

Abstract
KCNE1 encodes the β-subunit of the slow component of the delayed rectifier K+ current. The Jervell and Lange-Nielsen syndrome is characterized by sensorineural deafness, prolonged QT intervals, and ventricular arrhythmogenicity. Loss-of-function mutations in KCNE1 are implicated in the JLN2 subtype. We recorded left ventricular epicardial and endocardial monophasic action potentials (MAPs) in intact, Langendorff-perfused mouse hearts. KCNE1−/− but not wild-type (WT) hearts showed not only triggered activity and spontaneous ventricular tachycardia (VT), but also VT provoked by programmed electrical stimulation. The presence or absence of VT was related to the following set of criteria for re-entrant excitation for the first time in KCNE1−/− hearts: Quantification of APD90, the MAP duration at 90% repolarization, demonstrated alterations in (1) the difference, ∆APD90, between endocardial and epicardial APD90 and (2) critical intervals for local re-excitation, given by differences between APD90 and ventricular effective refractory period, reflecting spatial re-entrant substrate. Temporal re-entrant substrate was reflected in (3) increased APD90 alternans, through a range of pacing rates, and (4) steeper epicardial and endocardial APD90 restitution curves determined with a dynamic pacing protocol. (5) Nicorandil (20 µM) rescued spontaneous and provoked arrhythmogenic phenomena in KCNE1−/− hearts. WTs remained nonarrhythmogenic. Nicorandil correspondingly restored parameters representing re-entrant criteria in KCNE1−/− hearts toward values found in untreated WTs. It shifted such values in WT hearts in similar directions. Together, these findings directly implicate triggered electrical activity and spatial and temporal re-entrant mechanisms in the arrhythmogenesis observed in KCNE1−/− hearts.
Keywords: Mice, Action potential duration, Ventricular arrhythmogenesis, Re-entry
Introduction
Ventricular tachycardia (VT) is an important cause of morbidity and mortality accounting for 80% of the annual 50,000–70,000 cases of sudden cardiac death (SCD) in the UK [36] and over 300,000–400,000 cases in the USA [77]. A subset of these deaths is caused by an inherited predisposition to VT of which congenital long QT syndrome (LQTS) forms an important genetically transmitted category with a prevalence recently estimated at one in 2,500 live births [8].
LQTS patients have been classified according to their mode of transmission into the autosomal recessive Jervell and Lange-Nielsen (JLN) syndrome [20] or the autosomal dominant Romano–Ward (RW) syndrome [43, 71]. Jervell and Lange-Nielsen in 1957 provided the first report of LQTS in their description of a Norwegian family with four out of six children having deaf-mutism, prolonged electrocardiographic QT intervals, syncopal episodes, and sudden death. JLN and RW syndromes are thus additionally differentiated phenotypically by the congenital bilateral sensorineural deafness that afflicts patients with JLN syndrome but not RW syndrome [20, 43, 71]. Since the first report of LQTS, a wide range of LQTS subtypes have been discovered (reviewed by [6]). All of these affect ion channels or proteins that interact with ion channels, predisposing patients to potentially lethal ventricular arrhythmias, syncope, SCD, characteristic prolonged electrocardiographic QT intervals, and in some cases, T wave abnormalities [8, 54]. Such conditions provide opportunities to study mechanisms of arrhythmia resulting from defined genetic abnormalities.
The KCNE1 gene encodes a 129-amino acid protein, minK, an accessory β-subunit that, together with KvLQT1 α-subunits (encoded by the KCNQ1 gene), form the slow component [5, 49] of the delayed rectifier K+ current in cardiac myocytes [37, 38]. Autosomal dominant heterozygous mutations in the KCNE1 gene may result in the long QT 5 syndrome [62]. More marked reductions in IKs resulting from homozygous or compound heterozygous mutations in KCNQ1 or KCNE1 can give rise to the characteristic JLN syndrome [35, 53, 61, 67]. JLN syndrome has, therefore, been classified into JLN1 and JLN2, distinguishing between associated mutations in KCNQ1 or KCNE1, respectively [6]. Loss-of-function mutations in KCNE1, which may give rise to LQT5 or JLN2 are thus associated with reduced K+ currents and compromised repolarization reserve.
Further experimental studies exploring for mechanisms of arrhythmogenesis and potential therapeutic approaches in JLN syndrome, with due attention to the gene in which the primary genetic abnormality resides, are thus of significant interest. Such gene-specific investigations of JLN syndrome at the experimental level are warranted, firstly, in view of the fact that the JLN1 and JLN2 variants, both particularly severe forms of LQTS [56], while clinically similar [67], have mutations in distinct genes. Secondly, JLN syndrome is rare, with an estimated incidence of 1.6 to six cases per million [10]. Thirdly, besides involving β-subunits rather than α-subunits of the channels concerned, KCNE1−/− mutations accounted for 9.5% of patients with JLN syndrome that could be successfully genotyped in a recent clinical review [56]. There is, therefore, limited knowledge about the mechanisms involved and possible therapeutic approaches in both JLN syndrome and JLN2 in particular. Finally, there are potential therapeutic implications for studying JLN syndrome according to the underlying genetic abnormality in view of a range of treatment options for LQTS, including pharmacological approaches such as β-blockers, as well as pacemakers, left cervicothoracic sympathectomy, and implantable cardiac defibrillators [42]. Nevertheless, as many as 51% of patients with JLN syndrome experience arrhythmic events despite current treatment options [56]. Together with current suggestions for genotype-targeted therapy [33], these considerations warrant correspondingly genotype-targeted investigations of arrhythmogenic mechanisms in the JLN variants.
Mouse models have previously provided good paradigms for a variety of genetic cardiac arrhythmia conditions [27]. They have also permitted systematic identification and separation of the contrasting contributions made by ectopic and re-entrant arrhythmogenic mechanisms, as well as spatial and temporal components contributing to re-entry [16, 17, 23, 25, 65]. A mouse model of JLN2 has been developed previously by targeted homozygous deletion of the KCNE1 gene [68]. This model replicates the primary abnormalities observed in the human JLN syndrome in showing inner ear defects [9] manifesting as shaker/waltzer movement disorders, as well as an increased tendency to ventricular arrhythmias compared to wild-type (WT) hearts [4, 66].
The present study accordingly examined potential transmural mechanisms for ventricular arrhythmogenesis in this mouse model of JLN2 at the isolated whole organ level. We made monophasic action potential (MAP) recordings for quantitative analyses of changes in action potential waveforms in isolated whole hearts. This thus complemented previous electrocardiographic studies in intact animals [9] in allowing for assessments of intrinsic cardiac properties independent of autonomic factors while preserving intercellular coupling and spatial electrophysiological characteristics. This permitted recordings of localized epicardial and endocardial waveforms in contrast to electrocardiographic studies that would have only represented voltage differences summed across the entire myocardium. Thus, it was possible to apply to KCNE1−/− hearts experimental tests applied on a previous occasion in a murine model for LQT3 [17]. This made it possible to identify tendencies for spontaneous arrhythmogenicity manifesting as triggered activity and spontaneous VT, as well as assessing for provoked arrhythmogenic tendencies [65]. It also made it possible to systematically apply and quantify criteria for spatial re-entrant substrates [3] in the form of altered ∆APD90 [25, 64] and local critical intervals for re-excitation [44]. Finally, we could quantify criteria for temporal substrates [72, 73] through a description of APD90 alternans and APD90 restitution properties [16, 45].
It was possible next to assess the effect of pharmacological manipulations on all of these criteria as well as upon the observed arrhythmogenicity. Such experiments involved comparison of these features in both KCNE1−/− and WT hearts in the presence or absence of nicorandil. Preliminary clinical reports had suggested that nicorandil may be antiarrhythmic in clinical LQTS [1, 13, 50, 58], though this has not been investigated in the setting of LQTS specifically due to KCNE1 mutations. Similar suggestions have also arisen from animal studies using experimental models of LQT1, LQT2, and LQT3 in the canine wedge [57], LQT1 in the guinea pig [76], and in a LQT model induced by disopyramide pretreatment [12] in all of which repolarization had been delayed by pharmacological manipulations but have not been investigated in any arrhythmogenic model based upon KCNE1 abnormalities.
