

Abstract
Purpose
Different pharmacokinetic methods for [18F]FDDNP studies were evaluated using both simulations and clinical data.
Procedures
Methods included two-tissue reversible plasma (2T4k), simplified reference tissue input (SRTM), and a modified 2T4k models. The latter included an additional compartment for metabolites (2T1M). For plasma input models, binding potential, BPND, was obtained both directly (=k3/k4) and indirectly (using volume of distribution ratios).
Results
For clinical data, 2T1M was preferred over 2T4k according to Akaike criterion. Indirect BPND using 2T1M correlated better with SRTM then direct BPND. Fairly constant volume of distribution of metabolites was found across brain and across subjects, which was strongly related to bias in BPND obtained from SRTM as seen in simulations. Furthermore, in simulations, SRTM showed constant bias with best precision if metabolites entered brain.
Conclusions
SRTM is the method of choice for quantitative analysis of [18F]FDDNP even if it is unclear whether labeled metabolites enter the brain.
Key words: [18F]FDDNP, PET, SUV, Reference tissue model, Plasma input model, Metabolites
Introduction
[18F] FDDNP has recently been introduced [1] as a positron emission tomography (PET) ligand for in vivo imaging of amyloid plaques and neurofibrillary tangles in the human brain. Plaques and tangles are thought to be the hallmark of Alzheimer’s disease (AD) [2], and early in vivo detection of these neuropathological lesions could be an important step in evaluating future treatment strategies for AD.
So far, only a few methods for quantification of [18F]FDDNP have been evaluated, such as residence time within a cerebral region relative to that in pons [3], standardized uptake value (SUV) [4], distribution volume ratio (DVR) [4] obtained with Logan analysis [5] using cerebellum as reference region, and several simplified reference tissue-based methods [6] also using cerebellum as reference region. Using Logan analysis, Kepe et al. [7] recently reported increased levels of [18F]FDDNP binding in neocortical regions compared with that in cerebellum in AD patients, whereas no difference in uptake between cerebellum and other regions was found in healthy controls (HC). In addition, Small et al. [8] found that global DVR values in HC were lower than in patients with mild cognitive impairment (MCI), which in turn were lower than in AD subjects. However, arterial sampling was not used in any of these studies. Arterial sampling is considered to be the gold standard, especially if pathological changes may also affect reference regions.
The purpose of the present study was to investigate which pharmacokinetic model could best be used for quantitative analysis of [18F]FDDNP studies. To this end, both simulated and clinical [18F]FDDNP data were used. Data were analyzed using various compartmental models based on plasma [9] and reference tissue [10–12] input data. In addition, a plasma input model was evaluated, which accounted for uptake of labeled metabolites in the brain. Finally, standard uptake value ratios with cerebellum (SUVr) were investigated.
Methods
Scanning Protocol
Clinical data were derived from ongoing patient studies consisting of 12 subjects (six HC, three MCI [13], and three AD) with ages ranging from 58 to 72 years. Mean age (±SD) was 66 ± 5, 68 ± 4, and 63 ± 6 for HC, MCI, and AD, respectively. AD patients were diagnosed with probable AD meeting NINCDS-ADRDA criteria [14]. The study was approved by the Medical Ethics Committee of the VU University Medical Centre, and each subject gave written informed consent prior to inclusion in the study. Clinical results are beyond the scope of the present study and will be reported elsewhere.
As part of the study protocol, each subject first underwent a T1-weighted magnetic resonance imaging (MRI) scan using a 1.5-T SONATA scanner (Siemens Medical Solutions, Erlangen, Germany). This MRI scan was performed to exclude anatomical abnormalities and for co-registration and segmentation purposes.
PET studies were performed using an ECAT EXACT HR+ scanner (CTI/Siemens, Knoxville, USA). The characteristics of this scanner have been described previously [15]. First, a 10-min transmission scan in 2D acquisition mode was performed. This scan was used to correct the subsequent emission scan for tissue attenuation. Next, a dynamic emission scan in 3D acquisition mode was performed following bolus injection of 168 ± 8 MBq [18F]FDDNP [16]. This dynamic emission scan consisted of 23 frames (1 × 15, 3 × 5, 3 × 10, 2 × 30, 3 × 60, 2 × 150, 2 × 300, 7 × 600 s) with a total scan duration of 90 min. All frames were reconstructed using FORE+ 2D filtered back projection [17] and a Hanning filter with a cutoff of 0.5 times the Nyquist frequency. Reconstructions included all standard corrections, such as normalization, and decay, dead time, attenuation, randoms, and scatter [18] corrections.
The protocol also included continuous arterial sampling, starting 2 min prior to injection and continuing up to 60 min, using a dedicated online detection system [19]. In addition, at set times (5, 10, 20, 40, and 60 min post-injection), arterial sampling was interrupted briefly for the withdrawal of discrete arterial samples. After each sample, the arterial line was flushed with heparinized saline in order to avoid clotting within the line. Finally, two manual samples were withdrawn at 75 and 90 min post-injection.
Arterial blood samples were used to determine plasma and whole blood radioactivity concentrations using a well counter cross calibrated against the PET scanner [20]. In addition, plasma parent tracer and metabolite concentrations were determined using solid phase extraction combined with high-performance liquid chromatography technology and equipped with off-line radioactivity detection [21]. An arterial whole blood curve was obtained by correcting the online sampler curve for decay, removing the flushing periods, and calibrating against the discrete (whole blood) sample data. Finally, a metabolite-corrected plasma curve was derived from this whole blood curve using both plasma to whole blood ratios and metabolite fractions obtained from the manual blood samples. To this end, the total fraction of labeled metabolites as function of time was fitted to a Hill-type function [22]. This function provided more accurate fits than other functions such as multi-exponentials. Arterial blood sampling was not available for six subjects (three HC and three MCI). Data from these subjects were only used for investigating reference tissue models.
Image Analysis
De-skulled T1-weighted MRI scans [23] were co-registered [24, 25] with a summed PET image (frames 3–12, 25 s–5 min post-injection). This summed PET image resembles a flow image, thereby maximizing cortical information. Time–activity curves were then generated using MR-based automatic delineation of regions of interest (ROI), as described by Svarer et al. [26]. For the purpose of the present study, only TACs from 17 regions (cerebellum, orbital frontal cortex, medial inferior frontal cortex, anterior cingulate cortex, thalamus, insula, caudate, putamen, superior temporal cortex, parietal cortex, medial inferior temporal cortex, superior frontal cortex, occipital cortex, sensory motor cortex, posterior cingulate cortex, enthorinal cortex, hippocampus), all averaged over left and right hemispheres, were analyzed. These anatomical regions were small and defined in gray matter only, thereby minimizing signal dilution due to partial volume effects as much as possible.
