarly, Akt1 promotes cell motility and MMP9 production via NF-κB (nuclear factor-κB) in fibrosarcoma cells [29]. Expression of a constitutively active Akt1 allele in squamous carcinoma cells induces epithelial to mesenchymal transition (EMT), a process that involves reduced intercellular adhesion and increased motility [30]. Furthermore, both Akt1 and Akt2 have been shown to promote invasion of human pancreatic cancer cells by up-regulating IGF-I receptor (IGF-IR) expression [31]. Thus, in a range of cell types, Akt signaling is generally associated with enhanced cell motility and invasion, although these data were collected primarily using over-expression of artificially activated Akt alleles.
3.2 Inhibition of breast cancer cell invasive migration by Akt1
In contrast to the above studies, several recent studies have demonstrated a somewhat surprising anti-migratory role for Akt1 in human epithelial breast cancer cell lines. Three independent studies originally revealed that overexpression of Akt1 inhibits invasive migration of various breast cancer cell lines, whereas silencing of Akt1 using siRNA enhances migration [14, 32, 33]. In our own studies we demonstrated that the inhibitory effect of Akt1 on invasive migration is mediated through the proteasomal degradation of the transcription factor NFAT (Nuclear Factor of Activated T cells) [32]. NFAT transcriptional activity is required for breast cancer cell migration [34], and a few motility genes have been identified as NFAT target genes. One of these is autotoxin, an autocrine motility factor that is upregulated in several carcinomas [35]. A separate study also demonstrated that cyclooxygenase-2 (COX-2), the enzyme that mediates the formation of prostanoids and is a target of several non-steroidal anti-inflammatory drugs, is also induced by NFAT and plays an important role in breast cancer cell invasion [36] (Fig. 1).
Fig. 1.
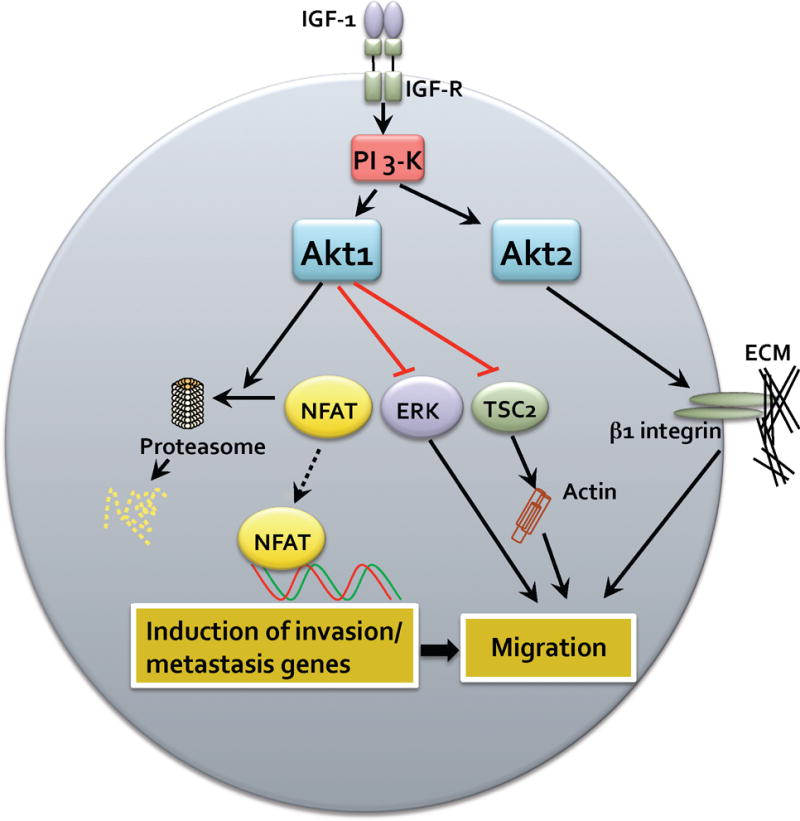
Mechanisms of carcinoma cell migration mediated by Akt. Breat carcinoma cells respond to IGF-1 leading to activation of PI 3-K and in turn phosphorylation and activation of Akt1 and Akt2. Akt1 can function to suppress carcinoma cell migration by various redundant mechanisms, inducing degradation of NFAT and attenuation of pro-invasion and metastasis genes. Akt1 can also suppress ERK and TSC2 activity leading to suppression of migration. In contrast, Akt2 functions to enhance carcinoma cell migration, in part by up-regulating β1 integrins and other yet to be determined mechanisms.
