

Abstract
Drug transporters are recognized as key players in the processes of drug absorption, distribution, metabolism, and elimination. The localization of uptake and efflux transporters in organs responsible for drug biotransformation and excretion gives transporter proteins a unique gatekeeper function in controlling drug access to metabolizing enzymes and excretory pathways. This review seeks to discuss the influence intestinal and hepatic drug transporters have on pharmacokinetic parameters, including bioavailability, exposure, clearance, volume of distribution, and half-life, for orally dosed drugs. This review also describes in detail the Biopharmaceutics Drug Disposition Classification System (BDDCS) and explains how many of the effects drug transporters exert on oral drug pharmacokinetic parameters can be predicted by this classification scheme.
Key Words: BDDCS, drug transporters, hepatic, intestinal, transporter/enzyme interplay
Introduction
Since the discovery of Pgp in 1976 (1), the importance of drug transporters in influencing the pharmacokinetics of orally dosed drugs has become increasingly evident (2–4). Drug transporters in both the gut and the liver can help control access of drugs to systemic circulation by dictating the amount of drug that enters the body from the gut lumen and influencing how much drug escapes first pass metabolism in both gut and liver. The Biopharmaceutics Drug Distribution and Classification System (BDDCS) can be a helpful tool in predicting the effects that drug transporters in the gut and liver will have on a drug’s pharmacokinetic profile (5). The BDDCS is a modification of the Biopharmaceutics Classification System (BCS) proposed by Amidon et al. (6) that is based on the permeability and solubility characteristics of a drug compound. This review seeks to briefly discuss intestinal and hepatic transporters, review the BDDCS, summarize the effects of transporters in both the gut and the liver on pharmacokinetic parameters of orally dosed drugs, and seek how to potentially predict some of these effects using the BDDCS.
Transporters
A large amount of work has identified and characterized intestinal and hepatic transporters in regards to tissue expression profiles, regulation, mechanisms of transport, substrate and inhibitor profiles, species differences, and genetic polymorphisms. A detailed review of this work is beyond the scope of this article, but further information can be found in several recent extensive reviews (7–20).
Both uptake and efflux transporters are important in determining oral drug disposition by controlling absorption and bioavailability. See Figs. 1 and 2 for their localization in enterocytes and hepatocytes and Table I for examples of substrates for those transporters expressed in the intestine and/or liver. The major uptake transporters responsible for xenobiotic transport belong to the two solute carrier (SLC and SLCO) superfamilies. The SLC superfamily members typically transport Type I organic anions (21), cations, and zwitterions (22), while the SLCO superfamily transports Type II organic anions (21). Members of the SLC and SLCO families use a variety of porter mechanisms (i.e. uniporter, antiporter, symporter), not all of which have been fully elucidated for each specific transporter. In general, these transporters do not utilize an energy source other than a chemiosmotic gradient created by translocation of ions across the membrane (7,9,23–25). The SLC superfamily encompasses a variety of transporters, including the organic anion transporters (OAT, SLC22A), the organic cation transporters (OCT, SLC22A), the electroneutral organic cation transporters (OCTN, SLC22A), the equilibrative nucleoside transporters (ENT, SLC29), the concentrative nucleoside transporters (CNT, SLC28), the apical Na+−dependent bile salt transporter (ASBT, SLC10), the monocarboxylate transporters (MCT, SLC16), and the peptide transporters (PEPT, SLC15). The SLCO family is made up of the organic anion transporting polypeptides (OATP).
Fig. 1.

Enterocyte with intestinal uptake (green) and efflux (blue) transporters. Multidrug resistance protein (MDR), Multidrug resistance associated protein (MRP), Breast cancer resistance protein (BCRP), Monocarboxylate transporter protein (MCT), Peptide transport protein (PEPT), Organic anion transporting polypeptide (OATP), Organic cation transporter (OCT), apical sodium-dependent bile acid transporter (ASBT), Concentrative nucleotide transporter (CNT), Electroneutral organic cation transporter (OCTN), Equilibrative nucleoside transporter (ENT). Adapted from Custodio et al. (58).
Fig. 2.

Hepatocyte with hepatic uptake (green) and efflux (blue) transporters. Multidrug resistance associated protein (MRP), Organic cation transporter (OCT), Organic anion transporter (OAT), Organic anion transporting polypeptide (OATP), sodium-taurocholate cotransporting polypeptide (NTCP), P-glycoprotein (Pgp), Multidrug resistance protein (MDR), Breast cancer resistance protein (BCRP), Bile salt export pump (BSEP). Adapted from Custodio et al. (58).
Table I.
Substrates and Inhibitors of Intestinal and Hepatic Transporters
| Substrates | Inhibitors | |
|---|---|---|
| Gut/Apical Gene (Protein) | ||
| SLC16A1 (MCT1) | lactate, pyruvate, benzoic acid (96) | AR-C117977 (111) |
| SLC15A1 (PEPT1) | ampicillin, captopril, valacyclovir (97) | 4-aminomethylbenzoic acid, Lys[Z(NO(2))]-Pro (112,113) |
| SLC15A2 (PEPT2) | ampicillin, captopril, valacyclovir (97) | cefadroxil, captopril (114) |
| SLCO2B1 (OATP2B1) | estrone-3-sulfate, atorvastatin (98) | rifampin, bromosulfophthalein (97) |