An assessment of the effects of nicorandil in a model for JLN2 is important as pharmacological agents that are antiarrhythmic in one particular LQTS subtype may not act similarly in other subtypes [55]. Nicorandil could potentially offset the delayed repolarization resulting from reduced delayed K+ current observed in JLN in view of previous descriptions of nicorandil-mediated reductions in myocardial APD90 in different cardiac preparations. The concentrations of nicorandil used in this study (100 nM to 20 µM) were based on established literature that has demonstrated such empirical actions of nicorandil in a variety of cardiac preparations. This may be through actions providing an enhanced repolarization reserve through increased K+ conductance [18, 22, 57, 60]. Nicorandil causes significant APD90 shortening at the cellular level at concentrations of 10 µM in left atrial muscle fibres [74], 1, 10, 30, and 100 µM in Purkinje fibres [18, 31, 75], and 10 and 100 µM in isolated guinea pig and rabbit ventricular myocytes [15, 22] independently of external [Na+] [18], and in the absence of alterations in Ca2+ currents [15, 22], maximum upstrokes of the action potential [18, 22] and maximum diastolic membrane potentials ([15, 18, 31] but see [74]). It also shortens APD90 at the tissue [57, 76] and whole organ [26] levels at 2 to 20 µM in canine LQT1, LQT2, and LQT3 pharmacological models [57], at 5 to 30 µM in guinea pig hearts in the absence or presence of chromanol 293B [76], at 20 µM in murine hypokalaemic hearts [26], and at 10 to 20 µM in murine Scn5a+/∆KPQ hearts modeling LQT3 [17].
The present findings establish empirically for the first time that nicorandil may have a useful antiarrhythmic role as reflected in its suppression of triggered activity, spontaneous VT, and provoked VT. They also demonstrate a parallel modification of recently introduced quantitative indicators of both spatial and temporal re-entrant substrates [16]. In so doing, these studies also demonstrate an existence of abnormal restitution properties in the KCNE1−/− system for the first time. Together, these findings implicate such features in the arrhythmogenic properties observed in KCNE1−/− hearts.
Materials and methods
Experimental animals
KCNE1−/− mice were supplied by Vetter et al. [68] and inbred upon the 129/Sv background. Breeding pairs of homozygotes and WTs were established and gave rise to lines of KCNE1−/− and WT animals. Mice were housed at 21 ± 1°C with 12 h light/dark cycles. Mice were fed sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and had free access to water at all times. Randomly selected male and female mice aged 5–7 months were used for experiments. All procedures conformed to the UK Animals (Scientific Procedures) Act 1986.
Heart isolation
A Langendorff-perfusion technique was used as previously described [14]. Mice were killed by cervical dislocation (Schedule 1: Animals (Scientific Procedures) Act 1986). Hearts were exposed by bilateral sternotomy, rapidly excised, and a small section (3–4 mm) of aorta was identified and cannulated in ice-cold, Krebs–Henseleit solution using a custom-made 21-gauge cannula and secured using an aneurysm clip (Harvard Apparatus, Edenbridge, Kent, UK). Krebs–Henseleit solution consisted of (in mM): NaCl 119, NaHCO3 25, KCl 4, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 10, and Na pyruvate 2, pH 7.4, bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, UK). The isolated hearts were then transferred within 120 s from excision to the Langendorff-perfusion system. They were perfused retrogradely with Krebs–Henseleit solution via the coronary arteries at a flow rate of 2.0–2.5 mL/min using a peristaltic pump (Watson-Marlow Bredel Model 505S, Falmouth, Cornwall, UK). The perfusate had been passed through two, 200- and 5-mm, filters (Millipore, Watford, UK) and then heated to 37°C by a water-jacketed heat-exchange coil and circulator (Ecolin Staredition E100, Lauda, Lauda-Königshofen, Germany). Healthy hearts regained a healthy pink colour and contracted spontaneously. Approximately 10% of hearts were discarded due to signs of ischaemia.
Perfused heart electrophysiology
MAPs were recorded from the epicardial and endocardial surfaces of the left ventricular free wall. Epicardial recordings were made with a MAP electrode (Linton Instruments, Harvard Apparatus, UK) designed for rodent hearts and positioned against the basal left ventricular epicardial surface. Endocardial recordings were made using a custom-made MAP electrode constructed from Teflon-coated silver wire of 0.25 mm diameter (Advent Research Materials, UK) and 99.99% purity. The final 1 mm of Teflon coating was removed from the electrode tips, which were then galvanically chlorided to avoid DC offset. An aperture was carefully made in the interventricular septum to allow the electrode to be positioned against the left ventricular endocardial free wall. MAP waveforms were quantified to determine APDx, the time taken for repolarization to x%, thus permitting the quantification of APD30, APD50, APD70, and APD90. MAPs met previously established criteria including a rapid upstroke, smooth contoured repolarization phase, and a stable baseline [28]. Peak depolarization (0% repolarization) was considered to be the most positive point of deflection above the baseline, while 100% repolarization was considered to be the point of return to baseline [25]. MAP signals were preamplified with a NL100AK head stage and band pass filtered (0.5 Hz to 1 kHz; Neurolog NL 125/6 Filter, Neurolog, Hertfordshire, UK). A micro 1401plus MKII laboratory interface (Cambridge Electronic Design, Cambridge, UK) was used to digitize these signals at a sampling frequency of 5 kHz. Spike2 software (Cambridge Electronic Design, Cambridge, UK) was used to analyze MAP waveforms.
Experimental protocol
Hearts were stimulated from the basal right ventricular epicardium with a bipolar platinum electrode with 1 mm interpole distance. Square-wave stimuli (Grass S48 Stimulator, Grass-Telefactor, Slough, UK) of 2 ms duration and three times the diastolic threshold [25] were applied at a basic cycle length (BCL) of 125 ms corresponding to murine whole heart physiological heart rates [41]. Hearts were initially paced for at least 10 min at a constant BCL of 125 ms to reach a steady-state. Following this initial equilibration period, both spontaneous and provoked arrhythmogenesis were assessed in the absence and presence of nicorandil. VT was defined as five or more consecutive premature MAPs [25]. Spontaneous arrhythmogenic properties were assessed in the absence of extrinsic pacing. Provoked arrhythmogenesis was assessed using a programmed electrical stimulation (PES) method adapted from clinical practice [51] and previously applied to murine hearts [14]. The PES protocol consisted of cycles of 8 S1 stimuli applied at a BCL of 125 ms, followed by a S2 extrastimulus. The S1-S2 stimulus interval began at 125 ms and decreased by 1 ms with each successive cycle of eight S1 stimuli. This extrasystolic protocol also enabled the quantification of ventricular effective refractory periods (VERPs) from the longest S1–S2 interval that failed to elicit a MAP. Action potential durations (APDs) and stimulus-depolarization conduction latencies were quantified during steady-state pacing at a 125 ms BCL [25, 64]. A dynamic pacing protocol (see the “Results” section) was used to assess temporal properties allowing the examination of APD90 alternans and APD90 restitution properties.
Pharmacological agents
Drug-containing solutions were made by adding nicorandil (Tocris Bioscience, UK) from 10 mM stock solutions made up in doubly distilled water, to Krebs–Henseleit solution, and serially diluted to the target concentrations (100 nM, 1 µM, 10 µM, and 20 µM).
Data analysis and statistics
Data are presented as the means±standard error of the means together with the number of hearts (n). Categorical data were compared using Fisher’s exact test (one-tailed), and continuous data were compared using analysis of variance (SPSS software) and Student’s unpaired t test as appropriate. P values less than 0.05 were considered significant. Curve fitting to experimental restitution data sets was performed using a Levenberg–Marquardt algorithm (Origin 8, OriginLab, MA, USA).
Results
Langendorff preparations of murine hearts were initially perfused using control Krebs–Henseleit solution and MAP recordings were then made following 10 min of such perfusion. Recordings made in the presence of nicorandil were similarly made following 10 min of perfusion with nicorandil-containing Krebs–Henseleit solution. Hearts were first investigated during intrinsic activity for assessment of spontaneous arrhythmogenic properties. They were then investigated for provoked arrhythmogenesis using a PES protocol [4]. MAP waveforms were recorded and assessed from the left ventricular epicardium and endocardium during steady-state 8-Hz pacing as on previous occasions in the mouse heart [16, 25]. Finally, dynamic repolarization properties were investigated using a dynamic pacing protocol.
Triggered activity and spontaneous arrhythmogenesis in intrinsically beating KCNE1−/− hearts
Intrinsically beating KCNE1−/− hearts perfused with control solution demonstrated triggered activity, observed as ectopic MAP waveforms in nine out of ten hearts (Fig. 1a). Such activity typically occurred immediately following or just prior to MAP repolarization in agreement with the timing of afterdepolarization phenomena described on earlier occasions [19, 23]. These abnormalities were absent in intrinsically beating WT hearts (n = 5) (Fig. 1b). We then tested the effect of nicorandil upon the occurrence of triggered activity. Nicorandil (20 µM) suppressed triggered activity in KCNE1−/− hearts (Fig. 1c), while in WT hearts, there remained a total absence of such triggered electrical activity (n = 7 and 5, KCNE1−/− and WT hearts, respectively) (Fig. 1d).