Studies by the Brugge laboratory demonstrated that Akt1 inhibits the motility of MCF10A cells by attenuating extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) activity [14] (Fig. 1). They also investigated the effects of Akt1 and ERK on EMT and interestingly found that EMT induced in Akt1 downregulated cells is not rescued by inhibition of ERK, suggesting that an alternative downstream target or pathway is responsible for the suppressive effects of Akt1 on EMT. A separate mechanism by which Akt1 might inhibit migration is the regulation of tumor suppressor Tuberous Sclerosis Complex 2 (TSC2). The Bissell laboratory showed that overexpression of a constitutively active Akt1 allele inhibits Rho-GTPase activity and breast cancer cell invasion with a concomitant phosphorylation and degradation of TSC2 [33] (Fig. 1). Thus, there are at least three signaling mechanisms which contribute to the inhibitory effects of Akt1 on breast cancer cell invasive migration. It remains to be determined whether these pathways act independently or synergistically to induce a transcriptional program that dampens breast cancer cell motility. Although the above studies were all performed using in vitro assays, more recent studies have begun to address the role of Akt isoforms in modulating invasion and metastasis in animal models of breast cancer progression.
3.3 Lessons from in vivo models
Several mouse models have been proven to be instrumental in exploring pathways involved in breast tumorigenesis and progression. In the polyomavirus middle T antigen (PyV mT) model, transgenic mice with mammary epithelium-specific expression of wild-type PyV mT rapidly develop highly metastatic mammary tumors by activating PI 3-K, Src family kinases and Shc pathways [37–39]. Mice expressing a mutant PyV mT uncoupled from PI 3-K develop mammary hyperplasias that are highly apoptotic [40]. In order to examine the role of Akt in breast tumorigenesis, Muller and colleagues crossed the mutant PyV mT strain with mice expressing a constitutively active Akt1 transgene [41]. Overexpression of activated Akt1 accelerates mammary tumorigenesis by providing an anti-apoptotic signal. Interestingly, it did not rescue the highly metastatic phenotype exhibited by wild-type PyV mT. In subsequent studies, the same group showed that in the ErbB2 receptor tyrosine kinase-mediated tumorigenesis model, even though activated Akt1 shortens the latency of multifocal mammary tumor development, it actually suppresses tumor invasion into the surrounding tissues [42]. These results provided the first hard evidence that Akt1 might function in an anti-invasive migration and metastatic role in vivo.
In contrast to the overexpression studies, experiments using Akt1-deficient mice have not yielded wholly consistent results. In one study, the invasive and metastatic potential of ErbB2-driven mammary tumors was shown to be higher in Akt1 knockout mice [43]. Yet in another study, Akt1 deficiency was found to reduce lung metastases, suggestive of a pro-invasive role for Akt1 [44]. In the same study it was further shown that Akt1 enhances migration of mammary epithelial cells in a paracrine fashion by stimulating expression and secretion of chemokines CXCL16 and MIP1γ. One possible explanation for these differential outcomes may lie in the different genetic backgrounds of the mice and tissue environments utilized. It should also be noted that in the above studies, Akt1 was ablated in all tissues and cellular compartments. Given the importance of PI 3-K and Akt signaling in the tumor stroma which contributes to progression (as discussed below), this may account for the differences observed. Thus one prediction is that more advanced mouse model studies using mammary-specific and conditional ablation of Akt isoform(s) will provide a more accurate account of exactly how Akt isoforms modulate breast cancer induction and progression to metastasis. Furthermore, transgenic mice harboring kinase-inactive ‘knock-in’ mutations should also be informative in dissociating kinase activity versus kinase-independent functions of a given Akt isoform.