| SLCO1A2 (OATP1A2) | estrone-3-sulfate, deltorphin II, fexofenadine (99) | rifampin, bromosulfophthalein (97) |
| SLC22A3 (OCT3) | cimetidine (97) | corticosterone, decynium22 (115) |
| SLC10A2 (ASBT) | bile salts (taurocholate) (100) | S-8921, SC-435 (116,117) |
| SLC28A1 (CNT1) | thymidine, zidovudine, zalcitabine (101) | adenosine (118) |
| SLC28A2 (CNT2) | guanosine, ribavirin (102) | thymidine (119) |
| SLC22A4 (OCTN1) | quinidine, verapamil (97) | cimetidine, pyrilimine, verapamil (120) |
| SLC22A5 (OCTN2) | quinidine, verapamil (97) | pyrilamine, TEA (121) |
| ABCB1 (Pgp) | digoxin, fexofenadine, indinavir, vincristine, colchicine, | GG918, verapamil (97) |
| topotecan, paclitaxel (97) | ||
| ABCC2 (MRP2) | indinavir, cisplatin, glutathione-, glucuronide-, sulfate-conjugates (97,103) | MK-571, cyclosporine (97,122) |
| ABCG2 (BCRP) | rosuvastatin, doxorubicin, sulfasalazine, topotecan, daunorubicin (97) | FTC, Ko-143 (123) |
| ABCC4 (MRP4) | topotecan, ceftizoxime, cefazolin, urate (104,105) | MK-571 (122) |
| Gut/Basolateral Gene (Protein) | ||
| SLCO3A1 (OATP3A1) | PGE1, PGE2, BQ-123, vasopressin (106) | |
| SLCO4A1 (OATP4A1) | deesterified unoprostone carboxylate (107) | |
| SLC22A1 (OCT1) | acyclovir, amantadine, desipramine, ganciclovir, metformin (97) | montelukast (124) |
| SLC22A2 (OCT2) | amantadine, cimetidine, memantine (97) | bromosulfophthalein (125) |
| ABCC1 (MRP1) | adefovir, indinavir (97) | MK-571, probenecid (122,126) |
| ABCC3 (MRP3) | etoposide, methotrexate, tenoposide (97) | MK-571, indomethacin (122,127) |
| ABCC4 (MRP4) | topotecan, ceftizoxime, cefazolin (105,106) | MK-571 (122) |
| ABCC5 (MRP5) | cGMP, methotrexate, mercaptopurine (108) | MK-571, sildenafil (122,128) |
| SLC16A1 (MCT1) | lactate, pyruvate, benzoic acid (96) | AR-C117977 (111) |
| SLC29A1 (ENT1) | adenosine (109) | nitrobenzylmercaptopurine riboside, dipyridamole (130,131) |
| SLC29A2 (ENT2) | ddC (2'3'-dideoxycytidine), AZT (3'-azido-3'-deoxythymidine), | nitrobenzylmercaptopurine riboside, dipyridamole (130,131) |
| ddI (2',3'-dideoxyinosine) (110) | ||
| Hepatic/Apical Gene (Protein) | ||
| ABCB1 (Pgp) | digoxin, fexofenadine, indinavir, vincristine, colchicine, | GG918, verapamil (97) |
| topotecan, paclitaxel (97) | ||
| ABCB4 (MDR3) | digoxin, paclitaxel, vinblastine (97) | cyclosporine, vinblastine, verapamil (132) |
| ABCC2 (MRP2) | indinavir, cisplatin, ciprofloxacin, glutathione-, glucuronide-, | MK-571, probenecid (122,126) |
| sulfate-conjugates (97,103) | ||
| ABCB11 (BSEP) | vinblastine (97) | cyclosporine, troglitazone (133) |
| ABCG2 (BCRP) | rosuvastatin, doxorubicin, sulfasalazine, topotecan, daunorubicin (97) | FTC, Ko-143 (123) |
| Hepatic/Basolateral Gene (Protein) | ||
| SLC22A1 (OCT1) | acyclovir, amantadine, desipramine, ganciclovir, metformin (97) | prazosin, verapamil (134) |
| SLCO1B1 (OATP1B1) | rifampin, rosuvastatin, methotrexate, pravastatin, thyroxine (97) | rifampin (86,97) |
| SLCO1B3 (OATP1B3) | digoxin, rifampin, methotrexate (97) | rifampin (86,97) |
| SLCO2B1 (OATP2B1) | estrone-3-sulfate, atorvastatin (98) | rifampin (86) |
| SLCO1A2 (OATP1A2) | estrone-3-sulfate, deltorphin II, fexofenadine (99) | rifampin (86) |
| SLC10A1 (NTCP) | rosuvastatin (97) | bosentan, propranolol, cyclosporine (135,136) |
| SLC22A7 (OAT2) | zidovudine (97) | pravastatin, probenecid, taurocholate (137,138) |
| ABCC3 (MRP3) | etoposide, methotrexate, tenoposide (97) | MK-571, indomethacin (122,127) |
| ABCC4 (MRP4) | topotecan, ceftizoxime, cefazolin (104,105) | MK-571 (122) |
| ABCC5 (MRP5) | cGMP, methotrexate, mercaptopurine (108) | MK-571, sildenafil (122,128) |
| ABCC6 (MRP6) | cisplatin, daunorubicin (97) | verapamil, indomethacin, benzbromarone (122,129) |
Efflux transporters expressed in the intestine and liver include P-glycoprotein (Pgp, ABCB1), bile salt export pump (BSEP, ABCB11), multidrug resistance proteins (MRP1- 6, ABCC1-6), and breast cancer resistance protein (BCRP, ABCG2), all members of the ATP-Binding Cassette superfamily (14,26). Members of this superfamily use ATP as an energy source, allowing them to pump substrates against a concentration gradient.
These transporters’ expression in the intestine and/or liver, the two major sites affecting how much drug will get into the body after an oral dose, makes factors affecting their function important determinants of oral drug pharmacokinetics. Regulatory elements controlling their protein levels, genetic polymorphisms leading to increased or reduced function, and coadministration with inhibitors are all important avenues by which a transporter’s ability to transport substrates is altered. While an extensive discussion of these factors is not the purpose of this review, Tables I, II, and III list commonly used transporter inhibitors, established polymorphisms shown to have an effect in human clinical studies, and known regulatory elements for some transporters.
Table II.
Regulatory Nuclear Hormone Receptors and Ligands
| Gene (Protein) | Nuclear Receptor | Ligand |
|---|---|---|
| ABCB1 (MDR1) | PXR, CAR | rifampin (139,140) |
| ABCC2 (MRP2) | PXR, CAR, FXR | rifampin, phenobarbital, chenodeoxycholic acid (141) |
| ABCC3 (MRP3) | PXR, CAR | rifampin, phenobarbital (142) |
| ABCC4 (MRP4) | CAR | rifampin (143) |
| ABCG2 (BCRP) | PPARg | rosiglitizone (144) |
| ABCB11(BSEP) | FXR | chenodeoxycholic acid (145) |
| SLC10A1 (NTCP) | GR | dexamethasone(146) |
| SLCO1A2 (OATP1A2) | PXR | rifampin (147) |
| SLCO1B1 (OATP1B1) | PXR | rifampin (142,148) |
| SLCO1B3 (OATP1B3) | FXR | chenodeoxycholic acid (149) |
Table III.