Kinetic Analyses of Clinical Data
Clinical data were analyzed using conventional plasma input and reference tissue-based algorithms [9]. As there is some concern that labeled [18F]FDDNP metabolites might cross the blood–brain barrier [27, 28], additional analyses were performed using a plasma input model accounting for metabolites entering the brain. In addition, average regional activity concentration ratios with the reference region (SUVr) were derived over the time intervals of 40–60, 60–90, and 80–90 min after injection. For all reference tissue models and SUVr cerebellar gray matter was used as reference region based on its relatively low levels of amyloid and neurofibrillary tangles [2].
Five different conventional compartmental models were evaluated: single-tissue (1T2k), two-tissue irreversible (2T3k) and two-tissue reversible (2T4k) plasma input models, and simplified (SRTM) [12] and full (FRTM) [10, 11] reference tissue models. Plasma input models contained one additional fit parameter for blood volume. The SRTM was used to estimate binding potential directly 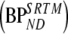 . The 2T4k plasma input model was used to estimate binding potential both directly
. The 2T4k plasma input model was used to estimate binding potential both directly 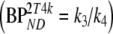 and indirectly using volume of distribution ratios
and indirectly using volume of distribution ratios 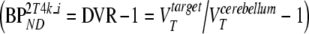 [11]. The latter approach will be indicated by 2T4ki.
[11]. The latter approach will be indicated by 2T4ki.
Based on the possibility of metabolites entering the brain [27, 28], also a modified 2T4k model was used. This model included an additional (parallel) single-tissue compartment for labeled metabolites (2T1M, Fig. 1). The metabolite input curve was based only on polar metabolites and ignores the minor fraction of other metabolites. The direct binding potential 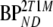 for this model was defined as k3/k4 (Fig. 1), and the volumes of distributions, VT and VTm, were defined as
for this model was defined as k3/k4 (Fig. 1), and the volumes of distributions, VT and VTm, were defined as 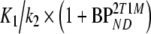 and K1m/k2m, respectively. Similar to 2T4ki, the binding potential was also estimated indirectly using the volume of distribution ratios with the cerebellum as reference region
and K1m/k2m, respectively. Similar to 2T4ki, the binding potential was also estimated indirectly using the volume of distribution ratios with the cerebellum as reference region 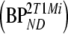 . All 2T1M fits were repeated after fixing VTm to the cerebellum value (=2T1Mfvtm). This model was also used to estimate binding potential both directly
. All 2T1M fits were repeated after fixing VTm to the cerebellum value (=2T1Mfvtm). This model was also used to estimate binding potential both directly 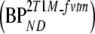 and indirectly using DVR − 1
and indirectly using DVR − 1 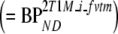 . The indirect methods for estimating BPND using 2T1M and 2T1Mfvtm will be indicated by 2T1Mi and
. The indirect methods for estimating BPND using 2T1M and 2T1Mfvtm will be indicated by 2T1Mi and 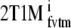 , respectively.
, respectively.
Fig. 1.

Schematic diagram of the model that includes a tissue metabolite compartment. BBB represents the blood–brain barrier; PParent and PMet are tracer plasma radioactivity concentrations (Bq ml−1) of parent and labeled metabolites, respectively; T is the radioactivity concentration in tissue, F+NSParent that of free and non-specifically bound parent in tissue, SParent that of bound parent in tissue; and F+NSMet that of labeled metabolites in tissue (Bq ml−1); K1 (ml·cm−3·min−1) and k2 (min−1) are rate constants, describing exchange of parent between plasma and tissue; k3 and k4 are rate constants (min−1), describing exchange of parent between free and bound compartments; K1m (ml·cm−3·min−1) and k2m (min−1) are rate constants, describing exchange of labeled metabolites between plasma and tissue.
Clinical Studies
Clinical results were evaluated in several ways. First, for all compartmental models (SRTM, FRTM, 1T2k, 2T3k, 2T4k, 2T1M, and 2T1Mfvtm), fitted TACs were evaluated using the Akaike criterion. Next, where possible, BPND values were estimated both directly and indirectly using VT ratios with cerebellum (i.e., 2T4ki, 2T1Mi and 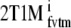 ). In addition, estimates of BPND were compared with
). In addition, estimates of BPND were compared with 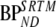 . Finally, SUVr − 1 values were estimated and compared with
. Finally, SUVr − 1 values were estimated and compared with  , given that at true equilibrium, SUVr corresponds with DVR.
, given that at true equilibrium, SUVr corresponds with DVR.
Simulation Studies
Conventional Simulations
Simulated time–activity curves (TACs) for target and reference regions were generated using a typical [18F]FDDNP plasma input function in combination with the two-tissue reversible plasma input model (2T4k). In the simulations, variations in binding, delivery, and fractional blood volume were investigated.
Kinetic parameters were based on typical 2T4k parameters obtained from clinical data. Default parameters for simulated reference and target tissue TACs are given by R1 and T1, respectively, as listed in Table 1. Target tissue parameters were varied with respect to binding potential (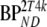 and
and 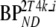 : T2–T5), fractional blood volume (Vb: T6, T7), and delivery (K1: T8–T15). One hundred TACs were generated for each run and noise was added to simulate an average noise level of 7.5% coefficient of variation (COV). Noise simulation was based on clinically derived typical values of total scanner true counts, frame lengths, and decay correction factors. A detailed description of the noise simulation used is given in [29].
: T2–T5), fractional blood volume (Vb: T6, T7), and delivery (K1: T8–T15). One hundred TACs were generated for each run and noise was added to simulate an average noise level of 7.5% coefficient of variation (COV). Noise simulation was based on clinically derived typical values of total scanner true counts, frame lengths, and decay correction factors. A detailed description of the noise simulation used is given in [29].
Table 1.