Fig. 1.

Representative MAP recordings from intrinsically beating KCNE1−/− hearts Langendorff-perfused with control Krebs–Henseleit solution demonstrating typical spontaneous triggered activity occurring as ectopic MAP waveforms (asterisk) in association with the onset of spontaneous VT (a) in contrast to their absence in similarly perfused intrinsically beating WT hearts (b). Treatment with 20 µM nicorandil reduced both triggered activity and VT in KCNE1−/− hearts (c). WT hearts treated with nicorandil did not show triggered activity or VT (d)
Following these preliminary observations of the effects of 20 µM nicorandil, we proceeded to investigate the effect of a range of nicorandil concentrations (100 nM, 1 µM, 10 µM, and 20 µM) upon triggered activity in intrinsically beating hearts. The incidence of such activity was determined from randomly selected 10 s sampling periods in each heart at any given nicorandil concentration from MAP recordings in each heart (consisting of 211 ± 67 and 337 ± 80 MAPs per heart, WT and KCNE1−/−, respectively). Nicorandil treatment was associated with a concentration-dependent reduction in the incidence of triggered activity in KCNE1−/− hearts. Thus, the proportion of ectopic MAP waveforms decreased from 28.1 ± 8.4% in hearts perfused with control solution (n = 10) to 20.9 ± 10.9% at 100 nM (n = 5, P > 0.05), 20.5 ± 10.9% at 1 µM (n = 5, P > 0.05), 2.2 ± 2.1% at 10 µM (P < 0.05; n = 5), and 0.9 ± 1.1 at 20 µM nicorandil (P < 0.01; n = 7) (all comparisons performed against untreated hearts). WT hearts did not show triggered activity anywhere in the range of nicorandil concentrations (n = 5 at each concentration).
These ectopic waveforms frequently occurred in association with runs of spontaneous VT, defined as five or more premature MAP waveforms [25], in intrinsically beating KCNE1−/− hearts (Fig. 1a). The incidence of VT was determined by analyzing MAP waveforms recorded from such intrinsically beating hearts. Spontaneous VT occurred in six out of ten untreated KCNE1−/− versus none out of five untreated WT hearts, demonstrating a significant tendency to spontaneous VT in KCNE1−/− compared to WT hearts (P < 0.05; Fisher’s exact test). In contrast, during treatment with nicorandil (20 µM), spontaneous VT was not seen in any KCNE1−/− or WT hearts (n = 7 and 5, for KCNE1−/− hearts and WT hearts, respectively), demonstrating a significant antiarrhythmic effect of nicorandil upon spontaneous VT (P < 0.05; Fisher’s exact test).
Provoked arrhythmogenesis in KCNE1−/− hearts
We then investigated tendencies to provoked arrhythmogenesis using an established PES protocol adapted from clinical practice [51] and successfully applied in murine cardiac systems [4, 41]. The protocol consisted of cycles of eight S1 stimuli at a frequency of 8 Hz, with each cycle followed by an extrastimulus, S2, initially 125 ms after the preceding S1 stimulus. This S1–S2 stimulus was then progressively reduced by 1 ms with each cycle until hearts were either refractory to the S2 or VT was initiated. PES resulted in VT in five out of five untreated KCNE1−/− hearts (Fig. 2a) in contrast to none of five untreated WT hearts that were totally refractory to such provocation (Fig. 2b). We then assessed the effect of a range of concentrations of nicorandil (100 nM, 1 µM, 10 µM, and 20 µM) on this arrhythmogenicity. PES resulted in VT in five out of five hearts at 100 nM and 1 µM nicorandil. Higher concentrations of nicorandil (10 and 20 µM) reduced this incidence to one out of five hearts (Fig. 2c), demonstrating a significant antiarrhythmic effect against PES-induced arrhythmogenesis (P < 0.05 versus untreated hearts; Fisher’s exact test). WT hearts remained refractory to provocation by PES at all concentrations used (Fig. 2d) (n = 5).
Fig. 2.

MAP recordings obtained during a PES protocol used to assess for provoked arrhythmogenic tendencies. They demonstrate the typical initiation of VT in untreated KCNE1−/− hearts observed in five out of five hearts (a) and the prevention of such VT by treatment with nicorandil (20 µM) in four out of five hearts (P < 0.05) (c). WT hearts were refractory to PES provocation whether untreated (b) or treated with nicorandil (d)
Previous reports have described sex-linked differences in arrhythmogenic phenotype in a number of congenital arrhythmic conditions [2]. Our study did not detect significant differences in findings between male and female KCNE1−/− hearts. Thus, triggered activity was seen in six of seven male and three of three female untreated hearts. Spontaneous VT was seen in four of seven males and two of three females. PES induced VT in three of three males and two of two females. This is consistent with a previous report [4] that investigated KCNE1−/− hearts with bipolar electrograms, but not MAP recordings, in which untreated KCNE1−/− hearts were all arrhythmic (five males and five females) with PES. Our results showed that nicorandil (20 µM) produced similar actions upon KCNE1−/− mice whether male or female. Triggered activity was observed in just one of five male and zero of two female nicorandil-treated hearts. Spontaneous VT was observed in zero of five males and zero of two females studied. VT was then provoked by PES in one of three male and zero of two female hearts. In contrast, none of two male and three female untreated WT hearts showed evidence for triggered activity or spontaneous VT. None of three male and two female untreated WT hearts showed PES-induced VT. Following the introduction of 20 µM nicorandil, none of two male and three female hearts showed either triggered activity or spontaneous VT, nor did any of two male or three female WT hearts show PES-induced VT. Furthermore, there was no evidence for sex-linked differences in VERP in either KCNE1−/− or WT hearts. Thus, male and female animals gave VERPs of 28.8 ± 2.7 and 30.5 ± 5.1 ms in KCNE1−/− hearts and 25.6 ± 2.5 and 27.5 ± 3.5 ms in WT hearts, respectively. Following the addition of nicorandil, the respective values were 32.0 ± 3.2 and 28.0 ± 9.0 ms in KCNE1−/− and 25.7 ± 0.5 and 29.5 ± 2.5 ms in WT hearts, respectively.
Epicardial and endocardial MAP waveforms in WT and KCNE1−/− hearts
Previous studies have investigated epicardial and endocardial action potential waveforms in murine models of inherited LQTS [64, 65] and hypokalaemic LQTS [23, 25] and suggested that alterations in the durations of epicardial waveforms compared to endocardial waveforms may be associated with altered arrhythmogenic tendencies. We, therefore, proceeded to quantify APDs in both untreated and nicorandil-treated hearts to investigate the underlying basis of arrhythmic phenomena for the first time in KCNE1−/− hearts. Representative epicardial and endocardial MAP waveforms in WT and KCNE1−/− hearts recorded in the absence and presence of 20 µM nicorandil are shown in Fig. 3. This demonstrates the typically prolonged epicardial MAP waveform in KCNE1−/− compared to WT hearts and also demonstrates the shortened epicardial MAP waveforms in both KCNE1−/− and WT hearts perfused with nicorandil. APDs at x% repolarization (APDx) were quantified during steady-state 8 Hz extrinsic pacing (Fig. 4) approximating typical physiological murine heart rates [41]. In the epicardium of untreated KCNE1−/− hearts, APD90 and APD70 were significantly longer than in the epicardium of untreated WT hearts. Thus, APD90 was 55.1 ± 1.0 versus 40. 9 ± 1.5 ms (P < 0.001; n = 5 and 5) and APD70 was 30.7 ± 1.9 versus 21.4 ± 1.4 ms (P < 0.01; n = 5 and 5) in KCNE1−/− and WT hearts, respectively. In contrast to these epicardial findings, there was no difference in endocardial APD between untreated KCNE1−/− and WT hearts. Endocardial APD90 was 51.3 ± 1.4 and 51.0 ± 1.2 ms in untreated KCNE1−/− and WT hearts, respectively (P > 0.05; n = 5 in both cases).
Fig. 3.

Representative MAP waveforms during steady-state 8-Hz pacing recorded from the left ventricular epicardia and endocardia of KCNE1−/− (a and c) and WT (b and d) hearts perfused with control solution (a and b) or 20 µM nicorandil (c and d). KCNE1−/− hearts had longer epicardial MAP waveforms (a) than WTs similarly perfused with control solution (b), and both were shortened by nicorandil (c and d) while endocardial MAPs were conserved in both (c and d)
Fig. 4.