Examples of Clinically Relevant Polymorphisms
| Gene (Protein) | SNP | Haplotype | Clinical Association |
|---|---|---|---|
| ABCB1 (Pgp) | 3435C>T | reduced digoxin exposure (150) | |
| 3435C>T | increased digoxin exposure (151) | ||
| 2677G>T | increased digoxin exposure (152) | ||
| C3435C>T, 2677G>A | decreased fexofenadine exposure (153) | ||
| ABCC2 (MRP2) | -1549G>A, -1019A>G, -24C>T, 1249G>A, | reduced irinotecan-associated diarrhea (154) | |
| IVS26, -34T>C, and 3972C>T | |||
| ABCG2 (BCRP) | 421C>A | reduced diflomotecan bioavailability (155) | |
| 421C>A | increased topotecan bioavailability (156) | ||
| 421C>A | decreased rosuvastatin exposure (157) | ||
| SLCO1B1 (OATP1B1) | 388A>G, 521T>C | increased pravastatin exposure (158) | |
| 521T>C | increased pravastatin exposure (159) | ||
| 521T>C | increased fexofenadine exposure (160) | ||
| 521T>C | increased repaglinide exposure (161) | ||
| 388A>G | 388A>G, 521T>C | increased pitavastatin exposure (162) | |
| 521T>C | increased rosuvastatin exposure (163) | ||
| 521T>C | increased atorvastatin exposure (163) | ||
| 521T>C | increased nateglinide exposure (164) | ||
| 521T>C | increased atrasentan exposure (165) | ||
| 388A>G | 388A>G, 521T>C | increased irinotecan exposure (166) | |
| 388A>G, 521T>C | increased ezetimibe exposure (167) | ||
| 521T>C | increased torsemide exposure (168) | ||
| SLCO1B3 (OATP1B3) | T334G, G699A | increased mycophenolic acid exposure (169) | |
| SLC22A1 (OCT1) | R61C, G401S, 420del | G465R, 420del; G174S, 420del | increased metformin exposure (65) |
The Biopharmaceutics Drug Disposition Classification System (BDDCS)
The BDDCS (5) is a modification of the BCS (6), in which compounds are categorized in four classes according to their permeability and solubility, characteristics that will greatly influence oral absorption. The BCS (See Fig. 3) was developed to aid in predicting in vivo pharmacokinetic behavior from in vitro solubility and in vitro or in vivo permeability measurements. The FDA, hoping to ease regulatory and financial burdens, enacted the BCS as a method for determining which compounds may receive a waiver for in vivo bioavailability and bioequivalence testing (27).
Fig. 3.

The Biopharmaceutics Classification System (BCS) as put forth by Amidon et al. (6) and defined by the FDA (21).
Class 1 compounds, having high solubility and permeability, formulated as immediate release solid dosage forms that display rapid dissolution may be eligible for the waiver. Solubility is relatively easily determined; however this is not the case with permeability. The FDA, in implementing the BCS, considers a highly permeable compound as one that achieves 90% absorption in humans (27). This is based on the finding that all of the compounds initially studied in human in situ gut permeability studies (28,29) exhibiting permeability equal to or greater than metoprolol (1.34 × 10−4 cm/sec; 17 of 29 compounds) show equal or more than 90% extent of absorption in humans. In contrast, those 12 compounds exhibiting permeability less than metoprolol were less than 90% absorbed in humans. This is an unfortunate coincidence with the potential for confusing applicability, since permeability is a rate parameter, whereas percent absorption is an extent parameter, and they are not interchangeable. A situation can be envisioned in which a compound has low permeability but achieves high absorption. For example, sotalol exhibits a low Papp in the apical to basolateral direction of 1.53 ± 0.29 (×10−6 cm/sec) in a Caco2 cell line yet has 95% absorption compared to other compounds with similar Papp values that show only 50% (atenolol) and 70% (acebutolol) absorption (30).
The recognition that BCS Class 1 and 2 compounds are mainly eliminated by metabolism while Class 3 and 4 compounds are mainly eliminated by biliary or renal excretion of unchanged drug led to the development of the BDDCS (See Fig. 4 and Table IV) by Wu and Benet (5), in which the permeability characteristic was replaced by route of elimination. It is believed that extent of metabolism (by oxidative Phase 1 and conjugative Phase 2 enzymes) is an appropriate surrogate for permeability since for a drug to get metabolized, it must get absorbed (31). Generally, a drug’s elimination route is much easier to determine than its extent of absorption or its permeability in humans. Currently, there are several methods for determining drug permeability detailed in the FDA CDER guidance (27). Human pharmacokinetic studies to determine mass balance or absolute bioavailability can be used, but they can be affected by high inter-subject variability and are expensive and time-consuming. Human in vivo intestinal perfusion studies, extensively reviewed by Lennernas et al. (32), are useful, and several methods are available to conduct these experiments, but they have the same drawbacks as pharmacokinetic studies. Intestinal perfusion studies with representative animal models either in vivo or in situ, or excised tissue studies with human or animal intestinal tissue are other options, as are in vitro cell studies in an epithelial cell line, such as Caco2 monolayers (27). While the use of animal models is less expensive and time-consuming than human studies, there are species differences in expression and substrate profiles for transporters and metabolic enzymes that can lead to misclassification of compounds (33–35). Indeed, the CDER guidance recommends that non-human systems be used only for compounds transported by passive processes (27). When using human cell lines in permeability studies, care must be taken to ensure that expression levels of transporters are representative of those seen in vivo (36–38); otherwise, discrepancies could arise between in vitro and in vivo results.
Fig. 4.

Biopharmaceutics Drug Disposition Classification System (BDDCS) as put forth by Wu and Benet (5).
Table IV.