Kinetic parameters used to generate reference (R) and target (T) tissue time activity curves (TAC)
| TAC | K1 (ml·cm−3·min−1) |  |
Vb | VT | 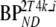 |
|---|---|---|---|---|---|
| R 1 | 0.35 | 1.63 | 0.050 | 8.7 | – |
| T 1 | 0.35 | 2.3 | 0.050 | 11 | 0.25 |
| T 2 | 0.35 | 1.7 | 0.050 | 8.9 | 0.03 |
| T 3 | 0.35 | 2.0 | 0.050 | 9.9 | 0.14 |
| T 4 | 0.35 | 2.6 | 0.050 | 12 | 0.37 |
| T 5 | 0.35 | 2.9 | 0.050 | 13 | 0.48 |
| T 6 | 0.35 | 2.3 | 0.025 | 11 | 0.25 |
| T 7 | 0.35 | 2.3 | 0.075 | 11 | 0.25 |
| T 8 | 0.21 | 2.3 | 0.050 | 11 | 0.25 |
| T 9 | 0.25 | 2.3 | 0.050 | 11 | 0.25 |
| T 10 | 0.28 | 2.3 | 0.050 | 11 | 0.25 |
| T 11 | 0.32 | 2.3 | 0.050 | 11 | 0.25 |
| T 12 | 0.39 | 2.3 | 0.050 | 11 | 0.25 |
| T 13 | 0.43 | 2.3 | 0.050 | 11 | 0.25 |
| T 14 | 0.46 | 2.3 | 0.050 | 11 | 0.25 |
| T 15 | 0.50 | 2.3 | 0.050 | 11 | 0.25 |
K1, k2, and k3 are rate constants, Vb is fractional blood volume, VT is volume of distribution, and 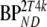 and
and 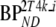 are binding potentials estimated directly (2T4k) and indirectly (2T4ki) using the reversible two-tissue model, respectively. The indirect calculation of BPND was performed using target to reference volumes of distribution ratios. During all simulations, k4 (0.032 min−1) and K1/k2 (3.3) were fixed
are binding potentials estimated directly (2T4k) and indirectly (2T4ki) using the reversible two-tissue model, respectively. The indirect calculation of BPND was performed using target to reference volumes of distribution ratios. During all simulations, k4 (0.032 min−1) and K1/k2 (3.3) were fixed
Simulated data were analyzed using conventional models as described above (1T2k, 2T3k, 2T4k, SRTM, FRTM, and 2T4ki) and SUVr methods over the time intervals 40–60, 60–90, and 80–90 min. Fits were evaluated by comparing goodness of fit according to the Akaike criterion. Next, models were evaluated by comparing bias and COV of estimated binding potential (where appropriate, also the indirect estimation through volumes of distribution). Bias was estimated in relative terms using 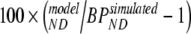 , where
, where 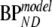 represents BPND estimated using the method of analysis under investigation and
represents BPND estimated using the method of analysis under investigation and 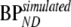 simulated BPND, which was set to either
simulated BPND, which was set to either 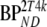 or
or 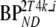 for direct and indirect methods, respectively.
for direct and indirect methods, respectively.
Metabolite Model
To simulate the effects of cerebral uptake of labeled metabolites, simulated TACs were generated using a typical [18F]FDDNP plasma input function in combination with the 2T1M model. Kinetic parameters were based on typical 2T1M parameters obtained from clinical data. Default parameters for simulated reference and target tissue TACs are given by R1 and T1, respectively, as listed in Table 2. Target tissue parameters were varied, including binding potential ( and
and 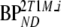 : T2–T9), delivery of metabolites (K1m: T10–T13), and volume of distribution of metabolites (VTm: T14–T21). One hundred TACs were generated for each run and noise was added to simulate an average noise level of 7.5% COV, as described above.
: T2–T9), delivery of metabolites (K1m: T10–T13), and volume of distribution of metabolites (VTm: T14–T21). One hundred TACs were generated for each run and noise was added to simulate an average noise level of 7.5% COV, as described above.
Table 2.
Kinetic parameters used to generate reference (R) and target (T) TAC
| TAC | K1 (ml·cm−3·min−1) | 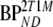 |
Vb | VT | 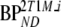 |
K1m (ml·cm−3·min−1) | VTm |
|---|---|---|---|---|---|---|---|
| R 1 | 0.34 | 0.65 | 0.05 | 4 | – | – | – |
| T 1 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.59 |
| T 2 | 0.34 | 0.8 | 0.05 | 4.37 | 0.09 | 0.50 | 0.6 |
| T 3 | 0.34 | 1.0 | 0.05 | 4.86 | 0.21 | 0.50 | 0.6 |
| T 4 | 0.34 | 1.2 | 0.05 | 5.34 | 0.33 | 0.50 | 0.6 |
| T 5 | 0.34 | 1.4 | 0.05 | 5.83 | 0.45 | 0.50 | 0.6 |
| T 6 | 0.34 | 1.6 | 0.05 | 6.31 | 0.58 | 0.50 | 0.6 |
| T 7 | 0.34 | 1.8 | 0.05 | 6.80 | 0.70 | 0.50 | 0.6 |
| T 8 | 0.34 | 2.1 | 0.05 | 7.53 | 0.88 | 0.50 | 0.6 |
| T 9 | 0.34 | 2.3 | 0.05 | 8.01 | 1.00 | 0.50 | 0.6 |
| T 10 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.00 | 0.6 |
| T 11 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.01 | 0.6 |
| T 12 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.03 | 0.6 |
| T 13 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.05 | 0.6 |
| T 14 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.1 |
| T 15 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.2 |
| T 16 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.3 |
| T 17 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.4 |
| T 18 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.5 |
| T 19 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.7 |
| T 20 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.8 |
| T 21 | 0.34 | 1.3 | 0.05 | 5.59 | 0.39 | 0.50 | 0.9 |
A two-tissue reversible compartmental model with an additional single-tissue compartment for metabolites was used. K1, k2, and k3 are rate constants, Vb is fractional blood volume, and VT is volume of distribution for [18F]FDDNP. 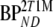 and
and 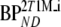 and are binding potentials estimated directly and indirectly using the metabolite model, respectively. The indirect calculation of BPND was performed using target to reference volumes of distribution ratios. K1m is a rate constant and VTm the volume of distribution for the metabolites. During all simulations, k4 (0.099 min−1) and K1/k2 (2.4) were fixed, and same, the K1m and k2m were used in reference and target tissues
and are binding potentials estimated directly and indirectly using the metabolite model, respectively. The indirect calculation of BPND was performed using target to reference volumes of distribution ratios. K1m is a rate constant and VTm the volume of distribution for the metabolites. During all simulations, k4 (0.099 min−1) and K1/k2 (2.4) were fixed, and same, the K1m and k2m were used in reference and target tissues
Simulated data were analyzed using 2T4k, 2T4ki, 2T1Mfvtm (i.e., 2T1M with VTm fixed to the simulated value), 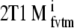 (i.e., indirect estimation through volumes of distribution), and SRTM. Bias and COV of binding potential obtained were compared between the various models. Bias for direct and indirect 2T1M models was defined in a similar way as for the standard models, i.e., using
(i.e., indirect estimation through volumes of distribution), and SRTM. Bias and COV of binding potential obtained were compared between the various models. Bias for direct and indirect 2T1M models was defined in a similar way as for the standard models, i.e., using 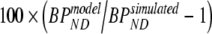 , where
, where 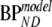 represents BPND estimated using the method of analysis under investigation and
represents BPND estimated using the method of analysis under investigation and 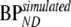 simulated BPND, set to either
simulated BPND, set to either 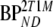 or
or 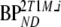 for direct and indirect models, respectively.
for direct and indirect models, respectively.