APD at 30%, 50%, 70%, and 90% repolarization quantified from MAP recordings made from the epicardium of KCNE1−/− (a) and WT (b) hearts during 8-Hz extrinsic pacing demonstrated a concentration-dependent reduction in APD90 and APD70 with nicorandil. In contrast, similarly obtained MAP recordings from the endocardium of KCNE1−/− (c) and WT (d) hearts demonstrated conserved APD values during perfusion with nicorandil (*P < 0.05; **P < 0.01; ***P < 0.001)
We also determined the effect of a range of concentrations of nicorandil (100 nM, 1 µM, 10 µM, and 20 µM) upon APD in KCNE1−/− hearts. In the epicardium of KCNE1−/− hearts, APD90 was reduced by nicorandil to 51.1 ± 0.9, 44.5 ± 2.2, and 38.9 ± 1.8 ms during treatment with 1, 10, and 20 µM nicorandil (P < 0.05, P < 0.001, and P < 0.001 versus untreated KCNE1 hearts at 1, 10, and 20 µM, respectively). In WT hearts, epicardial APD90 was also shortened by nicorandil to 35.6 ± 1.5 and 33.8 ± 0.9 ms at 10 and 20 µM nicorandil, respectively (10 µM, P < 0.05; 20 µM, P < 0.001; n = 5 in all cases). In contrast, endocardial APD90 was not changed by nicorandil in either KCNE1−/− or WT hearts. Thus, during perfusion with 20 µM nicorandil, endocardial APD90 was 53.5 ± 1.5 and 50.6 ± 1.2 ms in KCNE1−/− and WT hearts, respectively (n = 5 in all cases; P > 0.05 versus untreated hearts of the same genotype).
These findings demonstrate preferential abnormalities in epicardial APD90 and conserved endocardial APD90 in parallel with increased arrhythmic tendencies in untreated KCNE1−/− hearts. These findings together complement previous observations in a model of LQT3 [64] and the hypokalaemic murine heart [25]. The present findings further show that nicorandil altered these KCNE1−/− MAP waveform characteristics toward those found in untreated WTs.
Transmural gradients of repolarization in WT and KCNE1−/− hearts
The ventricular wall has been shown to possess heterogeneities in APD, not only in the murine heart [69] but also in larger hearts such as the canine heart [3]. It has been suggested that such regional differences in APD are due to heterogeneities in spatial patterns of repolarization [3] resulting from transmural gradients of ion channel expression [52]. In the murine system, the transmural gradient of repolarization, ∆APD90, given by the difference between endocardial and epicardial APD90, has been used to explore for spatial substrates for re-entrant excitation [24, 25].
We initially determined epicardial and endocardial activation times, which reflect the time taken from ventricular electrical stimulation to peak depolarization. We were able to quantify activation times from MAP recordings by measuring the latent period between electrical stimulation and the peak of the MAP. Activation times were 17.3 ± 1.3 and 16.4 ± 1.7 ms in the epicardium and endocardium of untreated KCNE1−/− hearts, respectively, and did not significantly differ. Activation times were 16.1 ± 1.1 and 17.9 ± 1.1 ms in the epicardium and endocardium of untreated WT hearts (n = 5 in all cases), respectively, which also did not differ significantly. Following treatment with 20 µM nicorandil, activation times in the epicardium and endocardium were 16.8 ± 1.2 and 17.4 ± 1.3 ms in KCNE1−/− hearts and 19.1 ± 1.8 and 15.7 ± 1.0 ms in the epicardium and endocardium of WT hearts (n = 5 in all cases), respectively, which also did not differ significantly. Finally, epicardial or endocardial activation times both did not differ between KCNE1−/− and WT hearts, whether in untreated hearts or in those treated with 20 µM nicorandil. Thus, changes in arrhythmogenicity were not associated with detectable changes in activation times. Furthermore, in the absence of significant differences in activation times, we were able to represent the transmural gradient of repolarization by ΔAPD90 as on previous occasions in the murine heart [16, 25].
Figure 5 shows the ∆APD90 values for KCNE1−/− and WT hearts in the absence and presence of nicorandil. In untreated KCNE1−/− hearts, ∆APD90 was −3.8 ± 1.7 ms (n = 5), significantly smaller (P < 0.001) than in untreated WT hearts where ∆APD90 was 10.9 ± 1.9 ms (n = 5). Nicorandil resulted in a concentration-dependent increase in ∆APD90 in KCNE1−/− hearts, reaching significance at 10 and 20 µM nicorandil, with ∆APD90s of 5.1 ± 2.9 and 14.6 ± 2.4 ms at 10 and 20 µM, respectively (10 µM, P < 0.05; 20 µM, P < 0.001). This concentration range was similar to that at which spontaneous and provoked arrhythmogenic phenomena were reduced and at which epicardial APD90 was shortened. In WT hearts, nicorandil also concentration-dependently increased ∆APD90 in WT hearts, reaching statistical significance at 10 and 20 µM. In WT hearts, ∆APD90 was increased to 17.3 ± 1.7 (n = 5) and 16.9 ± 1.5 ms (n = 5) at 10 and 20 µM nicorandil (P < 0.05 in both cases), respectively.
Fig. 5.

The transmural gradient of repolarization, ∆APD90, determined from endocardial and epicardial MAP recordings during 8-Hz extrinsic pacing through a range of nicorandil concentrations in KCNE1−/− (a) and WT hearts (b) (*P < 0.05; ***P < 0.001)
Critical intervals for local re-excitation in WT and KCNE1−/− hearts
Recent studies in the hypokalaemic murine heart have suggested that the interval between APD90 and VERP reflects the likelihood for local re-excitation and that reductions in this interval may be antiarrhythmic [44]. We accordingly investigated this relationship in KCNE1−/− hearts for the first time. Figure 6 shows such critical intervals over a range of nicorandil concentrations in hearts paced at 8 Hz. In untreated KCNE1−/− hearts, the epicardial critical interval was significantly greater than in untreated WT hearts, 25.3 ± 3.3 versus 14.7 ± 2.2 ms in KCNE1−/− and WT hearts, respectively (P < 0.05; n = 5). We then investigated the effect of nicorandil upon such intervals (Fig. 6). Nicorandil produced a concentration-dependent reduction in local critical intervals for re-excitation, reaching significance at 20 µM nicorandil with a critical interval of 8.2 ± 3.8 ms (P < 0.01 versus untreated KCNE1−/− hearts; n = 5). Nicorandil also produced concentration-dependent reductions in critical intervals in WT hearts, reaching significance at 20 µM with a critical interval of 7.5 ± 2.4 ms (P < 0.05 versus untreated WT hearts; n = 5). Thus, the enhanced arrhythmogenicity in KCNE1−/− hearts compared to WT hearts was accompanied by larger critical intervals for local re-excitation. Furthermore, nicorandil produced reductions in local critical intervals in parallel with its antiarrhythmic effect in KCNE1−/− hearts and alongside the lack of arrhythmia inducibility in WT hearts.
Fig. 6.

Comparison of epicardial APD90, VERP, and local critical intervals for re-excitation measured during steady 8-Hz extrinsic pacing over a range of nicorandil concentrations in the epicardium of KCNE1−/− (a) and WT (b) hearts (*P < 0.05; **P < 0.01; ***P < 0.001). APD90 data set from Fig. 5
The magnitude of APD90 alternans in WT and KCNE1−/− hearts
Alternations in APD90 have been associated with a variety of arrhythmias in human [54] and animal studies [45]. We used a dynamic pacing protocol to assess APD90 properties over a range of BCLs [29]. This protocol consisted of cycles consisting of 100 stimuli at each BCL, starting with a BCL of 190 ms and reduced by 5 ms decrements with each successive cycle down to a final BCL of 65 ms. The final 50 action potentials within each cycle of 100 action potentials at any particular BCL yielded steady-state recordings that were analyzed to determine APD90 values, as in a previous study in the hypokalaemic murine heart [45]. Firstly, in all hearts, alternans in both epicardial and endocardial MAPs increased as BCL decreased (Fig. 7). Secondly, successive values of APD90 alternans in untreated KCNE1−/− hearts (Fig. 7, squares) tended consistently to fall above corresponding points obtained in WT hearts (Fig. 7, triangles) through the investigated range of BCLs. This trend applied for both epicardial (Fig. 7a) and endocardial values (Fig. 7b). Finally, treatment with 20 µM nicorandil reduced such differences in alternans between KCNE1−/− and WT hearts in both epicardium (Fig. 7c) and endocardium (Fig. 7d).
Fig. 7.

