

Abstract
New generation sequencers have been developed with a strong impact on genomics. These sequencers are based on a principle different from the Sanger method, and can sequence one to several million templates in a single run, albeit read length is relatively small. The current large-scale efforts are: 1) complete genome sequencing of 1,000 individuals, the primary objective of which is identification of rare SNP variants, not identified by the international HapMap project; 2) large-scale sequencing of cancer genomes to construct a complete catalog of genomic changes. These sequencers are also being applied in the identification of new infectious agents. Steady increase in data production capacity and decrease of cost will definitely make the sequencers a powerful diagnostic tool, especially for screening of all genetic diseases. On the contrary, statistical problems inherent to large data sets need to be solved before application to specific problems in medical science.
Keywords: Massive parallel analysis, the 1000 genomes project, the cancer genome atlas
Introduction
In the field of genomics, the next generation DNA sequencer is currently the hottest topic. These new sequencers can produce over 100 times more data compared to the most sophisticated capillary sequencers based on the Sanger method. The rapid developments of machines and bioinformatics are making the goal a “1,000 dollar genome sequence”, i.e., sequencing individual human genomes at a cost of $ 1, 000 each. The entire scene of biomedical science may change when the goal has been reached.
In this review, I summarize the principle of the next generation sequencers, current applications, and their future prospects in medical science. The first generation sequencers refer to those based on the Sanger method, the second generation sequencers are those based on massive parallel analysis, and the third generation sequencers are those based on single molecule sequencing in addition to massive parallel analysis. Because the current excitement comes from the second-generation sequencers, I will show their basic principle first.
Principle of the second generation sequencer
Three second generation sequencers are commercially available: Roche FLX [1], Illumina Genome Analyzer (GA) [2], and Lifetechnologies' SOLiD [3, 4]. Those machines are widely distributed, and their performance has been well characterized. All sequencers are based on a similar principle.
Use PCR products from single molecules as templates. With FLX and SOLiD, PCR amplification is performed on microbeads using emulsion PCR so that PCR products from a single molecule are attached to a single bead. With FLX, each bead is located in a picoliter well. With GA, PCR amplification is performed on a slide glass, making “clusters” of PCR products derived from single molecules [5]. Cluster formation is more sophisticated because theoretically a higher density of templates can be achieved.
Sequence by repetitive reaction. Information of 1-2 bases from a large number of templates is obtained by a single reaction, where the bases are discriminated by a fluorescent dye. Each time, a fluorescent image of the entire field, i.e., all templates, is captured with a CCD camera so that all analyzed bases are recorded. After clearing out the dyes, the same cycle is continued until no further base information can be obtained.
A schematic representation of the representtative sequence principle is shown in Figure 1. Each sequencer employs different principles of reaction including:
Pyrosequencing [6] (FLX). When an extension reaction occurs, one dNTP is added, and pyrophosphate (PPi) is released. ATP sulfurylase quantitatively converts PPi to ATP in the presence of adenosine 5′ phosphosulfate. This ATP acts as fuel to the luciferase-mediated conversion of luciferin to oxyluciferin that generates visible light in amounts that are proportional to the amount of ATP. The light produced in the luciferase-catalyzed reaction is detected by a camera and is analyzed in a program.
Reversible terminator (GA). Using a fluorescent dye-labeled terminator, the single base extension reaction is performed. Then, the fluorescent dye and the blocking group are chemically removed, and the next extension reaction is performed. The terminators are similar to those reported in [7].
Sequencing by ligation (SOLiD). This reaction utilizes the base discrimination ability of DNA ligase. Two bases adjacent to the ligation point are used for sequencing. One cycle consists of ligation of oligonucleotides, and cleavage and removal of the extended product. The cycles are repeated until no detectable fluorescent signals are obtained. One of the earliest examples of sequencing by ligation is described in [8].
Figure 1.