Classification of 172 Drugs by BDDCS adapted from Wu and Benet (5)
| Class 1: High Solubility/Extensive Metabolism | Class 2: Low Solubility/Extensive Metabolism | ||
|---|---|---|---|
| Abacavir | Ketoprofen | Albendazole | Ketoconazole |
| Acebutolol | Ketorolac | Amiodarone | Lansoprazole |
| Acetaminophen | Labetolol | Atorvastatin | Lopinavir |
| Albuterol | Levamisole | Azathioprine | Lovastatin |
| Allopurinol | Levodopa | Azithromycin | Mebendazole |
| Amitryptiline | Lidocaine | Carbamazepine | Mefloquin |
| Antipyrine | Meperidine | Carvediol | Nalidixic acid |
| Buspirone | Metoprolol | Chlorpromazine | Naproxen |
| Caffeine | Metronidazole | Cisapride | Nelfinavir |
| Chloramphenicol | Midazolam | Clofazamine | Nevirapine |
| Chlorpheniramine | Minocycline | Cyclosporine | Nifedipine |
| Codeine | Misoprostol | Danazol | Oxaprozin |
| Colchicine | Morphine | Dapsone | Phenytoin |
| Cyclophosphamide | Phenobarbital | Diclofenac | Piroxicam |
| Desipramine | Phenylalanine | Diflunisol | Praziquantel |
| Dexamethasone | Prednisolone | Efavirenz | Raloxifene |
| Diazepam | Primaquine | Felodipine | Rifampin |
| Diltiazem | Promazine | Flurbiprofen | Ritonavir |
| Diphenhydramine | Promethazine | Glipizide | Saquinavir |
| Disopyramide | Propranolol | Glyburide | Sirolimus |
| Doxepin | Pyrazinamide | Griseofulvin | Spironolactone |
| Enalapril | Quinidine | Haloperidol | Sulfamethoxazole |
| Ergonovine | Quinine | Ibuprofen | Tacrolimus |
| Ergotamine | Rosiglitizone | Indinavir | Tamoxifen |
| Ethinyl estradiol | Salicyclic acid | Indomethacin | Terfenadine |
| Fluoxetine | Sotolol | Itraconazole | Warfarin |
| Glucose | Theophylline | Ivermectin | |
| Hydralazine | Valproic acid | ||
| Imipramine | Verapamil | ||
| Isoniazid | Zidovudine | ||
| Isosorbid dinitrate | |||
| Class 3: High Solubility/Poor Metabolism | Class 4: Low Solubility/Poor Metabolism | ||
| Acyclovir | Ganciclovir | Acetazolamide | |
| Amiloride | Hydrochlorothiazide | Aluminum hydroxide | |
| Amoxicillin | Lamivudine | Amphotericin | |
| Atenolol | Levofloxacin | Chlorothiazide | |
| Atropine | Lisinopril | Chlorthalidone | |
| Bidisomide | Lithium | Ciprofloxacin | |
| Bisphosphonates | Lomefloxacin | Colistin | |
| Captopril | Metformin | Digoxin | |
| Cefazolin | Methotrexate | Furosemide | |
| Cetirizine | Metoclopramide | Neomycin | |
| Chloroquine | Nadolol | Nystatin | |
| Cimetidine | Neostigmine | Ofloxacin | |
| Cloxacillin | Penicillins | Phenazopyridine | |
| Dicloxacillin | Pitavastatin | Talinolol | |
| Doxycycline | Pravastatin | ||
| Ephedrine | Pyridostigmine | ||
| Erythromycin | Ranitidine | ||
| Ethambutol | Riboflavin | ||
| Famotidine | Tetracycline | ||
| Fexofenadine | Trimethoprim | ||
| Fluconazole | Valsartan | ||
| Folinic acid | Zalcitabine | ||
The use of human liver microsomes, microsomes generated from Sf9 insect cells expressing recombinant metabolic enzymes, and isolated human and animal hepatocytes are widely used to help determine if a drug will be eliminated by metabolism. Simple incubation experiments will determine if a compound is a substrate for metabolizing enzymes and can be followed by in vivo animal and human studies to determine the importance of metabolism as a route of elimination. Information on elimination route is obtained by measuring parent drug and metabolite levels in plasma and urine collected during Phase 1 human pharmacokinetic studies.
In the BDDCS framework, if a drug is ≥ 90% metabolized (a conservative estimate that matches the 90% absorbed criteria currently used in BCS), then based on solubility, the drug can be designated as Class 1 or 2. The ≥ 90% absorption of BCS (21) and the recommendation of Benet et al. (23) to add ≥ 90% metabolized as an alternative basis for allowing a waiver of in vivo bioequivalence are regulatory issues. Wu and Benet (5) recommend for transporter predictions utilizing ≥ 70% metabolism for Class 1 and 2 drugs and <70% metabolism for Class 3 and 4 drugs. They point out that in fact very few marketed drugs fall into the 30 to 70% metabolism category and that most drugs are either extensively metabolized (≥70%) or markedly poorly metabolized (<30%).
The main difficulty in utilizing BCS to make predictions about drug disposition, and particularly the effect of transporters, relates to the fact that the FDA has promulgated a regulatory definition of permeability that differs from the scientific definition. The FDA defines a drug as BCS Class 1 when the extent of absorption (a thermodynamic measure) is ≥90%, and then states that this is a highly permeable (a kinetic measure) compound even when measured permeability is low. This ambiguity is reflected in the recent paper of Chen and Yu (39). Our laboratory is in the process of addressing this issue in more detail (40). Talinolol is an excellent example of the BCS/BDDCS difference. Talinolol is listed in all BCS compilations as a Class 2 drug because it has a high extent of absorption. However, its measured permeability is low (41). BDDCS classifies talinolol as a Class 4 drug because it is not metabolized, and thus predicts its poor permeability and it being a substrate for both an uptake and efflux transporter as demonstrated by Shirasaka et al. (41).
One of the major misconceptions of the scientific community is that BCS and BDDCS allow prediction of the extent of absorption. Because FDA has confounded permeability and extent of absorption, BCS cannot predict absorption. That is, a drug is considered to be highly absorbed in BCS as a result of the absorption measurement, independent of whether permeability is high or low. In contrast, if a drug is extensively metabolized via Phase 1 and Phase 2 enzymes, BDDCS predicts it will be highly absorbed. However, if a drug is poorly metabolized, BDDCS provides no predictions of the extent of absorption; it may be high (in some cases because of transporters) or low. BDDCS, however, tells you that transporters must be evaluated.
The BDDCS allows for some general predictions regarding the role of transporters in oral drug disposition based upon the drug’s classification (Fig. 5). The relevant transporters in terms of BDDCS class in Fig. 5 reflect what Wu and Benet observed for the great majority of drugs in each class. Of course, such a simple categorization in only four classes for all drugs cannot be expected to be universally true with no exceptions, as stated by Wu and Benet (5). It is even more difficult to hypothesize universal scientific explanations for these findings. However, those proposed by Wu and Benet (5) as possible explanations are as follows: Class 1 drugs are highly permeable and highly soluble, allowing them to pass through the gut lumen unaided by transporters. This will occur even if a Class 1 compound is a substrate in vitro for an uptake or efflux transporter expressed in the gut. This is hypothesized to occur because the gut is sufficiently leaky, so that the high permeability of these drugs does not give the transporter sufficient access to the drug or, alternatively, the drug’s physicochemical properties allow it to achieve a high enough concentration in the lumen and within enterocytes that it will saturate any such transporter and be independent of transporter effects (5). Class 2 drugs are also highly permeable, so they will generally be able to enter enterocytes by passive diffusion, unaided by uptake transporters. However, due to low solubility limiting lumenal concentration, they are unlikely to saturate efflux transporters. Consequently, Class 2 compounds can be effluxed out of enterocytes and subject to efflux transporter effects that can influence bioavailability and absorption rate.