APD90 alternans recorded over a range of BCLs from the epicardium (a and c) and endocardium (b and d) of untreated (a and b) and nicorandil-treated (20 µM) (c and d) KCNE1−/− (a and c) and WT (b and d) hearts
APD90 restitution properties in WT and KCNE1−/− hearts
Increases in the steepness of APD90 restitution curves have been reported to associate with enhanced arrhythmogenicity in both clinical reports [40, 63] and experimental systems [29, 45]. APD90 restitution properties have been investigated by plotting APD90 versus the preceding diastolic interval (DI) [39]. We accordingly proceeded to investigate restitution properties in KCNE1−/− hearts and constructed APD90 restitution curves using experimental data obtained from both epicardium and endocardium using a dynamic pacing protocol for the first time in this system, thereby allowing comparison with other murine arrhythmic models to which this method has been applied [16, 45]. Furthermore, we then tested the effect of nicorandil on APD90 restitution properties in any model of JLN for the first time.
Figure 8 shows restitution curves obtained from the epicardium of KCNE1−/− and WT hearts perfused with either control solution or after treatment with 20 µM nicorandil. Similarly obtained restitution curves obtained from the endocardium of KCNE1−/− and WT hearts perfused with either control solution or after treatment with 20 µM nicorandil are shown in Fig. 9.
Fig. 8.

Epicardial restitution curves recorded from the epicardium of KCNE1−/− (a and c) and WT (b and d) hearts perfused with control solution (a and b) or 20 µM nicorandil (c and d)
Fig. 9.

Endocardial restitution curves recorded from the endocardium of KCNE1−/− (a and c) and WT (b and d) hearts perfused with control solution (a and b) or 20 µM nicorandil (c and d)
Such curves were fitted with a monoexponential curve [16, 45] of the form:
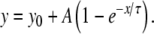 |
1 |
Empirical data points were fitted using a Levenberg–Marquardt algorithm [16, 45]. The gradients of the fitted functions are also shown in Figs. 8 and 9, given by the following function:
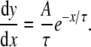 |
2 |
The parameters used in curve fitting for the epicardial and endocardial restitution data sets are shown in Tables 1 and 2.
Table 1.
Parameters used in curve fitting to experimentally obtained epicardial APD90 restitution data sets
| n | y0 (ms) | A (ms) | τ (ms) | χ2 | CDI (ms) | Maximum gradient | |
|---|---|---|---|---|---|---|---|
| KCNE1−/− | 6 | −75.71 ± 13.56 | 142.78 ± 12.75 | 29.06 ± 2.22 | 0.164 | 46.26 ± 6.48 | 1.32 ± 0.18 |
| KCNE1−/−+20 µM nicorandil | 6 | −4.18 ± 6.56 | 68.33 ± 2.80 | 68.48 ± 16.28 | 0.152 | −0.15 ± 0.05 | 0.55 ± 0.04 |
| WT | 6 | −7.04 ± 2.53 | 63.77 ± 1.62 | 53.06 ± 3.95 | 0.095 | 9.76 ± 1.06 | 0.59 ± 0.02 |
| WT+20 µM nicorandil | 6 | 10.6 ± 1.5 | 50.0 ± 4.0 | 136.2 ± 27.8 | 0.060 | −136.54 ± 40.86 | 0.27 ± 0.04 |
Table 2.
Parameters used in curve fitting to experimentally obtained endocardial APD90 restitution data sets
| n | y0 (ms) | A (ms) | τ (ms) | χ2 | CDI (ms) | Maximum gradient | |
|---|---|---|---|---|---|---|---|
| KCNE1−/− | 6 | 104.89 ± 21.81 | 159.31 ± 20.84 | 30.25 ± 2.78 | 0.654 | 50.26 ± 9.27 | 1.11 ± 0.21 |
| KCNE1−/−+20 µM nicorandil | 6 | −22.97 ± 6.30 | 86.38 ± 4.45 | 50.87 ± 5.93 | 0.152 | 26.94 ± 4.65 | 0.76 ± 0.07 |
| WT | 6 | −13.81 ± 4.37 | 81.31 ± 2.21 | 63.07 ± 7.50 | 0.178 | 16.02 ± 2.73 | 0.69 ± 0.04 |
| WT+20 µM nicorandil | 6 | 6.89 ± 1.44 | 66.89 ± 1.28 | 80.50 ± 6.61 | 0.081 | −14.91 ± 1.75 | 0.54 ± 0.03 |
Restitution curves in KCNE1−/− and WT hearts show contrasting critical diastolic intervals and maximum gradients
The slopes of restitution curves obtained under the different experimental conditions were quantified and summarized in Tables 1 and 2. This analysis showed that, in the absence of nicorandil, KCNE1−/− hearts possessed steeper gradients of restitution curves than WT hearts as assessed by both their critical diastolic intervals (CDIs) and their maximum gradients. CDI, the DI at which the gradient equals unity [45], was calculated from the fitted function. Untreated KCNE1−/− hearts showed larger epicardial CDIs than WTs, 46.26 ± 6.48 versus 9.76 ± 1.06 ms (P < 0.001; n = 6 in both cases). Similarly, larger CDIs were observed in the endocardium of KCNE1−/− hearts having a CDI of 50.26 ± 9.27 versus 16.02 ± 2.73 ms in WT hearts (P < 0.01; n = 6 in both cases). We therefore demonstrate larger CDIs in KCNE1−/− hearts compared to WT hearts and confirm this in both epicardium and endocardium.
Maximum gradients of APD90 restitution curves were then compared. In the epicardium of KCNE1−/− hearts, the maximum gradient was 1.32 ± 0.18, significantly larger than in WT hearts in which the maximum gradient was 0.59 ± 0.02 (P < 0.01; n = 6 in both cases). In the endocardium, the maximum gradient was also larger in KCNE1−/− hearts than in WTs hearts, though this did not reach statistical significance, with gradients of 1.11 ± 0.21 versus 0.69 ± 0.04 in KCNE1−/− and WT hearts, respectively (n = 6 in both cases).
Alterations in CDIs and maximum gradients correlate with changes in arrhythmogenic tendencies in both KCNE1−/− and WT hearts
In view of the observed antiarrhythmic actions of nicorandil in KCNE1−/− hearts, we then quantified the effects of nicorandil (20 µM), the highest concentration at which significant antiarrhythmic effects were observed, upon CDIs and maximum gradients for the first time in any model of JLN. Nicorandil significantly reduced the CDI in the epicardium of KCNE1−/− hearts (Fig. 10a) from 46.26 ± 6.48 to −0.15 ± 0.05 ms (P < 0.001; n = 6 in both cases). It had a similar action upon the CDI in the endocardium of KCNE1−/− hearts, shortening the CDI from 50.26 ± 9.27 to 26.94 ± 4.65 (P < 0.05; n = 6 in both cases). The maximum gradient was also significantly reduced in both the epicardium, from 1.32 ± 0.18 to 0.55 ± 0.04 (P < 0.01), and appreciably but not significantly in the endocardium, from 1.11 ± 0.21 to 0.76 ± 0.07 (P > 0.05) (Fig. 10c). In WT hearts, nicorandil reduced the epicardial CDI (Fig. 10b), from 9.76 ± 1.06 to −136.54 ± 40.86 ms (P < 0.05; n = 6). Similarly, nicorandil reduced the CDI in the endocardium of WT hearts from 16.02 ± 2.73 to −14.91 ± 1.75 ms (P < 0.001; n = 6). It also decreased the maximum gradient of restitution curves in WT hearts (Fig. 10d). This occurred in the epicardium, from 0.59 ± 0.02 to 0.27 ± 0.04 (P < 0.001; n = 6), and in the endocardium, from 0.69 ± 0.04 to 0.54 ± 0.03 (P < 0.05; n = 6) in untreated and nicorandil-treated WT hearts, respectively.
Fig. 10.

CDI (a and b) and maximum gradients (c and d) derived from restitution curves from the epicardium and endocardium of KCNE1−/− (a and c) and WT (b and d) hearts perfused with control solution or 20 µM nicorandil (*P < 0.05; **P < 0.01, ***P < 0.001)
Thus, the enhanced arrhythmogenicity observed in KCNE1−/− hearts was associated with steeper epicardial and endocardial restitution curves compared to WT hearts, while reductions in arrhythmogenicity by nicorandil were accompanied by reductions in such gradients.