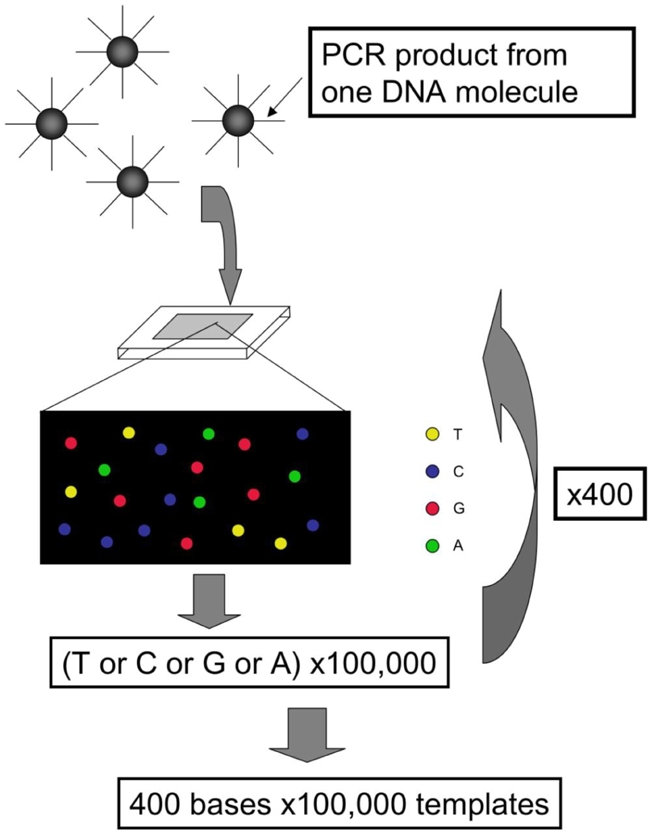
Schematic presentation of the principle of the second-generation sequencer. This scheme is based on Roche FLX.
The current benchmarks of the sequencers are summarized in Table 1. In brief, FLX produces long reads (∼400 bases), but the number of templates per run is moderate (∼1,000,000). GA and SOLiD produce short read (50∼75 bases), but are characterized by the large number of templates per run (100,000,000∼85,000,000). Their performance is increasing rapidly.
Table 1.
Comparison of sequencers (January, 2009). It should be noted that the throughput of each sequencer is improving rapidly
| ABI 3730xI | Roche GS FLX | Illumina GA | ABI SOLiD | |
|---|---|---|---|---|
| Bases/template | ∼1100 | ∼400 | ∼75 | 50 |
| Templates/run | 96 | 1,000,000 | 40,000,000 | 85,000,000 |
| Data production/day | 1 MB/day | 400MB/run/7.5hr | 3,000MB/run/6.5 days | 4,000MB/run/6 days |
| Maximum samples | 96 | 16 regions/plate | 8 channels/flowcell | 16chambers/2 slides |
| Sequence reaction | Sanger method | pyrosequencing | Reverse terminator | ligation sequencing |
The third generation sequencer-single molecule sequencing
Pacific Bioscience Inc. is developing a sequencer based on a new principle, which should be categorized as third generation. This DNA sequencer uses single DNA molecules as templates. The main characteristic of this sequencer is real-time monitoring of nucleotide incorporation with DNA polymerase. The major drawback of the second-generation sequencers from the Sanger method is short read of templates. Not like the second-generation sequencer, this sequencer can obtain reads of several kilobases from a single template. This sequencer is based on the following three technical components.
Zero mode waveguide [9]. A slide glass is coated with a thin aluminum layer. The aluminum layer has many small holes, with a diameter d ∼50 nm. Because the light, whose wave length is greater than 1.7 × d, is evanescent, the illuminating light exists only in the entrance of the hole. Because no propagation mode exists, these guides are referred to as “zero mode wave guide.” To enable real-time monitoring of DNA polymerase, the concentration of substrates (deoxynucleotide triphosphate, dNTP) should be more than micromole. However, other technologies require a much lower concentration for detection of fluorescence. Zero mode waveguide is the first technique solving this problem.