In addition to BPND, in metabolite simulations, also VT values were estimated using 2T4k and the 2T1Mfvtm models. Bias in estimated VT for the 2T4k model was defined as 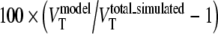 , where
, where  is
is 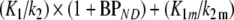 . Bias in estimated VT for the 2T1Mfvtm model was defined as
. Bias in estimated VT for the 2T1Mfvtm model was defined as 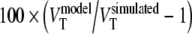 , where
, where 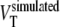 is (K1/k2) × (1 + BPND).
is (K1/k2) × (1 + BPND).
Results
Time–Activity Curves
Typical parent [18F]FDDNP and polar metabolite plasma curves are shown in Fig. 2a and typical time–activity curves for cerebellum and frontal cortex in Fig. 2b. The average (N = 5) fractions of [18F]-labeled metabolites in plasma as function of time are shown in Fig. 3, fitted using a Hill type function. Very rapid metabolism of [18F]FDDNP can be seen, with metabolite-related [18F] activity primarily due to polar metabolites (Fig. 3). The polar metabolites are composed of N-dealkylated fragments with similar chromatogram retention times as primarily fluoroethanal and with a smaller fractions of fluoroacetic acids [28].
Fig. 2.

Typical [18F]FDDNP decay corrected time activity curves from an AD subject for a metabolite-corrected parent compound and polar metabolites in plasma and b cerebellum and frontal cortex (ctx) gray matter.
Fig. 3.

Fraction of [18F]-labeled polar metabolites (dark dashed line) and parent compound (dark line) in plasma as function of time, averaged over seven subjects, together with standard deviations (dashed light gray lines at one SD from average).
Clinical Studies
Fig. 4 show typical fits using various compartmental models. In general, poor fits were seen for the 1T2k model (Fig. 4a). Metabolite models showed slightly better fits (Fig. 4b) than all conventional models (Fig. 4a). Good fits were seen for the reference tissue models (Fig. 4c). Similarly, the Akaike criterion gave preference to 2T1M models above other plasma input models (2T1Mfvtm 37.3%, 2T1M 31.7%, 2T4k 26.2%, 2T3k 4.8%, and 1T2k 0% preference, respectively). With respect to reference tissue models, the Akaike criterion had strong preference for SRTM (88%).
Fig. 4.

Decay corrected frontal cortex gray matter time–activity curve from an AD subject (=data) with fits using a conventional plasma input, b metabolite, and c reference tissue models.
Average values of the various parameters, together with observed range, for 2T4k, 2T1M, and SRTM models are given in Tables 3, 4, and 5 for typical AD regions, i.e., for regions previously shown to be involved in AD [8]. These regions were used in order to accurately estimate typical kinetic parameters for AD subjects, as the latter parameter estimates were used in the simulations.
Table 3.
Average (±SD) values of the kinetic parameters together with their range given between brackets for the reversible two-tissue compartment model
| Data | K1 | k2 | k3 | 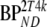 |
VT | 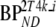 |
Vb |
|---|---|---|---|---|---|---|---|
| HC ctx | 0.25 ± 0.03 (0.2–0.3) | 0.06 ± 0.01 (0.04–0.08) | 0.02 ± 0.01 (0.01–0.05) | 2.14 ± 0.71 (1–4) | 14 ± 6 (7–25) | 0.22 ± 0.18 (−0.04–0.7) | 0.03 ± 0.01 (0.01–0.05) |
| AD ctx | 0.36 ± 0.08 (0.3–0.5) | 0.06 ± 0.01 (0.04–0.08) | 0.02 ± 0.01 (0.01–0.04) | 1.76 ± 0.71 (1–3) | 17 ± 5 (9–26) | 0.23 ± 0.20 (−0.03–0.6) | 0.05 ± 0.03 (0.02–0.11) |
| AD + HC cer | 0.35 ± 0.09 (0.3–0.5) | 0.07 ± 0.01 (0.06–0.09) | 0.02 ± 0.02 (0.01–0.06) | 1.67 ± 0.56 (1–3) | 13 ± 5 (6–20) | – | 0.03 ± 0.02 (0.01–0.07) |
These consisted of several typical AD cortical (ctx, i.e., orbital frontal cortex, medial inferior frontal cortex, anterior cingulate cortex, superior temporal cortex, parietal cortex, medial inferior temporal cortex, superior frontal cortex, enthorinal cortex, hippocampus) and cerebellar (cer) gray matter regions from healthy controls (HC) and AD subjects. K1 and K1m are given in ml·cm−3·min−1, k2 and k3 in min−1. 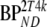 and
and 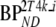 are binding potentials estimated directly and indirectly, respectively, using the reversible two-tissue plasma input model.
are binding potentials estimated directly and indirectly, respectively, using the reversible two-tissue plasma input model. 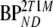 and
and 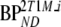 are binding potentials estimated directly and indirectly, respectively, using the metabolite model.
are binding potentials estimated directly and indirectly, respectively, using the metabolite model. 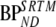 is the binding potential estimated using the simplified reference tissue model
is the binding potential estimated using the simplified reference tissue model
Table 4.
Average (±SD) values of the kinetic parameters together with their range given between brackets for the 2T1M metabolite model
| Data | K1 | k2 | k3 | 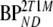 |
VT | 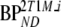 |
Vb | K1m | VTm |
|---|---|---|---|---|---|---|---|---|---|
| HC ctx | 0.28 ± 0.05 (0.2–0.4) | 0.33 ± 0.30 (0.09–0.68) | 1.06 ± 0.75 (0.01–2.43) | 3.07 ± 2.89 (0.4–11) | 3.70 ± 0.62 (3–5) | 0.03 ± 0.14 (−0.18–0.44) | 0.03 ± 0.01 (0.01–0.05) | 0.08 ± 0.10 (0.01–0.5) | 0.66 ± 0.07 (0.49–0.76) |
| AD ctx | 0.41 ± 0.10 (0.3–0.6) | 0.24 ± 0.15 (0.09–0.73) | 0.54 ± 0.50 (0.05–1.84) | 2.06 ± 1.65 (0.6–8) | 5.19 ± 0.86 (4–7) | 0.06 ± 0.11 (−0.2–0.3) | 0.04 ± 0.02 (0.01–0.09) | 0.16 ± 0.27 (0.03–0.93) | 0.65 ± 0.06 (0.55–0.78) |
| AD + HC cer | 0.39 ± 0.13 (0.3–0.6) | 0.28 ± 0.16 (0.10–0.53) | 0.49 ± 0.47 (0.04–1.30) | 1.89 ± 1.42 (0.5–4) | 4.24 ± 1.07 (3–6) | – | 0.02 ± 0.02 (0.01–0.05) | 0.34 ± 0.46 (0.02–1.00) | 0.60 ± 0.04 (0.53–0.67) |
These consisted of several typical AD cortical (ctx, i.e., orbital frontal cortex, medial inferior frontal cortex, anterior cingulate cortex, superior temporal cortex, parietal cortex, medial inferior temporal cortex, superior frontal cortex, enthorinal cortex, hippocampus) and cerebellar (cer) gray matter regions from healthy controls (HC) and AD subjects. K1 and K1m are given in ml·cm−3·min−1, k2 and k3 in min−1. 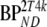 and
and 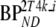 are binding potentials estimated directly and indirectly, respectively, using the reversible two-tissue plasma input model.
are binding potentials estimated directly and indirectly, respectively, using the reversible two-tissue plasma input model. 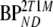 and
and 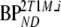 are binding potentials estimated directly and indirectly, respectively, using the metabolite model.
are binding potentials estimated directly and indirectly, respectively, using the metabolite model.  is the binding potential estimated using the simplified reference tissue model
is the binding potential estimated using the simplified reference tissue model
Table 5.