Discussion
The experiments described in this study characterized arrhythmic properties and their possible mechanisms at the whole organ level by comparing isolated perfused KCNE1−/− with WT hearts. The primary abnormality in the KCNE1−/− murine system is a loss-of-function, targeted deletion of the gene encoding the minK K+ channel accessory β-subunit. In common with patients with the corresponding JLN syndrome [20], the KCNE1−/− mouse is known to possess inner ear defects, manifesting as deafness and shaker/waltzer behavior, attributed to deficient transepithelial K+ secretion in the stria vascularis and vestibular dark cells of the inner ear. Histological appearances in the inner ears of KCNE1−/− mice [68] were also shown to be similar to those observed in post-mortem inspections of the human condition [11]. Previous experiments in patch-clamped neonatal cardiac myocytes, rather than whole heart investigations, had suggested alterations in cardiac electrophysiology involving late as opposed to early (Ito) K+ current in the KCNE1−/− cardiac system [9, 30]. These empirically demonstrated reductions in the rapid component of the delayed rectifier current (IKr) [30] relative to WT and a total absence of the slow component, IKs [9, 30], albeit from relatively low levels of detection in WTs. Correspondingly, mRNA for minK was detectable at low levels in WT but totally absent from KCNE1−/− myocytes [9]. In contrast, early outward currents (Ito) remained conserved [30] as were mRNA levels [9] for the alpha subunit KvLQT1 (which together with minK is responsible for IKs [5, 21, 49]), Kv1.5 (the alpha subunit responsible for IKur [70]) and mERG (the murine protein corresponding to the human alpha subunit protein, HERG, that underlies IKr [48]).
This paper complements those cellular studies. It reports that intact, perfused KCNE1−/− mouse hearts recapitulated cardiac features of the JLN2 variant of JLN syndrome [20] in their enhanced susceptibility to VT. It then related these properties to the presence of triggered activity and to criteria for re-entrant excitation that enabled the detection and separation of underlying re-entrant substrates. We then both empirically tested the efficacy of nicorandil as an antiarrhythmic agent in the KCNE1−/− murine model and related this to actions on such re-entrant criteria for the first time. In doing so, we empirically tested the idea that arrhythmogenicity could be reduced by the action of nicorandil, which, at the higher concentrations used in this study, has been shown to shorten cardiac APDs in other cardiac preparations (see the “Introduction” section) possibly through acting upon K+ conductance [18, 22, 57, 60], independent of external Na+, and in the absence of alterations in Ca2+ currents [15, 22]. It has been suggested for use as a potentially antiarrhythmic pharmacological agent in LQTS, supported by preliminary clinical studies in patients with LQT1 [58] and familial [1] and idiopathic [13, 50] LQTS where the genotype had not been determined. Experimental studies in a hypokalaemic murine model [26], a genetic murine LQT3 model [17], a pharmacological guinea pig LQT1 model [76], and pharmacological canine LQT1, LQT2, and LQT3 models are also consistent with potential antiarrhythmic roles for nicorandil [7, 57]. However, neither this antiarrhythmic action nor its possible mechanisms at the level of intact hearts have been specifically tested in any system modelling loss-of-function mutations in KCNE1, whether obtained through pharmacological or genetic modification or whether in experimental or clinical situations. We used concentrations (10 and 20 µM) comparable to those employed in earlier reports of nicorandil-mediated reductions in repolarization durations or suppressed arrhythmogenesis in other cardiac systems whether at the cellular [15, 18, 22, 31, 74, 75], tissue [57, 76], or whole organ levels [17, 26, 76].
Our experiments demonstrated both spontaneous and provoked arrhythmogenesis in isolated perfused KCNE1−/− hearts. They complement earlier reports that demonstrated altered electrocardiographic QT–heart rate relationships in intact anesthetized animals compatible with arrhythmogenic consequences [9] (but see [30]). A further report did not observe provoked arrhythmogenesis on the basis of optical recordings in Langendorff-perfused hearts. However, that study [47] adopted left ventricular apical rather than right ventricular stimulation, used longer (200 ms instead of 125 ms) BCLs, employed differing PES or burst stimulus protocols, and used C57Black6 rather than 129/Sv animals. Nevertheless, the arrhythmogenic properties described in the present paper closely agree with previous studies that had employed stimulation protocols and murine strains [4] that were comparable both to those used in this study and to those used to explore other murine models [25, 45]. These contrasts are consistent with the suggestion that arrhythmogenic propensity may require particularly sensitive provocation techniques for their detection [51].
The present experiments additionally applied a set of criteria previously developed in the murine system [16, 25, 44, 64]. They were applied in both KCNE1−/− and WT hearts, in the absence and presence of nicorandil. This application yielded parameters that were used to detect the presence or absence of both spatial re-entrant substrate, reflected in ∆APD90 and critical intervals for local re-excitation, and temporal re-entrant substrate, reflected in APD90 alternans and APD90 restitution properties.
Firstly, we demonstrated that intrinsically beating KCNE1−/−, but not WT hearts, showed spontaneous, nicorandil-sensitive triggered activity that was accompanied by longer runs of spontaneous VT. These had not been sought in previous reports [4, 9, 30, 47] which had studied electrocardiographic records, bipolar electrograms, or optical traces, rather than the MAP records used in this study. Nevertheless, these observations agree with and extend a recent description of MAP records which showed spontaneous nifedipine-sensitive abnormalities of repolarization [66]. The present studies go on to demonstrate that nicorandil suppressed both the triggered activity and the spontaneous VT.
Secondly, in contrast to WT, KCNE1−/− hearts showed evidence for abnormal spatial patterns of repolarization. This took the form of increased epicardial, though conserved endocardial, APD90s. This would in turn perturb transmural gradients of repolarization by resulting in reduced or even negative ∆APD90s. These findings could be obtained from the measurements of both endocardial and epicardial MAP, thereby adding to reports that were confined to either electrocardiographic or optical epicardial recordings [4, 9, 30, 47]. These results in KCNE1−/− hearts complement findings in other arrhythmogenic murine systems modeling human LQT3 [64] and acquired hypokalaemic LQTS [25] which also show prolonged epicardial relative to endocardial APD90. Altered spatial patterns of repolarization have also been demonstrated in pharmacological models of LQTS involving the arterially-perfused canine wedge [3]. Such alterations may contribute to re-entrant arrhythmogenesis in view of the relatively short distance across the thickness of the ventricular free wall [3]. We went on to demonstrate that nicorandil increased ∆APD90 by reducing the epicardial APD90 while conserving the endocardial APD90 not only in KCNE1−/− but also in WT hearts. It thus corrected the alteration in KCNE1−/− hearts in the direction of and consequently approximating WT values that had been observed in the absence of nicorandil. Such region-dependent actions of nicorandil upon epicardial rather than endocardial APD90 have been observed in a canine ischaemic model [32] as well as in hypokalaemic murine hearts [26].
Thirdly, KCNE1−/− hearts demonstrated larger local critical intervals for re-excitation than WT hearts. This paralleled their enhanced tendencies to spontaneous and provoked arrhythmogenesis. Such intervals were derived from the difference between APD90 and refractory period and have also been correlated with arrhythmogenic propensity in hypokalaemic murine hearts [44]. Treatment with nicorandil significantly reduced these critical intervals in both KCNE1−/− and WT hearts in parallel with reductions in arrhythmic tendency in KCNE1−/− hearts.
Fourthly, both KCNE1−/− and WT hearts demonstrated enhanced tendencies to APD90 alternans as derived from differences in successive odd and even APD90 values with decreasing BCL. However, such alternans was consistently greater in KCNE1−/− than in WT hearts in both epicardium and endocardium. Such trends have also been observed in murine models for LQT3 and hypokalaemic acquired LQTS [17, 45]. These findings also complement a previous electrocardiographic study that reported a development of T wave alternans (TWA) during isoproterenol-mediated increases in heart rate. The latter study did not observe differences between WT and KCNE1−/− hearts, but had studied relatively small numbers of anesthetized, intact mutant (two mice) and WT preparations (three mice) [9]. Furthermore, the properties shown by our isolated denervated cardiac preparations agree with and add to previous findings obtained under comparable conditions but confined to a single basic cycle length [66]. Previous analysis of human action potential waveforms using MAP electrodes has directly correlated APD alternans with TWA in LQTS [59]. In general, APD alternans is thought to underlie the TWA that has been observed in clinical electrocardiographic traces [34]. The latter in turn has been associated with enhanced arrhythmogenic propensity not only in LQTS [54] but also in a wide variety of other arrhythmic settings, including ischaemia, acidosis, and pharmacological agents (reviewed in [34]). Our results go on to additionally demonstrate that nicorandil reduced APD alternans in KCNE1−/− hearts to levels similar to WT hearts in both epicardium and endocardium.