Passivation of aluminum surface using polyphosphonate chemistry [10]. Aluminum surface is protected with polyvinylphosphonic chemistry from attachment of DNA polymerase. Thus, DNA polymerase molecules only attached to the silica surface, i.e., at the bottom of the holes, eliminating possible background fluorescent light.
Use of dNTPs whose terminal phosphate moieties are conjugated with fluorophores [11]. These fluorescence-labeled dNTPs release fluorescence when incorporated into DNA, and then lose the fluorophores. Thus, these dNTPs enable real-time monitoring of incorporation of nucleotides.
Characteristics of sequence data generated by the second-generation sequencers
Because the third generation sequencer has yet to be commercialized, this review further focuses on the second-generation sequencers. It should be noted that there is plenty of room for improvement in throughput of GA and SOLID. With these systems, templates can be accumulated at a much higher density. On the contrary, Roche FLX has limitations. Because the fluorescent dye, i.e., oxyluciferin, diffuses into the reaction solution, each template bead must be separated in an individual well. This feature limits the template density
The surrounding situation of the second-generation sequencers is different from that of the first-generation sequencers. The most important factor is the completion of the human genome project. As shown above, a major drawback of the next-generation sequencers from the previous sequencers is the short read length: 350 bases (FLX) and 50-75 bases (GA, SOLiD), compared to > 800 bases with first-generation sequencers. The short read length is a considerable disadvantage for de novo sequencing. In de novo sequencing, it is necessary to construct a complete sequence from a large number of short sequence pieces. If the one read length is short, the short pieces make only small overlaps, making it difficult to construct contigs. Thus, the second-generation sequencers, especially GA and SOLiD, are not intended for de novo sequencing. However, in the human genome, the short pieces may be assembled into large sequences, being matched with the reference human genome sequence. In this way, the second-generation sequencers can produce complete genome sequences of individuals. The major genome centers now challenge two targets, i.e., the genomes of individuals and cancer genomes.
Sequencing genomes of individuals
For several years, single nucleotide polymerphism (SNP) and its application to human genetics has been the most intensive area in genomics. SNP was at first intensively collected using sequences obtained during the human genome project. These SNPs (roughly 100 million) were organized by haplotypes identified by the international HapMap project [12]. Consequently, about 50,000 tag SNPs representing haplotypes, were obtained. Genetic loci associated with a number of common diseases have been identified using the above tag SNP set through genome-wide association studies (GWAS). Accumulating results, however, show that GWAS generally failed to identify most of the genetic background of common diseases. A series of articles has been recently published to review the results from various viewpoints [13-15]. There are now a number of discussions to determine the research direction, i.e., continuation of GWAS or turning the research direction to complete sequencing of individual human genomes. Because the SNP markers used in GWAS are based on the international HapMap project, they detect allele variants whose frequencies are over 5 %. Therefore, rare variants (0.1 - 5 %) cannot be detected in GWAS. Proponents of the genome sequencing argue that genetic association may be found with rare variants, not detected by the current tag SNPs, and the complete genome sequences of a large number of individuals will uncover the more detailed view of variations. Currently, the “1,000 genomes” project (http://www.1000genomes.org), an international project to sequence genomes of 1,000 individuals, is ongoing. The outcome of the projects will be an important resource for human genome variation, but the direct objective is identification of rare variants to extend current GWAS.