Fig. 5.

Transporter effects predicted by BDDCS following oral dosing.
Of the two explanations above for high permeability, only the first, i.e., that the hepatocyte is sufficiently leaky, so that the high permeability of these drugs does not give the transporter sufficient access to the drug, seems reasonable for Class 1 drugs. At this time, we have no good explanation for why certain (but not all) Class 2 drugs are substrates for hepatic uptake and efflux transporters. Perhaps the explanation of Fagerholm (42) that difference in the degrees of “high” permeability dictate whether the compound will be a substrate for transporters is correct, but this requires further investigation. Class 3 compounds have high solubility but low permeability, while Class 4 compounds have both low solubility and low permeability. For both of these classes, drug is unable to get into enterocytes unless aided by an uptake transporter. Even for highly soluble Class 3 drugs, the need for uptake transport will limit the intra-enterocyte drug concentration, making saturation of efflux transporters unlikely. Therefore, both Class 3 and Class 4 drugs may be subject to uptake and efflux transporter effects.
Oral Bioavailability (F) and Exposure (Area Under the Concentration-Time Curve)
Oral bioavailability is defined as the fraction of a dose administered that is absorbed into systemic circulation intact (43). It is a function of the fraction of the dose that is absorbed in the enterocytes (FA), the fraction of the absorbed dose that passes through the gut membranes intact (FG), and the fraction passing through the liver intact (FH) before reaching the systemic blood supply (Eq. 1). Transporters will help modulate F of drugs by playing a role in how much drug gets across the gut membranes and how much drug is taken up into hepatocytes.
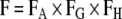 |
1 |
Bioavailability is proportional to the area under the concentration-time curve (AUC), which is a measure of drug exposure (Eq. 2). Consequently, transporter effects on F will result in a corresponding effect on AUC. Changes in AUC are easier to determine than those on F, since both in vivo oral and IV studies are needed to quantitate absolute bioavailability. Many drugs are only available in either oral or IV formulations, but not both.
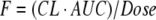 |
2 |
The concept of bioavailability relates to Eq. 2. For an IV dose, bioavailability is assumed to be complete, i.e. F = 1. Thus, assuming clearance (CL) to be constant between the IV and oral dosings, oral F may be determined by comparing exposure for oral and IV doses, while correcting for any differences in dose between the two studies. In situ and ex situ experiments with individual organs, such as the intestine or liver, using perfusion techniques are very useful for studying transporter effects on F, and coupling results from these organ experiments with whole animal and human AUC data can help elucidate the mechanisms involved in altering both bioavailability and drug exposure.
In the gut, Class 1 and 2 compounds have favorable permeability characteristics, allowing them to enter enterocytes unaided by apical uptake transporters. As explained above, due to their high solubility and rapid permeability, Class 1 compounds also readily bypass efflux transporters or, alternatively, the high concentrations likely saturate apical efflux transporters, and, therefore, Class 1 drug AUC and F will not be influenced by efflux transporters even if they are substrates in vitro. Cao et al. (44) demonstrated this in situ with an intestinal rat perfusion model in which they determined verapamil and propranolol permeabilities and showed that they were independent of Pgp expression level, which increased 6-fold along the intestinal tract from the duodenum to the ileum. While the status of propranolol as a Pgp substrate is unclear, it is well recognized that verapamil is a good Pgp substrate. Although AUC and F of Class 1 compounds are independent of apical efflux transporters, such efflux transporters will influence AUC and F of Class 2 compounds. Due to their low solubility, the low concentration of Class 2 compounds should not saturate apical efflux transporters as Class 1 compounds can. Therefore, apical efflux transporters will be able to pump drug back out into the lumen where it may re-enter enterocytes, thus allowing repeated exposure to drug metabolizing enzymes within the enterocytes. This interplay between apical gut efflux transporters and metabolizing enzymes within enterocytes has been well-characterized using in vitro and in vivo systems (45,46). Cummins et al. used both a Caco2 cell line overexpressing CYP3A4 (45) and a rat intestinal perfusion model (46) to demonstrate the importance of Pgp in influencing metabolism of K77, a cysteine protease inhibitor and dual substrate of Pgp and CYP3A4. In brief, both the cell line studies and the intestinal perfusion studies showed that Pgp inhibition increased intracellular K77 concentrations or appearance in mesenteric blood while decreasing polarized transport. No corresponding changes were seen for the CYP3A4 substrates felodipine (45), a Class 2 compound that is not a substrate of Pgp, or midazolam (46), a Class 1 compound not influenced by Pgp in the gut. The effects of inducing Pgp have been shown to yield opposite effects to that for Pgp inhibition in vivo with the Class 2 (BCS)/Class 4 (BDDCS) compound talinolol. Schwarz et al. (47) induced gut Pgp by pretreatment with St. John’s Wort and saw a significant reduction (25%) in F compared to the water control. Likewise, Westphal et al. (48) saw a significant (~20%) reduction in F for talinolol when Pgp was induced with rifampin. While the interplay between Pgp and CYP3A4 has been extensively studied, this phenomenon may also occur with other enterocytic drug metabolizing enzymes (CYP3A4, CYP2C9, CYP2C19, CYP2C8, CYP2D6, esterases, epoxide hydrolases, UGT1A1, UGT1A7–10, SULT1E1, SULT2A1, SULT1A3, N-acetyltransferases, glutathione-S-transferases) (49), and other apical efflux transporters (i.e., BCRP, MRP2). One such example is provided by Sesink et al. (50). Using Mrp2 knockout mice and the specific Bcrp1 inhibitor FTC, they showed in an in situ intestinal perfusion model that inhibiting apical efflux of quercetin glucuronides by Bcrp1 increased plasma quercetin levels more than 2-fold after enzymatic hydrolysis of the plasma samples. Another example is provided by Su et al. (51) in which rat ileal perfusions were performed with topotecan in the presence and absence of novobiocin, a Bcrp1 inhibitor. With novobiocin, a ∼ 50% decrease in intestinal secretion was seen with a corresponding increase in permeability, indicating that Bcrp1 inhibition influences topotecan absorption and bioavailability. Currently it is difficult to study BCRP and MRP2 interactions in human clinical studies due to the lack of specific inhibitors that can be used in people. Also, many drugs are substrates for multiple efflux transporters. Pgp and BCRP have extensive substrate overlap, and, therefore, one transporter may compensate when the other is inhibited. In preclinical studies, the use of knockout animals can circumvent this problem, but an effective strategy for human studies remains to be found. More studies with these other gut enzymes and efflux transporters are needed to fully characterize the importance of their potential interplay in influencing AUC and F of substrate drugs.