Average (±SD) values of the kinetic parameters together with their range given between brackets for the simplified reference tissue model, derived from clinical data
| Data | R1 | k2 | 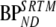 |
|---|---|---|---|
| HC ctx | 0.89 ± 0.06 (0.7–1.0) | 0.08 ± 0.02 (0.03–0.11) | 0.06 ± 0.04 (−0.07–0.12) |
| AD ctx | 0.87 ± 0.08 (0.7–1.0) | 0.06 ± 0.02 (0.03–0.11) | 0.07 ± 0.04 (0.00–0.14) |
Fig. 5 shows high correlation (R2 = 0.95) between K1m and k2m values from the 2T1M model over all regions and subjects, indicating an almost constant VT for metabolites. Table 6 shows correlation coefficients obtained from linear regression analyses of various outcome measures with 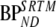 . As expected, high correlation was found between BPND estimates of SRTM and FRTM (R2 = 0.99). In addition, good correlation was obtained for SUVr over 40–60 min (R2 = 0.93). Note that the 2T1Mi models, i.e., including a compartment for metabolites entering the brain, correlated better with SRTM than the conventional 2T4ki model (Table 6 and Fig. 6).
. As expected, high correlation was found between BPND estimates of SRTM and FRTM (R2 = 0.99). In addition, good correlation was obtained for SUVr over 40–60 min (R2 = 0.93). Note that the 2T1Mi models, i.e., including a compartment for metabolites entering the brain, correlated better with SRTM than the conventional 2T4ki model (Table 6 and Fig. 6).
Fig. 5.

Correlation between metabolite influx (K1m) and efflux (k2m) rate constants for clinical [18F]FDDNP data from several regions of interests and subjects. These rate constants were obtained using a modified two-tissue reversible model, including an additional single-tissue compartment for metabolites.
Table 6.
Outcome parameters (slope, intercept, and Pearson product moment correlation coefficient R2) for linear regression of different models versus 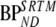
| Model | Slope | Intercept | R2 |
|---|---|---|---|
| FRTM | 0.97 | 0.01 | 0.99 |
| 2T4k | −4.11 | 2.21 | 0.15 |
| 2T4ki | NAa | NA | NA |
| 2T1M | NA | NA | NA |
| 2T1Mi | 2.66 | −0.11 | 0.68 |
| 2T1Mfvtm | NA | NA | NA |
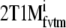 |
2.59 | −0.09 | 0.58 |
| SUVr40–60 | 1.14 | 0.03 | 0.93 |
| SUVr60–90 | 0.64 | 0.06 | 0.69 |
| SUVr80–90 | 0.46 | 0.06 | 0.43 |
In the case of SUVr methods, SUVr − 1 was taken as an estimate of BPND
aNA indicates poor correlation, i.e., R2 < 0.10
Fig. 6.

Correlation of BPND from 2T1Mifvtm and 2T4ki, respectively, with 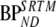 for clinical [18F]FDDNP data obtained from several regions of interest and subjects. 2T4ki represents the reversible two-tissue plasma input model and 2T1Mifvtm the modified reversible two-tissue model, which includes an additional parallel single-tissue compartment for metabolites. For both models, BPND was estimated indirectly through calculation of distribution volume ratios.
for clinical [18F]FDDNP data obtained from several regions of interest and subjects. 2T4ki represents the reversible two-tissue plasma input model and 2T1Mifvtm the modified reversible two-tissue model, which includes an additional parallel single-tissue compartment for metabolites. For both models, BPND was estimated indirectly through calculation of distribution volume ratios.
Simulation Studies
Conventional Simulations
In general, SRTM produced better fits than FRTM according to the Akaike criterion (∼85% preference). According to the same criterion, 2T4k showed better fits than both 1T2k and 2T3k in all simulations (∼96% preference). At lower K1 values, however, preference for 2T4k reduced in favor of 2T3k (max. 33% preference for 2T3k). Visual inspection showed poor fits for the 1T2k model, similar to what was seen in clinical fits, and therefore, it was excluded from further evaluation.
When simulating different levels of binding the 2T4k model (direct estimation of BPND) showed good overall accuracy (5% bias BPND), but mediocre precision (24% COV BPND). Results for reference tissue-based models, including SUVr, are summarized in Table 7. In general, all reference tissue-based methods showed increased bias at lower levels of binding, but results were more stable at higher levels. Therefore, bias estimates were averaged only over the stable part, i.e., for 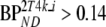 . In contrast, absolute differences with simulated
. In contrast, absolute differences with simulated 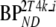 values were more stable and are shown over the full range of simulated
values were more stable and are shown over the full range of simulated 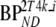 values (Table 7). Average bias (Table 7) over the stable range of
values (Table 7). Average bias (Table 7) over the stable range of 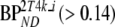 was lowest for indirect 2T4k (2T4ki). 2T4ki and SRTM both showed relatively constant bias and high precision for higher simulated BPND levels. Finally, best accuracy and precision for SUVr was obtained with SUVr40–60.
was lowest for indirect 2T4k (2T4ki). 2T4ki and SRTM both showed relatively constant bias and high precision for higher simulated BPND levels. Finally, best accuracy and precision for SUVr was obtained with SUVr40–60.
Table 7.