It is important to confirm whether the second-generation sequencers can identify SNP equally as well as the Sanger method. Two Caucasian individual genomes have been determined before the “1,000 genomes” project. One that was obtained by the Sanger method [16], identified 2.8 million known SNPs and about 0.74 million novel SNPs. The other that was sequenced with GS20, a previous model of FLX [17], identified 2.72 million known and 0.61 million novel SNPs. Pilot experiments of the 1,000 genome project determined genomes of two individuals with GA [18, 19]. The sequence of a male Yoruba identified 3.8-4.1 million SNPs, 73.6% of which were in dbSNP [18]. The sequence of an Asian individual identified 3 million SNPs, 73.5% of which were in dbSNP [19]. Recently, a new study compared the second-generation sequencers and a Sanger sequencer from the view point of GWAS [20]. In general, the second-generation sequencers had very high sensitivity, i.e., identification of SNPs, but relatively low specificity. This tendency was more prominent with GA and SOLiD, because of short sequence reads: errors were more common in repeated sequence regions, probably due to errors during sequence assembly. The other obstacle is biases in representation among genomic regions. To obtain complete coverage of a genomic region, it is necessary to obtain more reads. These results suggest that the next-generation sequencers are useful for SNP studies, if enough reads are obtained.
Still the complete human genome sequencing is expensive. In addition, a huge computational load is required. Instead, sequencing of all protein coding regions, named “exome”, is regarded as a cost-effective approach [21]. SNPs or mutations in coding regions are more informative and likely to be linked to diseases than those in non-coding regions. One of the examples is a study on pancreatic cancer described below [22].
Sequencing of cancer genomes
The objective of projects, such as The Cancer Genome Atlas (http://cancergenome.nih.gov), to sequence cancer genomes is a complete list of genomic changes contributing to carcinogenesis. These projects hypothesize that there would be undiscovered genes contributing to carcinogenesis, and they will accompany genomic changes such as mutations, copy number variations and translocations. Epigenetic events have also been known to contribute to carcinogenesis, and may be incorporated into the projects. Unbiased exploration of such events would substantially contribute to understanding of cancer, and lead to identification of new target molecules.
Several pilot experiments using the first generation sequencers have been performed. Due to limited throughput of the first generation sequencers, several early studies focused on specific gene families, such as tyrosine kinases, which were often activated by somatic mutations. An organized study was performed at the Welcome Trust Sanger Institute [23]. In that study, somatic mutations were classified into “driver” and “passenger” mutations. “Driver” mutations are defined as that conferring growth advantage, and “passenger” mutations are defined as those without any biological effects. Overall selection pressure by all the substitute mutations was calculated: 1.29 (95% confidence interval, 1.10-1.51; P=0.0013).197The other study examined the majority of the transcribed genes (18,191 genes) with eleven breast and eleven colorectal cancer tissues [24]. This study revealed that there were a large number of mutations with rare incidence, in addition to a small number of genes with mutations of high incidence. Both studies suggested that known somatic mutations were only a small fraction of mutations in cancer genomes, and more systematic analysis of the cancer genome, i.e., complete genome sequencing of a large number of cancer tissues, is necessary. These studies were followed by two studies on glioma [25, 26]. Both studies accompanied measurements of copy number variation by genome arrays and gene expression profiling [25] by microarrays or SAGE [26]. One of the studies found recurrent mutations at the active site of isocitrate dehydrogenase 1 (IDH1) in 12% of glioblastoma patients [26]. This result suggests that there would be additional important mutations not discovered so far.
Comparison of a cancer genome with the corresponding germline genome is very informative. One study analyzed the whole genome of malignant cells and normal cells from a single acute myelogenous leukemia (AML) patient [27]. The whole genome analysis revealed that the AML genome had only eight heterozygous, non-synonymous somatic mutations, all of which were novel. Another study to sequence all coding regions on a genome of familial pancreatic cancer identified that mutations in PALB2 was responsible for the disease, validated with 96 additional samples [22]. Both studies could pinpoint out a small number of candidate genes, demonstrating the accuracy and thoroughness of the whole-genome approach.