3.4 Positive role for Akt2 in breast cancer cell invasion
In direct contrast to the findings with Akt1, Akt2 has been shown to enhance the migratory and invasive phenotypes of breast carcinoma cells. Akt2, but not Akt1 or Akt3, has been shown to up-regulate β1 integrins and promote adhesion and invasion of breast cancer cells in vitro and also metastasis in vivo [45] (Fig. 1). In addition, the Brugge group showed that unlike Akt1, Akt2 promotes MCF10A cell migration in vitro and in 3D cultures [14]. In a separate study, upregulation of Akt2 by the basic helix-loop-helix transcription factor Twist was shown to promote breast cancer cell migration and invasion [46]. Thus, both in vitro and in vivo Akt1 and Akt2 clearly function in an opposing manner in the regulation of invasive migration leading to metastatic dissemination, although this does appear to depend on the assay used and the genetic background. What is not clear at this point, however, is the nature of the mechanism(s) that are responsible for this distinction. For example, although some Akt1 and Ak2 isoform-specific substrates have been identified such as the cell cycle regulator p21 CIP1 [47], none so far have provided a mechanism which accounts for isoform specificity in the context of invasive migration. Given that several Akt inhibitors are currently in clinical trials for targeted cancer therapy (e.g., triciribine and perifosine [48]) it will be important to determine the efficacy and specificity of such inhibitors in breast cancer progression, including metastasis. Akt1 and Akt2 chemical inhibitors with good selectivity have been developed, though these have not proven to be successful in clinical trials due to toxicity [49]. Nonetheless, these Akt inhibitors coupled with phospho-proteomic shRNA screens should prove to be invaluable for the identification of specific Akt substrates in cancer cells.
3.5 Opposing functions of Akt in fibroblast cell motility
Opposing results for Akt isoform-mediated fibroblast cell migration have also been reported. shRNA silencing studies performed in fibroblasts have shown that, in contrast to what has been found in breast cancer cells, Akt1 enhances whereas Akt2 retards cell migration in vitro [50]. In fibroblasts, the Rac/PAK pathway was shown to be one mechanism by which Akt1 and Akt2 mediates these opposing phenotypes. By swapping functional domains of Akt1 and 2, the linker region between the PH and catalytic kinase domains was demonstrated to be critical for the differential migratory phenotypes of Akt1 and Akt2 in fibroblasts. Although the specific mechanism by which the linker region determines specificity has not been identified, one possibility is that Akt isoforms bind to distinct scaffolding proteins in the linker region, thus facilitating access to different substrates. Similar studies with domain swapping have not been performed in epithelial cells, though it is very likely that the linker region is also involved. Thus, presently there is a considerable paucity of knowledge concerning the precise mechanism(s) which account for cell-type dependency of Akt1 and Akt2 on invasive migration. It is likely that by interacting with different scaffolding proteins, adaptors and lipids, distinct pools of Akt isoforms reside in different subcellular locations. Although compartmentalization of Akt1–3 has not been extensively characterized, experiments with breast cancer cells plated on collagen IV have shown that whereas Akt1 is predominantly localized in the nucleus, Akt2 is excluded from the nucleus and found primarily at the basal end of the cell proximal to ECM [45]. It is therefore very likely that differences in the subcellular distribution of Akt isoforms restrict substrate accessibility thereby conferring functional selectivity in various cell types.
4. Akt and regulation of the tumor-associated stroma
The tumor-associated stroma contains a variety of mesenchymal cells including fibroblasts, endothelial cells and immune cells. Signals from stroma can induce tumor formation, stimulate carcinoma cell growth as well as facilitate their dissemination. Although Akt1 acts as a negative regulator in breast cancer cell migration, recent studies have shown that it has positive effects on various components of the tumor microenvironment.