BDDCS Class 3 and 4 drugs, which have low permeability, require apical uptake transporters to help them cross the luminal barrier, since their low permeability limits diffusion into enterocytes. Clinical studies have shown that inhibition of apical gut uptake transporters can also influence plasma levels of substrate drugs. Glaeser et al. (52) showed that administration of grapefruit juice concomitantly or 2 h prior to dosing the Class 3 compound fexofenadine, an OATP1A2 substrate, decreased fexofenadine plasma levels compared to water control, as well as dosing of grapefruit juice 4 h prior to fexofenadine dosing. Since fexofenadine, like all BDDCS Class 3 drugs, is not extensively metabolized, the authors concluded that the effect was due to inhibition of OATP1A2 uptake of fexofenadine. Kato et al. (53) demonstrated in mouse pharmacokinetic and Ussing chamber studies that celiprolol absorption was significantly decreased by bromosulfophthalein (BSP), a broad organic anion transport inhibitor. They used mdr1a/1b knockout mice to avoid any confounding effects of Pgp and demonstrated decreased plasma levels of celiprolol upon BSP coadministration in the pharmacokinetic studies. They confirmed that the effect was due to decreased absorption in Ussing chamber studies by showing that BSP significantly reduced A to B permeability. Induction of intestinal uptake transporters conversely should increase the AUC and F of their substrates. A rat pharmacokinetic study by Koitabashi et al. (54) demonstrated that administration of orange juice significantly increased the AUC of orally dosed pravastatin while having no effect on IV pravastatin, indicating a change in F. They further investigated mRNA and protein levels of rat Oatp1a1 and Oatp1a4 and saw significant increases in both for each transporter. In addition to the rat studies, Koitabashi et al. (54) also ran a human study to examine the effect of orange juice on oral pravastatin. As with the rats, a significant increase in AUC was seen, although a corresponding IV study was not possible to provide evidence that the change was due to increased F.
Class 3 and 4 drugs also have the potential to be influenced by efflux transporters, since they are unlikely to attain intracellular concentrations high enough to saturate efflux transporters. Dahan et al. (55) examined the effect of intestinal Pgp on two Class 3 compounds, famotidine and cimetidine. They determined in rat in situ intestinal perfusion studies that differential Pgp expression in the proximal jejunum versus the distal ileum led to different famotidine and cimetidine permeabilities in these segments with significantly decreased permeability seen in the distal segment compared to the proximal segment, corresponding to the higher Pgp expression observed in the distal segment. Upon Pgp inhibition by verapamil, a significant increase in permeability for both compounds was seen in the distal ileum segment, while no significant difference was seen in the proximal jejunum. The distal ileum permeability approached the permeability seen in the proximal jejunum segment, effectively making permeability constant along the small intestine in the presence of verapamil. Evidence supporting the effect of apical gut efflux on BDDCS Class 4 drug absorption also exists for digoxin, a Pgp substrate. A human clinical study by Greiner et al. (56) showed that induction of Pgp by rifampin decreased the absorption of orally dosed digoxin, even though digoxin is not metabolized by CYPs in humans. Subjects received either IV or oral digoxin before and after Pgp induction. Pgp induction was examined by immunohistochemistry and Western blot analysis of duodenum samples and was shown to increase 3.5-fold after ~2 weeks of rifampin dosing. F was significantly decreased (19%) after Pgp induction. Further work is required to fully characterize the effect of gut apical transporters on Class 3 and 4 drugs.
The influence of intestinal basolateral uptake and efflux transporters has not yet been examined in depth to determine their influence on AUC and F; however, we can predict what effects they may have based on what we predict for the effects of apical transporters. Table V summarizes the effects of intestinal drug transporters on AUC by BDDCS class.
Table V.
Predicted Effect of Intestinal Drug Transporters on Exposure (AUC) by BDDCS Class

The BDDCS is also useful for making predictions regarding food effects on orally dosed drugs. The AUC and F of many compounds are greatly affected by concomitant food intake, and the FDA recommends that high fat meals (800–1000 cal: 50–65% from fat, 25–30% from carbohydrates, 15–20% from protein) be used in food-effect studies in the guidance “Food Effect Bioavailability and Fed Bioequivalence Studies” (57). Many factors are believed to contribute to these food effects, including changes in gastric emptying time, bile flow, pH of the intestine, splanchnic blood flow, and gut wall metabolism. A variety of evidence exists supporting food effects on transporters as well and is described in detail in a review by Custodio et al. (58). The BDDCS is able to predict what is seen clinically in food effect studies. These predictions are explained if it holds that high-fat meal components inhibit apical gut uptake and efflux transporters. Fig. 6 summarizes the effects of high fat meals on F and tmax by BDDCS Class (5,58).
Fig. 6.

Predicted high fat meal effects by BDDCS classification after Wu and Benet (5).
Drug that makes it across the intestinal barrier passes into the hepatic portal vein and travels to the liver, the final major site for first-pass effects, before reaching the systemic circulation. Drug transporters will play a role in determining how much drug enters hepatocytes and is exposed to hepatic drug metabolizing enzymes and biliary excretion. In hepatocytes, basolateral uptake transporters function to take drug up from the sinusoidal blood into hepatocytes while basolateral efflux transporters pump drug back out into the sinusoidal blood. Apical transporters face the bile ducts; therefore, apical efflux transporters will pump drug into the bile.
The major predictive difference for the BDDCS system between the gut and the liver relates to the importance of uptake transporters for BDDCS Class 2 drugs. It is possible that the intestinal membrane may be “leakier” than the hepatic membrane, allowing more drug to diffuse across the intestinal barrier than the hepatic barrier. For example, while BDDCS Class 2 compounds do not require uptake transporters to get into enterocytes, it has been shown that hepatic uptake transporters can be an important determinant of drug entry into hepatocytes and could thereby affect F through the first pass effect and AUC through the combination of the first pass effect and subsequent passages through the liver. Tirona et al. (59) demonstrated that the Class 2 compound rifampin could be transported by OATP1B1 and, to a lesser extent, OATP1B3 expressed in HeLa cells. Su et al. (60) determined that OATP1A2 is able to transport saquinavir, also a Class 2 compound, in oocytes and in HepG2 cells, a human hepatocellular liver carcinoma cell line. The AUC and F of Class 3 and 4 drugs, due to their low permeability, will also be influenced by hepatic uptake transporters. Watanabe et al. (61) investigated the importance of hepatic uptake transporters on the plasma levels of the Class 3 compound pravastatin. They developed a physiologic based pharmacokinetic (PBPK) model that incorporated hepatic sinusoidal uptake and demonstrated the sensitivity of plasma levels to a change in uptake. Decreasing uptake transporter activity to 1/3 of control led to simulated pravastatin plasma levels 271% of the control, while increasing uptake transporter activity to 3x the control led to simulated plasma levels 14% of control. Hepatic basolateral efflux transporters (i.e., MRP3, 4, 5, 6) may influence AUC and F since they provide an avenue of exit from hepatocytes. As in the intestine, it is possible that these membrane-bound transporters can interact with drug before it interacts with metabolizing enzymes associated with the endoplasmic reticulum. Altering these transporters’ function could affect the AUC and F of their substrates by altering first pass hepatic metabolism or biliary excretion. Theoretically, the apical efflux transporters could also affect AUC and F by helping to control intra-hepatocyte drug levels, although this effect would be dependent on the drug’s ability to get back out of the hepatocyte into sinusoidal blood either by diffusion or by basolateral efflux. A summary of the effects of hepatic drug transporters on AUC is given in Table VI and explicated in the next section.