The above early studies strongly suggest that the large-scale cancer genome projects would definitely contribute to our understanding of genetic changes in cancer. However, contribution to medicine is a different problem. The rationale to justify the large investments for these projects is identification of molecular targets and subsequent developments of anti-cancer drugs. The proponents of the projects argue that newly identified mutations will be effective targets for anti-cancer drug development. This reflects the current trend of anti-cancer drug development: a large number of molecular target drugs are now being developed or during clinical trials with expectations to improve cancer therapy. However, when the above cancer genome projects were finished, the current trend and enthusiasm might be finished. Already, there is controversy among scientists on the future prediction of molecular target drugs [28, 29]. So far, all molecular target drugs except imatinib extend overall survival only several months. Molecular target therapy might turn out to be not attractive as it is: pharmaceutical companies might lose interest. In any case, the resulting data will be valuable as a resource for cancer research.
Discovery of new infectious agents
The third important application of the second-generation sequencers is identification of infectious agents. RNA or DNA of human tissues or cells infected by a specific infectious agent such as a virus, bacterium, contain the human genome sequences as well as sequences of the infectious agent. Sequencing a large number of RNA or DNA pieces from an infected sample, the resulting sequences contain those derived from the infectious agent as well as from the human genome. Now that the complete human genome sequence has been obtained, subtraction of the human genome sequence should theoretically yield sequences of the infectious agent. This idea is not new. In 2002, a computational experiment was performed, by searching the human genome sequences for expressed tag sequences (EST) of human origin using data in the public database [30]. Among sequences not matching the human genome, more than 50 sequences matching virus genomes were identified. The same group performed a model experiment with tissues of post-transplant lymphoproliferative disorder (PTLD), and successfully recovered Epstein-Bar virus sequences, the known agent of PTLD [31]. These studies suggested the plausibility of the above experimental strategy.
In spite of the potential strength of the strategy, the high cost of DNA sequencing has prevented real application. Due to the decreased cost of sequencing by the second-generation sequencer, two studies using FLX appeared in 2008. One study focused on patients who died of febrile illness after visceral organ transplantation [32]. Unbiased transcript sequencing from liver and kidney, and subsequent data analysis revealed infection of a new arena virus. The other study focused on Merkel cell carcinoma, a rare type of skin cancer [33]. Sequencing of nearly 400,000 transcripts identified sequences similar to known polyoma viruses, Further analysis revealed a new polyoma virus sequence named Merkel cell polyoma virus.
Application to gene expression profiling
The sequencers can be applied to gene expression profiling, i.e., a genome-scale analysis of gene expression. Sequencing a large number of transcripts purified from a tissue or cell, and subsequently matching them to the human reference genome reveals the identity of each transcript. The expression level of the gene can be determined from the number of times each gene sequence appeared. This approach of gene expression profiling has been named digital gene expression profiling, and was originally initiated in the early stage of the human genome project [34]. Later, a new technique named serial analysis of gene expression (SAGE) [35], appeared. In SAGE, a small tag (SAGE tag), with a size of 9 to 21 bases, is obtained from each transcript, and tens of tags are concatemerized, and read with a sequencer. With SAGE, from a single read, frequency information of tens of transcripts can be obtained. Even still with SAGE, it was not practical to process a large number of samples due to low throughput of the sequencers based on the Sanger method. With the next-generation sequencers, digital expression profiling has finally become a plausible method comparable to microarrays. Its major advantage over microarray is straightforward standardization of the data. In digital expression profiling, data is just molecular counts. In contrast, the data obtained by microarray analysis is expression level against some standard, and it is difficult to compare data from different experimental series. However, for laboratory use, i.e., comparison of global gene expression among samples of interest, digital expression profiling does not have clear advantage over microarrays.
Discussion for future applications
In this review, the principle of the next generation sequencers, and their major research areas have been described. As shown above, the current applications are centered on continuation of works already started before appearance of the second-generation sequencers, and mainly restricted to experts in genomics. However, one of the most important aspects of this technical revolution should be the easy access to the large sequence data by scientists in other areas and doctors. For the wide spread use, sequence data will soon be available from outsourcing companies, but data analysis will still remain a difficult task. Development of software systems easily accessible to nonexperts is essential for utilization of large sequence data.