Average bias and absolute difference (diff.) with simulated BPND over a range of simulated 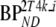 for various reference tissue models
for various reference tissue models
| Model | Bias (%) | SD in bias (%) | Diff. | SD in diff. |
|---|---|---|---|---|
| 2T4ki | 1 | 17 | 0.041 | 0.033 |
| SRTM | −12 | 16 | 0.053 | 0.062 |
| FRTM | −13 | 23 | 0.094 | 0.175 |
| SUVr40–60 | −1 | 14 | 0.034 | 0.026 |
| SUVr60–90 | 16 | 15 | 0.054 | 0.037 |
| SUVr80–90 | 22 | 33 | 0.095 | 0.072 |
In addition, corresponding SD are given. In the case of SUVr methods, SUVr − 1 was taken as an estimate of BPND. For bias, the range was set from 0.25 to 0.48 due to the large relative biases at lower specific binding. For absolute difference, the range was set from 0.03 to 0.48
All models were less sensitive to changes in Vb. At a fixed level of binding (BPND = 2.3, 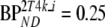 ), variations in derived BPND due to changes in simulated Vb were smaller than 5% for all compartmental models. For SUVr methods, bias (±COV) in approximated BPND (i.e., SUVr − 1) depended on the actual time interval used and changed from 21 ± 16% to 10 ± 18%, from 30 ± 22% to 28 ± 21%, and from 31 ± 41% to 34 ± 45% for SUVr40–60, SUVr60–90, and SUVr80–90, respectively, when varying Vb of the target region from 0.025 to 0.075.
), variations in derived BPND due to changes in simulated Vb were smaller than 5% for all compartmental models. For SUVr methods, bias (±COV) in approximated BPND (i.e., SUVr − 1) depended on the actual time interval used and changed from 21 ± 16% to 10 ± 18%, from 30 ± 22% to 28 ± 21%, and from 31 ± 41% to 34 ± 45% for SUVr40–60, SUVr60–90, and SUVr80–90, respectively, when varying Vb of the target region from 0.025 to 0.075.
Delivery differences affected all compartmental methods, including plasma input models. For the direct 2T4k model, bias in BPND varied from 10% to −2% when varying K1 of the target region from 0.212 to 0.496. Results for indirect 2T4k and SRTM are shown in Fig. 7. Results from FRTM are not included, as BPND bias was as high as 250%. Bias in BPND (±COV), based on SUVr − 1, ranged from −27 ± 18% to 3 ± 17%, from 25 ± 20% to −3 ± 18%, and from 48 ± 36% to −7 ± 39% for SUVr40–60, SUVr60–90, and SUVr80–90, respectively, when varying K1 of the target region from 0.212 to 0.496.
Fig. 7.

Bias of BPND for different compartment models for simulated [18F]FDDNP data (obtained using 2T4k) as function of variable target influx rates (K1 target). Error bars represent SD in bias. K1 in reference region was fixed to 0.35, and K1/k2 was fixed to 3.3 for both target and reference regions. 2T4ki and SRTM represent indirect (using volumes of distribution) two-tissue reversible and simplified reference tissue models, respectively.
Metabolite Simulations
Metabolite simulations were limited to 2T4k, 2T1Mfvtm, and SRTM. SRTM provided the most precise estimation of BPND over the range of simulated K1m with a COV of 4.0 ± 0.4%. Although there was a strong negative bias (−42 ± 7%) for SRTM, it was very constant (Fig. 8a). 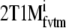 provided a more accurate estimate of BPND (5 ± 7% bias), but precision (COV of 37 ± 20%) was much poorer than for SRTM (Fig. 8a). 2T1Mfvtm, 2T4k, and 2T4ki provided lower precision and higher bias in estimated BPND over the range of simulated K1m (Fig. 8a, b). However, VT estimates with the 2T1Mfvtm model were much more accurate and had higher precision (Fig. 8c).
provided a more accurate estimate of BPND (5 ± 7% bias), but precision (COV of 37 ± 20%) was much poorer than for SRTM (Fig. 8a). 2T1Mfvtm, 2T4k, and 2T4ki provided lower precision and higher bias in estimated BPND over the range of simulated K1m (Fig. 8a, b). However, VT estimates with the 2T1Mfvtm model were much more accurate and had higher precision (Fig. 8c).
Fig. 8.

Bias of a indirect and reference tissue model BPND, b direct BPND, and cVT estimates from different compartment models. Error bars represent SD in bias. Simulated [18F]FDDNP data were generated using the metabolite plasma input model (2T1M) with variable metabolite influx rates (K1m).
Simulations over a range of different binding levels showed strong negative bias (−46 ± 4% with COV of 7 ± 6%) for SRTM, but again, resulting BPND estimates were the most stable amongst all models tested (Fig. 9a). Similarly, most accurate results were obtained with indirect 2T1Mfvtm (Fig. 9a). Precision of both SRTM and  improved with increasing BPND. BPND estimates obtained with 2T1Mfvtm, 2T4k, and 2T4ki were not precise and strongly biased (Fig. 9). Finally, Fig. 10a shows nearly linearly increasing (negative) bias in BPND obtained with SRTM for increasing VTm. In contrast, Fig. 10b illustrates that this bias was independent of K1m for K1m > 0.1.
improved with increasing BPND. BPND estimates obtained with 2T1Mfvtm, 2T4k, and 2T4ki were not precise and strongly biased (Fig. 9). Finally, Fig. 10a shows nearly linearly increasing (negative) bias in BPND obtained with SRTM for increasing VTm. In contrast, Fig. 10b illustrates that this bias was independent of K1m for K1m > 0.1.
Fig. 9.

Bias of a indirect (and reference tissue) and b direct BPND estimates from different compartment models for simulated [18F]FDDNP data generated using the metabolite plasma input model (2T1M) with various target tissue binding potentials. Error bars represent SD in bias.
Fig. 10.

Bias of BPND estimated with the simplified reference tissue model (SRTM) for simulated [18F]FDDNP data generated using the metabolite plasma input model (2T1M) as function of a various VTm values and b various metabolite influx rates (K1m). Error bars represent SD in bias.
Discussion
Clinical Studies
Due to rapid plasma clearance and metabolism [18F]FDDNP scans, it proved to be difficult to obtain reliable measurements of metabolite fractions at later time points. As a result, only incomplete plasma data were available for six of the subjects, and data from these subjects were only used for evaluation of reference tissue models. Although only six subjects with complete plasma input data remained, this seems to be sufficient for this initial evaluation of quantitative (plasma input) models. Clearly, however, the difficulties in obtaining reliable plasma metabolite data should be taken into account when selecting a method for routine clinical studies.
Clinical [18F]FDDNP data were analyzed using conventional models, models correcting for possible metabolites entering the brain, and activity ratios. Clearly, due to the very nature of clinical data, absolute statements about accuracy and precision cannot be given. Therefore, the various methods were compared using the Akaike criterion. In addition, resulting BPND values were compared with SRTM, as this model showed stable results during simulations.
On the basis of the Akaike criterion, plasma input models with a compartment for metabolites were preferred over conventional plasma input models, and SRTM was preferred over FRTM. Good correlations with SRTM were obtained only for FRTM, SUVr40–60, and plasma input models that included a compartment for metabolites entering the brain, such as 2T1Mi and 2T1Mifvtm (Fig. 6). For most other methods, correlations with SRTM were poor (R2 < 0.1).