Fifthly, application of a dynamic pacing protocol [29] to KCNE1−/− hearts permitted quantification of MAP waveforms in the steady-state achieved following introduction of a range of given BCLs. This approach successfully demonstrated that both epicardial and endocardial APD90 restitution curves from KCNE1−/− hearts showed greater slopes than did WT hearts in parallel with their arrhythmogenic tendencies. Establishing that such properties exist in isolated hearts free of autonomic innervation, as performed in this study, also complements a previous electrocardiographic study that reported abnormal adaptation of QT interval to heart rate in intact anesthetized KCNE1−/− mice [9]. However, this could not examine the relationship between APD90 and DI. Our findings contrast with measurements that had been obtained using extrasystolic stimuli rather than exploring a range of BCLs using a dynamic pacing protocol [47, 66]. Nevertheless, our results are likely to provide closer representations of APD since our use of contact MAP electrodes rather than optical measurements permitted both epicardial and endocardial recordings and yielded APD90 rather than APD75 values. This enabled relationships between APD90 and DI rather than APD75 and BCL to be investigated. This in turn permitted the graphical analysis introduced by Nolasco and Dahlens that permitted us to establish whether there existed a CDI at which APD90 varied with DI in a manner compatible with re-entrant arrhythmogenesis [39, 72]. The present studies consequently add to previous demonstrations of temporal re-entrant substrate in other arrhythmogenic murine heart models for LQT3 and Brugada syndrome [17, 46] and hypokalaemic VT [45]. Our results also demonstrated that nicorandil treatment reduced these gradients in both KCNE1−/− and WT hearts in parallel with its reduction of arrhythmogenic tendency.
KCNE1−/− hearts thus provided us with a valuable arrhythmogenic model exhibiting not only spontaneous but also PES-induced VT. They showed accompanying spontaneous triggered activity as well as measurable spatial and temporal heterogeneities in MAP waveform that could potentially underlie such arrhythmogenic tendencies. The present experiments went on to confirm such a hypothesis by demonstrating that an introduction of nicorandil produced a parallel abolition of all these properties. This was consistent with a participation of both triggered activity and spatial and temporal re-entrant substrate in the properties observed.
Acknowledgements
Funding was provided by the Wellcome Trust, Medical Research Council, British Heart Foundation, Helen Kirkland Trust, and Papworth Hospital, UK. SSH was supported by a Medical Research Council Capacity Building Studentship. MJK acknowledges the generous support of the Avrith Fund. Sandeep S. Hothi and Glyn Thomas have contributed equally to this work.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Aizawa Y, Uchiyama H, Yamaura M, Nakayama T, Arita M (1998) Effects of the ATP-sensitive K channel opener nicorandil on the QT interval and the effective refractory period in patients with congenital long QT syndrome. J Electrocardiol 31:117–123 [DOI] [PubMed]
- 2.Antzelevitch C (2006) Brugada syndrome. Pacing Clin Electrophysiol 29:1130–1159 [DOI] [PMC free article] [PubMed]
- 3.Antzelevitch C, Oliva A (2006) Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Intern Med 259:48–58 [DOI] [PMC free article] [PubMed]
- 4.Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CLH (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552:535–546 [DOI] [PMC free article] [PubMed]
- 5.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G (1996) KvLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature 384:78–80 [DOI] [PubMed]
- 6.Chiang C-E, Roden DM (2000) The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol 36:1–12 [DOI] [PubMed]
- 7.Chinushi M, Kasai H, Tagawa M, Washizuka T, Hosaka Y, Chinushi Y, Aizawa Y (2002) Triggers of ventricular tachyarrhythmias and therapeutic effects of nicorandil in canine models of LQT2 and LQT3 syndromes. J Am Coll Cardiol 40:555–562 [DOI] [PubMed]
- 8.Crotti L, Celano G, Dagradi F, Schwartz P (2008) Congenital long QT syndrome. Orphanet J Rare Dis 3:18 [DOI] [PMC free article] [PubMed]
- 9.Drici M-D, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G, Barhanin J (1998) Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ Res 83:95–102 [DOI] [PubMed]
- 10.Fraser GR, Froggatt P, James TN (1964) Congenital deafness associated with electrocardiographic abnormalities, fainting attacks and sudden death: a recessive syndrome. Q J Med 33:361–385 [PubMed]
- 11.Friedmann I, Fraser GR, Froggatt P (1966) Pathology of the ear in the cardio-auditory syndrome of Jervell and Lange-Nielsen (recessive deafness with electrocardiographic abnormalities). J Laryngol Otol 80:451–470 [DOI] [PubMed]
- 12.Fujimoto Y, Kusano KF, Morita H, Hong K, Yamanari H, Ohe T (2000) Nicorandil attenuates both temporal and spatial repolarization alternans. J Electrocardiology 33:269–277 [DOI] [PubMed]
- 13.Fujimoto Y, Morita H, Fukushima KK, Ohe T (1999) Nicorandil abolished repolarisation alternans in a patient with idiopathic long QT syndrome. Heart 82:e8 [DOI] [PMC free article] [PubMed]
- 14.Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CL (2005) Paced electrogram fractionation analysis of arrhythmogenic tendency in ∆KPQ Scn5a mice. J Cardiovasc Electrophysiol 16:1329–1340 [DOI] [PubMed]
- 15.Hiraoka M, Fan Z (1989) Activation of ATP-sensitive outward K+ current by nicorandil (2-nicotinamidoethyl nitrate) in isolated ventricular myocytes. J Pharmacol Exp Ther 250:278–285 [PubMed]
- 16.Hothi S, Gurung I, Heathcote J, Zhang Y, Booth S, Skepper J, Grace A, Huang C (2008) Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflügers Archiv 457:253–270 [DOI] [PMC free article] [PubMed]
- 17.Hothi SS, Booth SW, Sabir IN, Killeen MJ, Simpson F, Zhang Y, Grace AA, Huang CLH (2009) Arrhythmogenic substrate and its modification by nicorandil in a murine model of long QT type 3 syndrome. Prog Biophys Mol Biol 98:267–280. doi:10.1016/j.pbiomolbio.2009.01.006 [DOI] [PubMed]
- 18.Imanishi S, Arita M, Kiyosue T, Aomine M (1983) Effects of SG-75 (nicorandil) on electrical activity of canine cardiac Purkinje fibers: possible increase in potassium conductance. J Pharmacol Exp Ther 225:198–205 [PubMed]
- 19.January CT, Fozzard HA (1988) Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev 40:219–227 [PubMed]
- 20.Jervell A, Lange-Nielsen F (1957) Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am Heart J 54:59–68 [DOI] [PubMed]
- 21.Jespersen T, Grunnet M, Olesen S-P (2005) The KCNQ1 potassium channel: from gene to physiological function. Physiology 20:408–416 [DOI] [PubMed]
- 22.Kakei M, Yoshinaga M, Saito K, Tanaka H (1986) The potassium current activated by 2-nicotinamidoethyl nitrate (nicorandil) in single ventricular cells of guinea pigs. Proc R Soc Lond B Biol Sci 229:331–343 [DOI] [PubMed]
- 23.Killeen MJ, Gurung IS, Thomas G, Stokoe KS, Grace AA, Huang CLH (2007) Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol (Oxf) 191:43–58 [DOI] [PMC free article] [PubMed]
- 24.Killeen MJ, Sabir IN, Grace AA, Huang CLH (2009) Dispersions of repolarization and ventricular arrhythmogenesis: Lessons from animal models. Prog Biophys Mol Biol 98:219–229. doi:10.1016/j.pbiomolbio.2008.1010.1008 [DOI] [PubMed]
- 25.Killeen MJ, Thomas G, Gurung IS, Goddard CA, Fraser JA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CLH (2007) Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol (Oxf) 189:33–46 [DOI] [PMC free article] [PubMed]
- 26.Killeen MJ, Thomas G, Olesen SP, Demnitz J, Stokoe KS, Grace AA, Huang CLH (2008) Effects of potassium channel openers in the isolated perfused hypokalaemic murine heart. Acta Physiol (Oxf) 193:25–36 [DOI] [PMC free article] [PubMed]
- 27.Killeen MJ, Thomas G, Sabir IN, Grace AA, Huang CL (2008) Mouse models of human arrhythmia syndromes. Acta Physiol (Oxf) 192:455–469 [DOI] [PubMed]
- 28.Knollmann BC, Katchman AN, Franz MR (2001) Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12:1286–1294 [DOI] [PubMed]
- 29.Koller ML, Riccio ML, Gilmour RF (1998) Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol 275:H1635–H1642 [DOI] [PubMed]
- 30.Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, Roden DM (1999) Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res 84:146–152 [DOI] [PubMed]