Considering the steady increase of sequence capacity and decrease of cost, application to diagnosis will be realized in the near future. Routine neonatal diagnosis will be replaced with the routine sequence of the entire genome or the exome. This new type of diagnosis would reveal affected alleles of all known genetic diseases, including genes currently screened in postnatal diagnosis. From the data of a couple, risks of genetic diseases in their children can be accurately predicted.
Application to diagnostics for genetic diseases is easily predictable, but how to apply the next generation sequencers to medical science is rather difficult to predict. Large-scale data production such as the human genome project, the “1,000 genomes” project and “The Cancer Genome Atlas”, are productive as far as construction of resources. However, utilization of large data sets to solve a specific problem is usually difficult as exemplified by GWAS. The obstacles are mainly statistical problems inherent to large data sets. One is multiplicity in statistical tests. In general, there is no method of choice to control multiplicity, and the method is chosen through practical applications. For example, Bonferroni correction is used with GWAS, and the q-value or FDR is used for gene expression analysis. Although the validity of each method has been confirmed with repeated use, it should be noted that some true positives must be excluded. In particular, GWAS detected loci representing only a fraction of the genetic background of common diseases. One possibility is that true positive loci may be excluded by stringent criteria set by Bonferroni correction.
Another problem is the “curse of dimensionnality”. The “curse of dimensionality” is the problem caused by the exponential increase in volume associated with adding extra dimensions to a space. This problem results in an increased number of samples needed for analysis. In the cancer classification problem, where cancer samples are classified into two classes by means of gene expression profiling, the “curse of dimensionality” is avoided by dimension reduction, i.e., reduction of the number of genes by gene selection. For example, when the initial data set contains 10,000 genes, the problem is to classify cancers in a 10,000 dimensional space. By selection of differentially expressed genes, classification is usually performed in the reduced dimensional space, requiring adequate number of samples. On the contrary, it is impractical to reduce the number of SNP markers in GWAS. Thus, the aim of GWAS is limited to discovery of individual loci associated with the disease. GWAS cannot identify association of a disease with a combination of more than two genes. This is due to the “curse of dimensionality”, but an alternative explanation is as follows. When the number of tag SNP markers is 50,000, the number of the combination of two genes would be 1,249,975,000. It is impractical to perform this huge number of statistical tests, because of requirement of far larger cohorts and very low threshold p-value. The next-generation sequencers do not solve these statistical problems. Complex diseases are most likely to be mediated by numerous loci (both coding and non-coding) that interact with many environmental factors. Some argue that the whole genome sequencing would be useful for identification of such loci, but the real obstacle would be the statistical problem, which suggests requirement of a huge number of samples. This would also be the case in cancer genome projects.
Probably the most reasonable application would be identification of genes responsible for familial disorders. Familial disorders with small pedigrees, which cannot be subjected to linkage analysis, would be good targets. The above example of familial pancreatic cancer is a good example.