Table VI.
Predicted Hepatic Effect of Drug Transporters on Exposure (AUC) by BDDCS Class

Clearance (CL)
Clearance is technically defined as a proportionality factor that relates the rate of elimination of drug to the systemic drug concentration (43). It must be kept in mind that metabolism that occurs in the gut and first pass in the liver does not contribute to CL, since that drug is lost before it reaches systemic circulation. For orally dosed drugs, CL and F are intimately associated and cannot be separated unless corresponding IV studies have been done. Since this review deals with orally dosed drugs, we will discuss transporter effects on CL/F in addition to the intrinsic clearance by the liver.
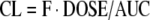 |
2a |
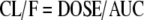 |
2b |
Gut transporters will exclusively affect F, thus CL will not change in response to induction or inhibition of gut transporters, only F will. For orally dosed drugs the liver and the kidneys are the major eliminating organs, and their clearances are additive.
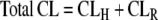 |
3 |
CLH can further be divided into uptake and elimination processes, with the latter divided into biliary excretory clearance and hepatic metabolic clearance (43). Both mechanisms can be affected by transporters.
Transporters in the liver and kidney can affect the CL of the portion of an orally administered drug that escapes the first-pass effect. Hepatic uptake transporters are expressed on the basolateral surface of hepatocytes facing the sinusoidal blood supply. Inhibiting these transporters prevents drug from being exposed to metabolizing enzymes within hepatocytes, decreasing the CLH of drugs that are substrates for these enzymes. For drugs that are not metabolized but are eliminated by biliary excretion, CLH may also decrease, since drug is prevented from getting into the bile through the hepatocytes. Conversely, induction of hepatic uptake transporters will increase the amount of drug able to get into hepatocytes and thus gain exposure to metabolizing enzymes or get pumped into the bile, thereby increasing CLH. CL/F will change in the same direction as CLH for each case, but the CL value is not separable from F based on oral data alone. In addition to basolateral uptake transporters, hepatocytes also express basolateral efflux transporters that pump drug back out into the sinusoidal blood. The effects of inhibiting or inducing these transporters will be opposite those for the basolateral uptake transporters. Inhibiting basolateral efflux will cause more drug to remain inside hepatocytes and will lead to increased CLH either by increased exposure to metabolizing enzymes or to efflux into the bile, while inducing them will lead to more drug being pumped out of hepatocytes and decreased CLH. Again, CL/F will change in the same direction as CLH.
Class 1 compounds, due to their high permeability, appear not to be influenced by basolateral hepatic uptake transporters. In the gut, Class 2 compounds are not strongly influenced by apical uptake transporters due to their high permeability; however, it has been shown that at least some of these compounds are influenced by basolateral hepatic uptake transporters. A human pharmacokinetic study by Lau et al. (62) showed that an IV dose of the OATP inhibitor rifampin significantly decreased CL/F of atorvastatin, presumably by decreasing basolateral uptake by OATP1B1 leading to decreased hepatic metabolism by CYP3A4. In vitro studies showed that 50 μM rifampin did not inhibit CYP3A4 metabolism of atorvastatin, and rifampin plasma levels in the human study were ∼15 μM, indicating that inhibition of hepatic metabolism was not a causal factor in reducing CL/F. Similar results were seen by Zheng et al. (63) in a human study examining the effect of IV rifampin on orally dosed glyburide, an OATP2B1 substrate. Upon multiple dosing of rifampin, but when rifampin was no longer present in the systemic circulation, glyburide CL/F was significantly increased compared to the control, due to induction of metabolic enzymes and transporters. As stated earlier, we have no good explanation for why some BDDCS class 2 compounds are substrates for hepatic uptake transporters but this is not seen for BDDCS class 1 compounds. It is difficult to imagine a scientific explanation based on a solubility effect in the liver. This is an area requiring further investigation.
As expected due to their low permeability potential, the CL/F of Class 3 and 4 drugs also can be influenced by basolateral hepatic uptake transporter effects. Studies with the Class 3 drugs pravastatin and pitavastatin have shown the effects of decreased function of OATP1B1 on CL/F. Deng et al. (64) performed human clinical studies looking at the effect of genotype of OATP1B1 (OATP1B1*1a homozygotes versus OATP1B1*15 homozygotes) on pitavastatin and pravastatin pharmacokinetics. They saw that CL/F for both compounds was significantly decreased in subjects with OATP1B1*15/*15 genotype compared to wildtype (2.07-fold and 2.88-fold for pravastatin and pitavastatin, respectively). Metformin, another Class 3 compound and a substrate for OCT1, has also shown transporter influence on pharmacokinetic parameters. Shu et al. (65) performed a clinical study to determine the effect of OCT1 genetic variation on metformin pharmacokinetics. Metformin was orally dosed in two subject groups, one group with wildtype OCT1 and the other composed of subjects carrying at least one reduced function allele. They found that the CL/F was significantly reduced in the variant allele group.
Hepatic apical efflux transporters responsible for pumping drug into the bile may also affect both hepatic metabolic clearance and biliary excretory clearance of their substrate drugs. Inhibiting apical efflux causes drug to remain trapped within hepatocytes. If the compound is a substrate for metabolizing enzymes (BDDCS Class 1 and 2 compounds), its metabolism will increase due to increased exposure to enzymes, and CLH will increase. If the drug is not subject to metabolic transformation but rather is eliminated by biliary excretion (Class 3 and 4 compounds), its CLH may decrease due to decreased excretion into bile. An alternative route of elimination is possible though, which may cause a situation to arise in which there is no net change in CL/F, but rather a change in the clearance profile seen as decreased biliary excretion and increased renal excretion if the drug is sufficiently hydrophilic to be eliminated in the urine. Induction of hepatic apical efflux transport could lead to increased biliary excretion but decreased metabolic transformation, making the effect on CLH dependent upon a drug’s substrate status for metabolizing enzymes. Work remains to be done to elucidate the effect of hepatic apical efflux transporters on CL/F separate from that of apical gut efflux transporters.