Specifically, in the case of the metabolite models, somewhat large values were seen for the micro parameters K1m and k2m (Fig. 5). However, the majority of observed K1m values are below 0.5 (many overlapping data at lower K1m), and K1m and k2m are well correlated. The higher K1m and k2m values are possibly the result of noise, as the higher values for K1m and k2m were generally seen for the smallest ROIs used. Moreover, the relative slow in-grow of metabolites, i.e., the metabolite input function shows a slow increase rather than a sharp peak, makes estimation of individual micro-parameters less precise. Note, however, that the macro-parameter VTm is reasonably stable across anatomical regions and subjects.
Observed improvements in fits and correlations for models that incorporate a labeled metabolite compartment, together with the reasonably constant VTm, suggest that metabolites enter the brain. This is in line with a previous assessment using multi-input spectral analysis [27] and initial animal studies where labeled metabolites of [18F]FDDNP were injected [28]. Additional studies are, however, needed to confirm whether this is indeed the case.
Simulation Studies
The standard 2T4k plasma input model was not a good model for estimating the level of specific [18F]FDDNP binding directly (i.e., as defined by k3/k4). Firstly, although BPND bias calculated relative to total binding potential showed good accuracy, it only had mediocre precision. Secondly, at lower regional delivery (=K1) levels, its bias was sensitive to the actual value of K1. This increased sensitivity to K1 is related to the kinetics of [18F]FDDNP, which, for a normal level of binding in regions with low delivery, are relatively slow. Under those conditions, within the time frame of a scan, tracer kinetics are best described by an irreversible compartment model, which is illustrated by the increased preference for the 2T3k model according to the Akaike criterion.
The 2T1Mfvtm model did not provide accurate estimates of (direct) BPND in the metabolite simulations (Figs. 8b and 9b). Although data were simulated using the same model, BPND was very sensitive to noise as a result of the large number of fit parameters. However, VT estimates obtained with the 2T1Mfvtm model were less sensitive to noise and provided highly accurate and precise VT values (Fig. 8c).
For plasma input models, more accurate and precise results were obtained with indirect estimates of BPND (2T4ki and 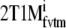 ) than with their direct counterparts (2T4k and 2T1Mfvtm), especially in the case of metabolite simulations. In most cases, these indirect methods showed lower bias than
) than with their direct counterparts (2T4k and 2T1Mfvtm), especially in the case of metabolite simulations. In most cases, these indirect methods showed lower bias than 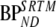 . This higher accuracy of 2T4ki (in conventional simulations) and
. This higher accuracy of 2T4ki (in conventional simulations) and  (in metabolite simulations) was, however, accompanied with lower precision compared with
(in metabolite simulations) was, however, accompanied with lower precision compared with 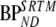 .
.
SRTM was better than FRTM, both in terms of bias and COV. In addition, in the majority of the simulations, it was preferred by the Akaike criterion. SRTM results were comparable in conventional and metabolite simulations. If no metabolites entered the brain, SRTM-derived BPND showed a constant negative bias, although it was affected by changes in delivery. On the other hand, when polar metabolites entered the brain, SRTM showed increasing negative bias with increasing VTm. However, clinical data showed an overall constant VTm (Fig. 5). This indicates that variable bias in SRTM-derived BPND due to variability in VTm will be limited. Simulations showed that for VTm fixed to the clinical estimated value (VTm = 0.6), this bias also was not sensitive to metabolite influx (Fig. 10b, K1m > 0.01, BPND bias ∼−46%). Furthermore, the bias found in these simulations could be related to the slope between 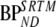 versus
versus 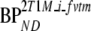 (slope = 2.59, Table 6; 100% × (1/2.59) = 39%) for the clinical studies, indicating a relatively constant metabolite contribution in practice. Thus, SRTM seems to be a useful method, irrespective whether labeled metabolites enter the brain, provided that VTm is relatively constant across the brain and between subjects, as actually suggested by the clinical results.
(slope = 2.59, Table 6; 100% × (1/2.59) = 39%) for the clinical studies, indicating a relatively constant metabolite contribution in practice. Thus, SRTM seems to be a useful method, irrespective whether labeled metabolites enter the brain, provided that VTm is relatively constant across the brain and between subjects, as actually suggested by the clinical results.
Among SUVr methods, over the range of simulated binding levels, only SUVr40–60 showed acceptable results, with much better accuracy (−1%) than SRTM (−12%). However, apart from a strong dependency on variations in delivery, SUVr results were also highly dependent on differences in blood volume fractions with only an acceptable accuracy for a specific time interval (SUVr60–90). In general, SUVr results varied widely between different time intervals which, together with the large uncertainties, indicate that this might not be the ideal method for clinical studies.
In general, SRTM showed best overall performance, although it showed some sensitivity to regional flow/delivery differences. As expected, SRTM, like all other reference tissue-based models, showed larger biases at lower regional binding levels. Important advantages of SRTM were its overall high precision and a relatively constant bias in BPND even in the presence of labeled metabolites. Although this bias was dependent on the actual value of VTm, clinical data indicated that this was constant across the brain and among subjects.
General Considerations
Amyloid plaques (and neurofibrillary tangles) do not behave like neuroreceptors. They have complex structures with multiple affinity sites for [18F]FDDNP. In addition, it is likely that access to amyloid sites will become more difficult with increasing plaque size [30]. Consequently, use of receptor-ligand models to assess amyloid load might provide an increasing underestimation with increasing load. In addition, quantification might be compromised by the possibility of labeled metabolites entering the brain, especially since the fraction of labeled (polar) metabolites in blood is high due to rapid metabolism of [18F]FDDNP.
Nevertheless, [18F]FDDNP may still be of value if a difference between MCI and healthy controls can be demonstrated, and it might be used to study pathological effects as a function of disease severity/progression [8]. In addition, at early stages, a stable receptor-ligand model is likely to provide a reasonable quantitative estimate of the pathological changes associated with AD and could therefore be an important tool in evaluating effects of therapy. Clearly, given its relatively small specific signal, the value of [18F]FDDNP can only be determined by large comparative clinical studies, such as the one by Small et al. [8]. From the present study, it follows that such a comparison should preferably be performed using SRTM as method of analysis, as it showed high precision and constant bias across the brain and among subjects even when metabolites enter the brain.
Conclusion
SRTM outperformed other models, providing BPND estimates with high precision. As clinical data indicated that the volume of distribution of labeled metabolites VTm was constant across the brain and among subjects, bias in SRTM-derived BPND will be constant and predictable. The main potential drawback is its sensitivity to variations in regional K1, although this did not appear to be a problem in the clinical studies.