References
- 1.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Pandey V, Nutter RC, Prediger E. Applied Biosystems SOLiD™ System: Ligation-Based Sequencing. In: Janitz M, editor. Next Generation Genome Sequencing: Towards Personalized Medicine. San Francisco: Wiley; 2008. pp. 29–41. [Google Scholar]
- 4.Nutter R. New Frontiers in Plant Functional Genomics Using Next Generation Sequencing Technologies. In: Kahl G, Meksem K, editors. The Handbook of Plant Functional Genomics, Concepts and Protocols. San Francisco: Wiley; 2008. pp. 431–444. [Google Scholar]
- 5.Adessi C, Matton G, Ayala G, Turcatti G, Mermod JJ, Mayer P, Kawashima E. Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucleic Acids Res. 2000;28:E87. doi: 10.1093/nar/28.20.e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 1996;242:84–89. doi: 10.1006/abio.1996.0432. [DOI] [PubMed] [Google Scholar]
- 7.Ruparel H, Bi L, Li Z, Bai X, Kim DH, Turro NJ, Ju J. Design and synthesis of a 3′-O-allyl photocleavable fluorescent nucleotide as a reversible terminator for DNA sequencing by synthesis. Proc Natl Acad Sci U S A. 2005;102:5932–5937. doi: 10.1073/pnas.0501962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, Roth R, George D, Eletr S, Albrecht G, Vermaas E, Williams SR, Moon K, Burcham T, Pallas M, DuBridge RB, Kirchner J, Fearon K, Mao J, Corcoran K. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- 9.Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 10.Korlach J, Marks PJ, Cicero RL, Gray JJ, Murphy DL, Roitman DB, Pham TT, Otto GA, Foquet M, Turner SW. Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc Natl Acad Sci USA. 2008;105:1176–1181. doi: 10.1073/pnas.0710982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 12.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 14.Hirschhorn JN. Genomewide association studies–illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 15.Kraft P, Hunter DJ. Genetic risk prediction–are we there yet? N Engl J Med. 2009;360:1701–1703. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- 16.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 18.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LI, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LI, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LI, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, Li H, Lu Y, Fang X, Liang H, Du Z, Li D, Zhao Y, Hu Y, Yang Z, Zheng H, Hellmann I, Inouye M, Pool J, Yi X, Zhao J, Duan J, Zhou Y, Qin J, Ma L, Li G, Yang Z, Zhang G, Yang B, Yu C, Liang F, Li W, Li S, Li D, Ni P, Ruan J, Li Q, Zhu H, Liu D, Lu Z, Li N, Guo G, Zhang J, Ye J, Fang L, Hao Q, Chen Q, Liang Y, Su Y, San A, Ping C, Yang S, Chen F, Li L, Zhou K, Zheng H, Ren Y, Yang L, Gao Y, Yang G, Li Z, Feng X, Kristiansen K, Wong GK, Nielsen R, Durbin R, Bolund L, Zhang X, Li S, Yang H, Wang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, Frazer KA. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ, McCombie WR. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 25.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, Cook L, Abbott R, Larson DE, Koboldt DC, Pohl C, Smith S, Hawkins A, Abbott S, Locke D, Hillier LW, Miner T, Fulton L, Magrini V, Wylie T, Glasscock J, Conyers J, Sander N, Shi X, Osborne JR, Minx P, Gordon D, Chinwalla A, Zhao Y, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson M, Baty J, Ivanovich J, Heath S, Shannon WD, Nagarajan R, Walter MJ, Link DC, Graubert TA, DiPersio JF, Wilson RK. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hait WN, Hambley TW. Targeted cancer therapeutics. Cancer Res. 2009;69:1263–1267. doi: 10.1158/0008-5472.CAN-08-3836. discussion 1267. [DOI] [PubMed] [Google Scholar]
- 29.Hambley TW, Hait WN. Is anticancer drug development heading in the right direction? Cancer Res. 2009;69:1259–1262. doi: 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- 30.Weber G, Shendure J, Tanenbaum DM, Church GM, Meyerson M. Identification of foreign gene sequences by transcript filtering against the human genome. Nat Genet. 2002;30:141–142. doi: 10.1038/ng818. [DOI] [PubMed] [Google Scholar]
- 31.Xu Y, Stange-Thomann N, Weber G, Bo R, Dodge S, David RG, Foley K, Beheshti J, Harris NL, Birren B, Lander ES, Meyerson M. Pathogen discovery from human tissue by sequence-based computational subtraction. Genomics. 2003;81:329–335. doi: 10.1016/s0888-7543(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 32.Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 33.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okubo K, Hori N, Matoba R, Niiyama T, Fukushima A, Kojima Y, Matsubara K. Large scale cDNA sequencing for analysis of quantitative and qualitative aspects of gene expression. Nat Genet. 1992;2:173–179. doi: 10.1038/ng1192-173. [DOI] [PubMed] [Google Scholar]
- 35.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]