4.1 Akt signaling during tumor angiogenesis
Vascular endothelial growth factor (VEGF), is a crucial growth and permeability factor for tumor angiogenesis and is frequently up-regulated in human solid tumors [10, 51, 52]. Akt has been shown to play a key role in multiple processes of VEGF-mediated angiogenesis, both in physiological and pathological contexts. In endothelial cells, Akt phosphorylates the substrate protein girdin thereby promoting VEGF-dependent cell migration, which is essential for sprouting, formation and branching of vessels during angiogenesis [53] (Fig. 2). In addition, VEGF enhances the survival of human endothelial cells via the Akt signaling pathway [54]. Akt activation in turn promotes VEGF expression in endothelial cells as well as tumor cells [55, 56] (Fig. 2), by a mechanism that depends at least in part on the transcription factors HIF-1 (hypoxia-inducible factor 1) and SP-1 (specificity protein-1) [56, 57]. The reciprocal interactions between Akt and VEGF provide a positive feedback loop facilitating the formation of the vasculature during tumor progression. Furthermore, overexpression of hyperactive Akt1 in endothelial cells leads to formation of enlarged and hyperpermeable vessels, thus recapitulating the abnormalities of tumor blood vessels [58]. Significantly, the pathological effects observed in the vasculature can be reversed by blocking the activity of either Akt or its downstream target mTOR [58]. These observations are in agreement with previous studies that showed that rapamycin inhibits angiogenesis and metastatic tumor growth [59]. Although the roles of individual Akt isoforms in angiogenesis have not explored, studies using Akt isoform-deficient mice have shown that Akt1, but not Akt2, is critical for VEGF-mediated angiogenesis [60]. The fact that Akt1 is the major isoform expressed in endothelial cells also supports the idea that this is the predominant isoform which mediates tumor angiogenesis [61]. Collectively, these studies provide concordant evidence for a positive regulatory role of Akt1 in angiogenesis. A recent study, however, demonstrated that in Akt1 null animals, blood vessels are immature and leaky [61]. The enhanced angiogenesis which results from ablation of Akt1 is accompanied with reduced levels of the vascular regulators thrombospondins 1 (TSP-1) and TSP-2. One possibility for the reported differences on the role of Akt1 in modulating tumor angiogenesis may reflect the distinct roles of Akt1 in different phases of angiogenesis. Clearly more studies are required to discern the precise role(s) of Akt isoforms prior, during and subsequent to the angiogenic switch. Moreover, whether mechanisms exist for the function of Akt in modulating vascular permeability, other than stimulation of VEGF expression, remains to be determined.
Fig. 2.
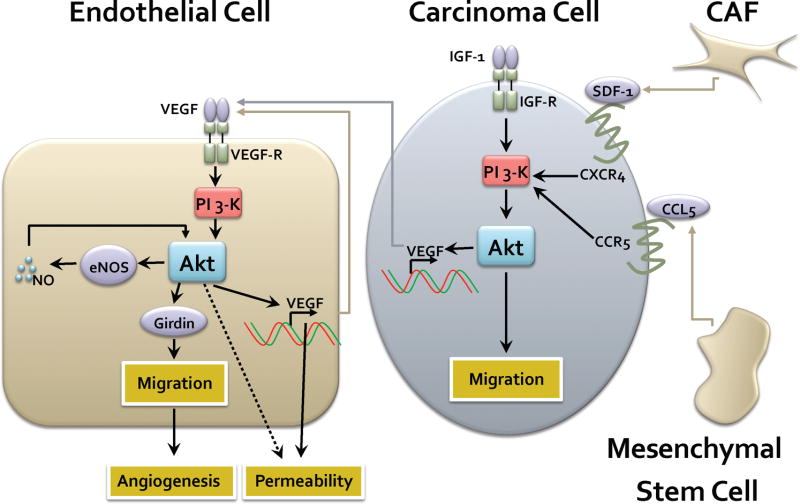
Paracrine and autocrine mechanisms of Akt-mediated tumor-stromal interactions. Carcinoma cells respond to a variety of ligands released by the tumor stroma, including SDF1 secreted by carcinoma-associated fibroblasts (CAF) and CCL5 secreted by mesenchymal stem cells. Acting through their cognate receptors this induces activation of the PI 3-K and Akt pathway leading to modulation of invasive migration. The Akt pathway also stimulates the transcriptional induction of VEGF which is then secreted and acts in a paracrine manner to stimulate its cognate receptor VEGF-R in endothelial cells. This in turn activates PI 3-K and Akt, with a variety of consequences. Akt phosphorylates the substrate girdin leading to enhanced endothelial cell migration. Akt can also further stimulate the transcriptional induction of VEGF leading to an autocrine loop which amplifies PI 3-K signaling. Akt can also directly stimulate eNOS (endothelial nitric oxidase synthase) to produce nitric oxide (NO), which can function in an amplification loop to further activate Akt. The net effect is enhancement of endothelial cell permeability, migration and angiogenesis.