Acknowledgments
The authors would like to thank the personnel of the Department of Nuclear Medicine & PET Research for tracer production and data acquisition. The authors would also like to thank the “American Health Assistance Foundation” (grant A2005-026), the “Internationale Stichting Alzheimer Research” (grant no. 05512), and the Netherlands Foundation of Scientific research (VIDI grant 016.066.309) for financial support.
Open Acces This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Barrio JR, Huang SC, Cole GM et al (1999) PET imaging of tangles and plaques in Alzheimer disease. J Nucl Med 40(Suppl. S):70P–71P (Abstract) 9935060
- 2.Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357 [DOI] [PubMed]
- 3.Shoghi-Jadid K, Small GW, Agdeppa ED (2002) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10:24–35 [PubMed]
- 4.Kepe V, Shoghi-Jadid K, Wu HM et al (2004) Global and regional [F-18]FDDNP binding as in vivo measure of Alzheimer’s disease. J Nucl Med 45(Suppl.):126–126
- 5.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL (1996) Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16:834–840 [DOI] [PubMed]
- 6.Wong KP, Kepe V, Small GW, Satyamurthy N, Barrio JR, Huang SC (2007) Comparison of simplified methods for quantitative analysis of [18F]FDDNP PET data. IEEE NSS-MIC, vol 4, pp 3146–3150 (Conference Proceeding)
- 7.Kepe V, Huang SC, Small GW, Satyamurthy N, Barrio JR (2006) Visualizing pathology deposits in the living brain of patients with Alzheimer’s disease. Methods Enzymol 412:144–160 [DOI] [PubMed]
- 8.Small GW, Kepe V, Ercoli LM et al (2006) PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med 355:2652–2663 [DOI] [PubMed]
- 9.Gunn RN, Gunn SR, Cunningham VJ (2001) Positron emission tomography compartmental models. J Cereb Blood Flow Metab 21:635–652 [DOI] [PubMed]
- 10.Hume SP, Myers R, Bloomfield PM et al (1992) Quantitation of carbon-11-labeled raclopride in rat striatum using positron emission tomography. Synapse 12:47–54 [DOI] [PubMed]
- 11.Lammertsma AA, Bench CJ, Hume SP et al (1996) Comparison of methods for analysis of clinical [C-11]raclopride studies. J Cereb Blood Flow Metab 16:42–52 [DOI] [PubMed]
- 12.Lammertsma AA, Hume SP (1996) Simplified reference tissue model for PET receptor studies. Neuroimage 4:153–158 [DOI] [PubMed]
- 13.Petersen RC, Doody R, Kurz A et al (2001) Current concepts in mild cognitive impairment. Arch Neurol 58:1985–1992 [DOI] [PubMed]
- 14.Mckhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical-diagnosis of Alzheimer’s disease: report of the Nincds-Adrda Work Group under the auspices of Department of Health and Human-Services Task Force on Alzheimers Disease. Neurology 34:939–944 [DOI] [PubMed]
- 15.Brix G, Zaers J, Adam LE et al (1997) Performance evaluation of a whole-body PET scanner using the NEMA protocol. J Nucl Med 38:1614–1623 [PubMed]
- 16.Klok RP, Klein PJ, van Berckel BN, Tolboom N, Lammertsma AA, Windhorst AD (2008) Synthesis of 2-(1,1-dicyanopropen-2-yl)-6-(2-[18F]-fluoroethyl)-methylamino-naphthalene ([18F]FDDNP). Appl Radiat Isot 66:203–207 [DOI] [PubMed]
- 17.Defrise M, Kinahan PE, Townsend DW, Michel C, Sibomana M, Newport DF (1997) Exact and approximate rebinning algorithms for 3-D PET data. IEEE Trans Med Imaging 16:145–158 [DOI] [PubMed]
- 18.Watson CC (2000) New, faster, image-based scatter correction for 3D PET. IEEE Trans Nucl Scien 47:1587–1594 [DOI]
- 19.Boellaard R, van Lingen A, van Balen SCM, Hoving BG, Lammertsma AA (2001) Characteristics of a new fully programmable blood sampling device for monitoring blood radioactivity during PET. Eur J Nucl Med 28:81–89 [DOI] [PubMed]
- 20.Greuter HNJM, van Ophemert PLB, Luurtsema G, Franssen EJF, Boellaard R, Lammertsma AA (2004) Validation of a multiwell gamma-counter for measuring high-pressure liquid chromatography metabolite profiles. J Nucl Med Technol 32:28–32 [PubMed]
- 21.Takkenkamp K, Luurtsema G, Greuter HN et al (2007) Plasma metabolites of [11C]PIB and [18F]FDDNP. J Label Comp Radiopharm 50(S1):S474 (Abstract)
- 22.Gunn RN, Sargent PA, Bench CJ et al (1998) Tracer kinetic modeling of the 5-HT1A receptor ligand [carbonyl-11C]WAY-100635 for PET. Neuroimage 8:426–440 [DOI] [PubMed]
- 23.Smith SM (2002) Fast robust automated brain extraction. Hum Brain Map 17:143–155 [DOI] [PMC free article] [PubMed]
- 24.Maes F, Collignon A, Vandermeulen D, Marchal G, Suetens P (1997) Multimodality image registration by maximization of mutual information. IEEE Trans Med Imaging 16:187–198 [DOI] [PubMed]
- 25.West J, Fitzpatrick JM, Wang MY et al (1997) Comparison and evaluation of retrospective intermodality brain image registration techniques. J Comput Assist Tomogr 21:554–566 [DOI] [PubMed]
- 26.Svarer C, Madsen K, Hasselbalch SG et al (2005) MR-based automatic delineation of volumes of interest in human brain PET images using probability maps. Neuroimage 24:969–979 [DOI] [PubMed]
- 27.Lubberink M, van Berckel BNM, Luurtsema G et al (2007) Multi-input spectral analysis for assessing cerebral uptake of labelled metabolites: validation and application to [11C]PIB and [18F]FDDNP studies. J Cereb Blood Flow Metab 27. BrainPET07 Abstract PS1-5M (Abstract)
- 28.Luurtsema G, Schuit RC, Takkenkamp K et al (2008) Peripheral metabolism of [18F]FDDNP and cerebral uptake of its labelled metabolites. Nucl Med Biol 35:869–874 [DOI] [PubMed]
- 29.Yaqub M, Boellaard R, Kropholler MA, Lammertsma AA (2006) Optimization algorithms and weighting factors for analysis of dynamic PET studies. Phys Med Biol 51:4217–4232 [DOI] [PubMed]
- 30.Shoghi-Jadid K, Barrio JR, Kepe V, Huang SC (2006) Exploring a mathematical model for the kinetics of beta-amyloid molecular imaging probes through a critical analysis of plaque pathology. Mol Imaging Biol 8:151–162 [DOI] [PubMed]