- 31.Lathrop DA, Nanasi PP, Varro A (1990) In vitro cardiac models of dog Purkinje fibre triggered and spontaneous electrical activity: effects of nicorandil. Br J Pharmacol 99:119–123 [DOI] [PMC free article] [PubMed]
- 32.Miyoshi S, Miyazaki T, Moritani K, Ogawa S (1996) Different responses of epicardium and endocardium to KATP channel modulators during regional ischemia. Am J Physiol Heart Circ Physiol 271:H140–H147 [DOI] [PubMed]
- 33.Moss AJ, Goldenberg I (2008) Importance of knowing the genotype and the specific mutation when managing patients with long-QT syndrome. Circ Arrhythmia Electrophysiol 1:219–226 [DOI] [PMC free article] [PubMed]
- 34.Myles RC, Burton FL, Cobbe SM, Smith GL (2008) The link between repolarisation alternans and ventricular arrhythmia: does the cellular phenomenon extend to the clinical problem? J Mol Cell Cardiol 45:1–10 [DOI] [PubMed]
- 35.Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15:186–189 [DOI] [PubMed]
- 36.National Institute for Health and Clinical Excellence (NICE) (2006) Implantable cardioverter defibrillators for arrhythmias. NICE, London
- 37.Noble D, Tsien RW (1969) Outward membrane currents activated in the plateau range of potentials in cardiac Purkinje fibres. J Physiol 200:205–231 [DOI] [PMC free article] [PubMed]
- 38.Noble D, Tsien RW (1969) Reconstruction of the repolarization process in cardiac Purkinje fibres based on voltage clamp measurements of membrane current. J Physiol 200:233–254 [DOI] [PMC free article] [PubMed]
- 39.Nolasco JB, Dahlen RW (1968) A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25:191–196 [DOI] [PubMed]
- 40.Pak H-N, Hong SJ, Hwang GS, Lee HS, Park S-W, Ahn JC, Ro YM, Kim Y-H (2004) Spatial dispersion of action potential duration restitution kinetics is associated with induction of ventricular tachycardia/fibrillation in humans. J Cardiovasc Electrophysiol 15:1357–1363 [DOI] [PubMed]
- 41.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA (2002) Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A 99:6210–6215 [DOI] [PMC free article] [PubMed]
- 42.Patel C, Antzelevitch C (2008) Pharmacological approach to the treatment of long and short QT syndromes. Pharmacol Ther 118:138–151 [DOI] [PMC free article] [PubMed]
- 43.Romano C, Gemme G, Pongiglione R (1963) Aritmie cardiache rare dell’eta’ pediatrica. Clin Pediatr 45:656–683 [PubMed]
- 44.Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang CL (2007) The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflügers Archiv 454:209–222 [DOI] [PMC free article] [PubMed]
- 45.Sabir IN, Li LM, Grace AA, Huang CL (2007) Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflügers Archiv 455:653–666 [DOI] [PMC free article] [PubMed]
- 46.Sabir IN, Li LM, Jones VJ, Goddard CA, Grace AA, Huang CL (2007) Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflügers Archiv 455(4):637–651 [DOI] [PMC free article] [PubMed]
- 47.Salama G, Baker L, Wolk R, Barhanin J, London B (2009) Arrhythmia phenotype in mouse models of human long QT. J Interv Card Electrophysiol 24:77–87 [DOI] [PubMed]
- 48.Sanguinetti M, Jiang C, Curran M, Keating M (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307 [DOI] [PubMed]
- 49.Sanguinetti MC, Curran ME, Zou A, Shen J, Specter PS, Atkinson DL, Keating MT (1996) Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKS potassium channel. Nature 384:80–83 [DOI] [PubMed]
- 50.Sato T, Hata Y, Yamamoto M, Morita H, Mizuo K, Yamanari H, Saito D, Ohe T (1995) Early afterdepolarization abolished by potassium channel opener in a patient with idiopathic long QT syndrome. J Cardiovasc Electrophysiol 6:279–282 [DOI] [PubMed]
- 51.Saumarez RC, Grace AA (2000) Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res 47:11–22 [DOI] [PubMed]
- 52.Schram G, Pourrier M, Melnyk P, Nattel S (2002) Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 90:939–950 [DOI] [PubMed]
- 53.Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Ruble C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H (1997) KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet 17:267–268 [DOI] [PubMed]
- 54.Schwartz PJ, Malliani A (1975) Electrical alternation of the T-wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am Heart J 89:45–50 [DOI] [PubMed]
- 55.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen L-SK, Colatsky TJ (1995) Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate: implications for gene-specific therapy. Circulation 92:3381–3386 [DOI] [PubMed]
- 56.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, Horie M, Schulze-Bahr E, Denjoy I (2006) The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 113:783–790 [DOI] [PubMed]
- 57.Shimizu W, Antzelevitch C (2000) Effects of a K+ channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the Long-QT syndrome. Circulation 102:706–712 [DOI] [PubMed]
- 58.Shimizu W, Kurita T, Matsuo K, Suyama K, Aihara N, Kamakura S, Towbin JA, Shimomura K (1998) Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation 97:1581–1588 [DOI] [PubMed]
- 59.Shimizu W, Yamada K, Arakaki Y, Kamiya T, Shimomura K (1996) Monophasic action potential recordings during T-wave alternans in congenital long QT syndrome. Am Heart J 132:699–701 [DOI] [PubMed]
- 60.Shivkumar K, Valderrabano M (2004) Use of potassium channel openers for pharmacologic modulation of cardiac excitability. J Cardiovasc Electrophysiol 15:821–823 [DOI] [PubMed]
- 61.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT (1997) Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med 336:1562–1567 [DOI] [PubMed]
- 62.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT (1997) Mutations in the hminK gene cause long QT syndrome and suppress lKs function. Nat Genet 17:338–340 [DOI] [PubMed]
- 63.Taggart P, Sutton P, Chalabi Z, Boyett MR, Simon R, Elliott D, Gill JS (2003) Effect of adrenergic stimulation on action potential duration restitution in humans. Circulation 107:285–289 [DOI] [PubMed]
- 64.Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL (2007) Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiol 578:85–97 [DOI] [PMC free article] [PubMed]
- 65.Thomas G, Killeen MJ, Grace AA, Huang CLH (2008) Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in ∆KPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol (Oxf) 192:505–517 [DOI] [PMC free article] [PubMed]
- 66.Thomas G, Killeen MJ, Gurung IS, Hakim P, Balasubramaniam R, Goddard CA, Grace AA, Huang CL (2007) Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J Physiol 578:99–114 [DOI] [PMC free article] [PubMed]
- 67.Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, Aslaksen B, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M (1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 6:2179–2185 [DOI] [PubMed]
- 68.Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J (1996) Inner ear defects induced by null mutation of the isk gene. Neuron 17:1251–1264 [DOI] [PubMed]
- 69.Wang Y, Cheng J, Joyner RW, Wagner MB, Hill JA (2006) Remodeling of early-phase repolarization: a mechanism of abnormal impulse conduction in heart failure. Circulation 113:1849–1856 [DOI] [PMC free article] [PubMed]
- 70.Wang Z, Fermini B, Nattel S (1993) Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res 73:1061–1076 [DOI] [PubMed]
- 71.Ward OC (1964) A new familial cardiac syndrome in children. J Ir Med Assoc 54:103–106 [PubMed]
- 72.Weiss JN, Chen P-S, Qu Z, Karagueuzian HS, Lin S-F, Garfinkel A (2002) Electrical restitution and cardiac fibrillation. J Cardiovasc Electrophysiol 13:292–295 [DOI] [PubMed]
- 73.Weiss JN, Karma A, Shiferaw Y, Chen P-S, Garfinkel A, Qu Z (2006) From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98:1244–1253 [DOI] [PubMed]
- 74.Yanagisawa T, Taira N (1980) Effect of 2-nicotinamidethyl nitrate (SG-75) on the membrane potential of left atrial muscle fibres of the dog. Increase in potassium conductance. Naunyn Schmiedebergs Arch Pharmacol 312:69–76 [DOI] [PubMed]
- 75.Yanagisawa T, Taira N (1981) Effect of 2-nicotinamidethyl nitrate (SG-75) on membrane potentials of canine Purkinje fibers. Jpn J Pharmacol 31:409–417 [DOI] [PubMed]
- 76.Yang Z, Shi G, Li C, Wang H, Liu KAI, Liu Y (2004) Electrophysiologic effects of nicorandil on the guinea pig long QT1 syndrome model. J Cardiovasc Electrophysiol 15:815–820 [DOI] [PubMed]
- 77.Zipes DP, Wellens HJJ (1998) Sudden cardiac death. Circulation 98:2334–2351 [DOI] [PubMed]