Similar to VEGF, Akt also collaborates with nitric oxide (NO)-dependent functions during angiogenesis. Akt phosphorylates and activates the endothelial NO synthase (eNOS) [62, 63] (Fig. 2). Activated eNOS in turn produces NO which mediates endothelial cell migration and angiogenesis via the Akt pathway [64]. Although the mechanism by which NO activates Akt is not fully understood, there is evidence that the cGMP-dependent kinase (PKG) plays a role in the process. Taken together, Akt positively impacts multiple mechanisms to promote and fine-tune the angiogenic process, which has obvious important consequences for tumor progression.
4.2 Akt and chemokine signaling in the stroma
In addition to growth factors that activate the PI 3-K and Akt pathway, chemokines and their receptors also play critical roles in tumor progression and metastasis. For instance, CXC chemokine receptor 4 (CXCR4) is highly expressed in human breast cancer cells. DNA microarray studies by Massague and co-workers have shown that bone-colonizing breast cancer cells express significantly higher levels of CXCR4 compared to parental cells [65]. siRNA-mediated knock-down of CXCR4 inhibits migration of breast cancer cells in vitro [66]. Consistent with this, blocking the interaction between CXCR4 and its ligand, stromal cell-derived factor 1 (SDF-1), significantly reduces metastases to the lung and regional lymph nodes [67]. Carcinoma-associated fibroblasts (CAFs), a major component of breast cancer stroma, secrete SDF-1 which in turn promotes growth of breast cancer cells by direct paracrine stimulation of CXCR4, and also stimulates angiogenesis via the recruitment of endothelial progenitor cells [68]. Another means through which SDF-1 enhances angiogenesis is via the Akt signaling pathway. Akt is phosphorylated upon SDF-1 stimulation [69], and SDF-1-induced expression of VEGF is blocked by PI 3-K inhibitors (Fig. 2). The critical roles of SDF-1 signaling have indeed made its receptor CXCR4 a prognostic marker of breast cancer, and current efforts are aimed at developing CXCR4 antagonists for anti-tumor therapeutics [70].
Mounting evidence suggests that in addition to SDF-1, the chemokine CCL5 is another important chemokine in tumor metastasis. Increased expression of CCL5 in tumors is directly correlated with more advanced stages of breast carcinoma [71]. Conversely, CCL5 is minimally detected in breast epithelial cells from patients with benign breast disorders. CCL5 contributes to breast tumor progression by inducing migration of monocytes into the tumor site as well as promoting expression of MMP9 by tumor-infiltrating monocytes [72]. CCL5 also exerts paracrine effects on breast cancer cells, because it is secreted from mesenchymal stem cells within the stroma and thereby activates its cognate receptor CCR5 on breast cancer cells augmenting their metastatic potency [73] (Fig. 2). Furthermore PI 3-K/Akt signaling is directly involved in CCL5-mediated breast cancer cell motility, as revealed by inhibitor studies. Once again, whether specific Akt isoforms are responsible for mediating the effects of CCL5, as well as the underlying molecular mechanisms, is yet to be determined.
In light of the importance of stromal compartments in breast cancer induction and progression, we propose that the paracrine effects of Akt on the stromal cells are as important as the cell-autonomous migratory functions of Akt. Studies using xenograft models in mice have made a major contribution in our understanding of stromal factors in breast tumorigenesis. However, injection of human breast epithelial cells into the mammary fat pads of mice does not fully recapitulate colonization that is seen in human patients. To circumvent this issue, Weinberg and colleagues have developed a human-in-mouse model in which human breast stromal fibroblasts are implanted into mammary glands of nude mice [74, 75]. Both the stromal fibroblasts and the breast cancer cells can be genetically modified to express different factors prior to implantation. This reconstruction model will likely be very useful in studying the role of Akt in the crosstalk between breast epithelial cancer cells and the stroma in vivo. Another appeal of this model is that it can be combined with live imaging technologies to monitor these highly complex molecular events in real-time.
5. Perspectives
Collectively, recent studies on the role of the PI 3-K and Akt pathway in human cancer have reaffirmed the importance of this pathway in disease progression. Similarly, studies on the role of Akt in modulating invasive migration and metastasis have informed the field that Akt isoform signaling is not redundant. Instead, there are clear isoform-specific functions for Akt1, Akt2 and Akt3 at the level of these phenotypes, and we speculate other phenotypes such as proliferation and survival as well. Clearly, however, our understanding of Akt isoform-specific signaling mechanisms is far from complete. There is surprisingly little information on the direct substrates of individual Akt isoforms, as well as the molecular determinants of compartmentalization of Akt isoforms which may ultimately confer functional selectivity. In this regard, isoform-specific siRNA and the recent development of isoform-specific inhibitors will afford invaluable tools and help to elucidate the relative contributions of each Akt isoform signal to invasive migration, metastasis and progression of breast cancer. Equally important is whether this specificity is restricted to breast cancer, or whether it is evident in other aggressive human solid tumors where the PI 3-K and Akt pathway is hyperactive, such as glioblastoma, as well as prostate and ovarian carcinoma.
Other studies have also demonstrated the importance of Akt signaling in the development of the tumor-associated stroma. However, studies on the roles of individual Akt isoforms in this process are limited. Given the fact that both the formation and progression of breast cancer require a well-orchestrated tumor-associated stroma, investigation of the function of specific Akt isoforms in the tumor microenvironment is warranted. It is also predicted that more sophisticated models of tumor progression in mice will help to unravel some of the complexities and contradictions that have been reported thus far regarding the role of the Akt pathway in tumor progression and metastasis. In this context it is important to note that most studies to date have made use of hyperactive alleles of Akt such as a membrane targeted mutant, or whole animal knockout strategies such as homologous recombination in vivo or siRNA in vitro. However, none of these molecular events are seen in pathological settings of disease progression in humans. Recent discoveries in the field will enable the development of more physiologically-relevant models to accurately define the contribution of Akt signaling in carcinoma cells and the stroma. For example, the use of epithelial and stromal-specific transgenes in vivo with naturally occurring mutations in the pathway, such as oncogenic PI 3- K, loss or mutations in PTEN, and amplification or oncogenic mutations in Akt isoforms will more closely model the molecular events which occur in disease and should shed considerable light on these questions. By the same token, tissue specific (mammary epithelium, stromal compartments etc) ablation of Akt1–3 using shRNA in vivo should also afford new insight.
As the Akt pathway is such a crucial signaling node in cancer cell survival and proliferation, considerable efforts have been made to develop pharmacological inhibitors of Akt for therapeutic intervention. Information regarding cell-type dependent isoform specificity in tumor progression to metastasis will be of obvious importance for determining effective regiments. For example, an Akt1 inhibitor may be beneficial in fibrosarcomas to inhibit tumor growth and metastasis as well as stromal induction. This would also be predicted to have minimal side effects on glucose homeostasis, which at least in mice is regulated primarily by Akt2. However, Akt1 inhibition may not be recommended, and may even be contraindicated in breast cancer patients, as it may promote invasive migration to metastasis. The continuing efforts to dissect the molecular mechanisms of Akt isoform-specific signaling will likely provide fundamental new insights for designing more effective therapeutics for cancer treatment.
Acknowledgments
Work in the laboratory is supported by the National Institutes of Health and the Susan G. Komen Breast Cancer Foundation. We thank members of the Toker laboratory and Geoffrey Lau for very helpful discussions. We apologize for not being able to cite all articles relevant to this review due to space constraints.
Abbreviations
- DNA-PK
DNA-dependent protein kinase
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- FOXO
forkhead transcription factors
- IGF-1
insulin-like growth factor-1
- ILK
integrin-linked kinase
- MMP
matrix metalloproteinase
- mTOR
mammalian target of rapamycin
- NFAT
nuclear factor of activated T cells
- PDK-1
phosphoinositide-dependent kinase-1
- PH
pleckstrin homology
- PKB
Protein kinase B
- PI 3-K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensing homolog
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. Science. 1991;254(5029):274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 2.Staal SP. Proc Natl Acad Sci U S A. 1987;84(14):5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. Cell. 1995;81(5):727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 4.Altomare DA, Testa JR. Oncogene. 2005;24(50):7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 5.Engelman JA, Luo J, Cantley LC. Nat Rev Genet. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 6.Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. Am J Pathol. 2001;159(2):431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Tenorio G, Stal O. Br J Cancer. 2002;86(4):540–545. doi: 10.1038/sj.bjc.6600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 9.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Nature. 2004;428(6980):332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 10.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Angiogenesis. 2008;11(2):109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellacosa A, Kumar CC, Di CA, Testa JR. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 12.Toker A, Yoeli-Lerner M. Cancer Res. 2006;66(8):3963–3966. doi: 10.1158/0008-5472.CAN-06-0743. [DOI] [PubMed] [Google Scholar]
- 13.Woodgett JR. Curr Opin Cell Biol. 2005;17(2):150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. J Cell Biol. 2005;171(6):1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mora A, Komander D, van Aalten DM, Alessi DR. Semin Cell Dev Biol. 2004;15(2):161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 16.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 17.Fayard E, Tintignac LA, Baudry A, Hemmings BA. J Cell Sci. 2005;118(Pt 24):5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- 18.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 19.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Nat Rev Drug Discov. 2005;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 20.Manning BD, Cantley LC. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Genes Dev. 2001;15(17):2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. J Biol Chem. 2001;276(42):38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 23.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Science. 2001;292(5522):1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 24.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. J Clin Invest. 2003;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de JR, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Mol Cell Biol. 2005;25(5):1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Development. 2005;132(13):2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- 27.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Dev Cell. 2005;9(3):389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Park BK, Zeng X, Glazer RI. Cancer Res. 2001;61(20):7647–7653. [PubMed] [Google Scholar]
- 29.Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J. Faseb J. 2001;15(11):1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- 30.Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN, Larue L. Cancer Res. 2003;63(9):2172–2178. [PubMed] [Google Scholar]
- 31.Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. Cancer Res. 2001;61(2):589–593. [PubMed] [Google Scholar]
- 32.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Mol Cell. 2005;20(4):539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, Bissell MJ. Proc Natl Acad Sci U S A. 2006;103(11):4134–4139. doi: 10.1073/pnas.0511342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jauliac S, Lopez-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. Nat Cell Biol. 2002;4(7):540–544. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 35.Chen M, O’Connor KL. Oncogene. 2005;24(32):5125–5130. doi: 10.1038/sj.onc.1208729. [DOI] [PubMed] [Google Scholar]
- 36.Yiu GK, Toker A. J Biol Chem. 2006;281(18):12210–12217. doi: 10.1074/jbc.M600184200. [DOI] [PubMed] [Google Scholar]
- 37.Courtneidge SA, Heber A. Cell. 1987;50(7):1031–1037. doi: 10.1016/0092-8674(87)90169-3. [DOI] [PubMed] [Google Scholar]
- 38.Courtneidge SA, Smith AE. EMBO J. 1984;3(3):585–591. doi: 10.1002/j.1460-2075.1984.tb01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dilworth SM, Brewster CE, Jones MD, Lanfrancone L, Pelicci G, Pelicci PG. Nature. 1994;367(6458):87–90. doi: 10.1038/367087a0. [DOI] [PubMed] [Google Scholar]
- 40.Webster MA, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ. Mol Cell Biol. 1998;18(4):2344–2359. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Mol Cell Biol. 2001;21(6):2203–2212. doi: 10.1128/MCB.21.6.2203-2212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Cancer Res. 2004;64(9):3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 43.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Cancer Res. 2007;67(1):167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 44.Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, Mueller SC, Ojeifo J, Chen WS, Hay N, Pestell RG. Proc Natl Acad Sci USA. 2007;104(18):7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Cancer Res. 2003;63(1):196–206. [PubMed] [Google Scholar]
- 46.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Cancer Res. 2007;67(5):1979–1987. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- 47.Heron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A, Lamb NJ. Mol Cell Biol. 2006;26(22):8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D, Cheng GZ, Lindsley CW, Yang H, Cheng JQ. Curr Opin Investig Drugs. 2005;6(12):1250–1258. [PubMed] [Google Scholar]
- 49.Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE. Biochem J. 2005;385(Pt 2):399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. J Biol Chem. 2006;281(47):36443–36453. doi: 10.1074/jbc.M600788200. [DOI] [PubMed] [Google Scholar]
- 51.Aoki T, Nagakawa Y, Tsuchida A, Kasuya K, Kitamura K, Inoue K, Ozawa T, Koyanagi Y, Itoi T. Oncol Rep. 2002;9(4):761–765. [PubMed] [Google Scholar]
- 52.Kirkpatrick K, Ogunkolade W, Elkak A, Bustin S, Jenkins P, Ghilchik M, Mokbel K. Curr Med Res Opin. 2002;18(4):237–241. doi: 10.1185/030079902125000633. [DOI] [PubMed] [Google Scholar]
- 53.Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, Costantini F, Murohara T, Takahashi M. Nat Cell Biol. 2008;10(3):329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- 54.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. J Biol Chem. 1998;273(46):30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 55.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Proc Natl Acad Sci USA. 2000;97(4):1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Genes Dev. 2000;14(4):391–396. [PMC free article] [PubMed] [Google Scholar]
- 57.Pore N, Liu S, Shu HK, Li B, Haas-Kogan D, Stokoe D, Milanini-Mongiat J, Pages G, O’Rourke DM, Bernhard E, Maity A. Mol Biol Cell. 2004;15(11):4841–4853. doi: 10.1091/mbc.E04-05-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, Dvorak HF, Benjamin LE. Cancer Cell. 2006;10(2):159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guba M, von BP, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK. Nat Med. 2002;8(2):128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 60.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. J Clin Invest. 2005;115(8):2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Nat Med. 2005;11(11):1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Nature. 1999;399(6736):597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 64.Kawasaki K, Smith RS, Jr, Hsieh CM, Sun J, Chao J, Liao JK. Mol Cell Biol. 2003;23(16):5726–5737. doi: 10.1128/MCB.23.16.5726-5737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 66.Chen Y, Stamatoyannopoulos G, Song CZ. Cancer Res. 2003;63(16):4801–4804. [PubMed] [Google Scholar]
- 67.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 68.Orimo A, Gupta PB, Sgroi DC, renzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 69.Liang Z, Brooks J, Willard M, Liang K, Yoon Y, Kang S, Shim H. Biochem Biophys Res Commun. 2007;359(3):716–722. doi: 10.1016/j.bbrc.2007.05.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burger JA, Kipps TJ. Blood. 2006;107(5):1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 71.Luboshits G, Shina S, Kaplan O, Engelberg S, Nass D, Lifshitz-Mercer B, Chaitchik S, Keydar I, Ben-Baruch A. Cancer Res. 1999;59(18):4681–4687. [PubMed] [Google Scholar]
- 72.Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, Wigler N, Keydar I, Ben-Baruch A. Cancer Res. 2002;62(4):1093–1102. [PubMed] [Google Scholar]
- 73.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Nature. 2007;449(7162):557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 74.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Proc Natl Acad Sci USA. 2004;101(14):4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Proia DA, Kuperwasser C. Nat Protoc. 2006;1(1):206–214. doi: 10.1038/nprot.2006.31. [DOI] [PubMed] [Google Scholar]