While work has been done on determining effects of hepatic basolateral efflux in in situ isolated perfused rat liver studies (66) and in vivo with intraperitoneal dosing (67), the effect of hepatic basolateral efflux transporters on oral drug pharmacokinetics has not been extensively studied. However, we can hypothesize what effects inhibition or induction will have. Inhibition of hepatic basolateral efflux will cause drug to be trapped within hepatocytes, similar to hepatic apical efflux inhibition, and will lead to increased exposure to drug metabolizing enzymes or to apical efflux. This may lead to an increase in hepatic metabolic or biliary excretory clearance. Induction of hepatic basolateral efflux will cause more drug to be pumped back into the sinusoidal blood, decreasing the hepatic contribution to CL/F, but depending on the drug’s hydrophilicity may be compensated for by increased renal CL.
Half-life (t1/2)
The elmination half-life is dependent upon CL and V (Eq. 4).
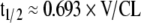 |
4 |
Whatever effects transporters exert on V and CL will influence the t1/2, either causing it to increase if the increase in V predominates, decrease if the increase in CL is more pronounced, or have no net effect if changes in V and CL cancel out each other. However, transporters in the gut may affect the absorption half-life, t1/2,abs (Eq. 5).
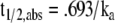 |
5 |
The rate constant of absorption, ka, may be influenced by gut transporters, and the BDDCS can be useful in predicting which drugs are likely to be affected. As stated before, Class 3 and 4 drugs are dependent on gut uptake transporters to get across the luminal membrane. If their uptake is inhibited, this is likely to decrease their absorption rate, driving down ka and increasing t1/2,abs. The opposite will hold true when uptake transporters are induced. The ka will increase and t1/2,abs will decrease as more drug is pumped into enterocytes. Parker et al. (68) ran a human study in which a single oral dose of digoxin was administered after 5 days pretreatment with grapefruit juice and saw a significant decrease in the digoxin ka compared to the water control arm (3.0 ± 2.4 to 1.2 ± 1.0 hr−1, p < 0.05). The plasma concentration vs. time profile also showed a later tmax. No other pharmacokinetic parameters were significantly different between the treatment groups. The ka and tmax results were not what would be expected if the interaction were due to Pgp inhibition (68,69), but rather what would be expected from inhibition of an intestinal uptake transporter. Although the specific OATP isoform responsible for intestinal uptake of digoxin has not been identified, the drug is known to be an OATP1B3 (70) substrate. The BDDCS predicts the clinical results seen by Parker et al., since digoxin is a Class 4 compound in humans and uptake transporter effects will be important for these compounds. Intestinal efflux transporters may also affect ka and t1/2,abs, especially for Class 2 compounds. If apical efflux transporters are inhibited, this will decrease the “cycling” effect that is seen when a drug is repeatedly pumped out and gains reentry into enterocytes, allowing the drug to exit basolaterally more quickly. In a study on the effects of ABCB1 genotype, Solas et al. (71) found that subjects dosed with indinavir (Class 2 compound) carrying one decreased-function allele, C3435CT, had a significantly higher ka (2-fold) compared to controls and carriers of two mutant alleles (they explain the lack of significant difference in the C3435TT group as possibly due to lack of statistical power with the small number of subjects). The t1/2,abs of Class 3 and 4 compounds may also be subject to apical efflux transporter effects. For the Class 3 compound fexofenadine, a Pgp substrate (72,73), Tannergren et al. (74) determined in human intestinal perfusion studies with the Gut-I-Loc system that coperfusion of fexofenadine with the Pgp inhibitor verapamil caused a significant increase in ka compared to the control (0.0030 ± 0.0012 min−1 to 0.0255 ± 0.0103 min−1 (P < .001)), which changes t1/2,abs. Decreasing the absorption rate often changes the appearance of the plasma concentration vs. time profile, even if no change in the extent of absorption as measured by area under the curve (AUC) is seen. The tmax can shift later and Cmax can decrease, causing the profile to appear flatter. Increasing the absorption rate constant may cause tmax to shift earlier and Cmax to increase, sharpening the plasma concentration vs. time profile. Class 1 drugs, for which transporter effects are minimal, should not show a significant change in the absorption rate constant or t1/2,abs.
Transporter effects on ka may also have implications for oral multiple dosing regimens. In a recent analysis, Sahin and Benet (75) defined a new half-life parameter, the operational multiple dosing half-life (t1/2,op), as “the half-life equal to the dosing interval at steady-state where the maximum concentration at steady-state is twice the maximum concentration found for the first dose and where the fall off to the trough concentration from the maximum concentration is consistent with this half-life.” They found that this t1/2,op parameter for some drugs may be quite sensitive to changes in ka. In simulations with diazepam, decreasing the ka from 2.77 h−1 to 0.347 h−1 led to a drastic increase in the t1/2,op, increasing it from about 13 h to 35 h. Diazepam is a Class 1 drug; therefore, we would not expect to see transporter effects on ka. However, further study is warranted to determine what effect varying ka has on the t1/2,op for Class 2, 3, and 4 compounds through transporter interactions.
Volume of Distribution (V)
Volume of distribution is defined as the “volume into which a drug distributes in the body at equilibrium (43)” and is a “direct measure of the extent of distribution (43).” Transporters in the gut will not affect V as it relates the concentration of drug in the systemic circulation to amount of drug in the body. Like CL, V for orally administered drugs cannot be separated from F, and V is reported as V/F. Very few drugs remain within physiologically realistic volumes (43), such as the plasma (3 L), extracellular water (16 L), or total body water (42 L). Instead they distribute into organs and tissues, greatly increasing their apparent V. Transporters may influence V by mediating their transport into and out of a variety of tissues and organs. The most widely studied sites thus far in relation to transporter influence on distribution are the liver and the brain, but the principles determined for these organs should hold for other organs and tissues expressing drug transporters. Several drugs whose site of action is the liver, including some statins, are concentrated in the liver by hepatic uptake transporters. If their hepatic uptake is inhibited, more drug remains in the plasma and less drug gets into the liver, decreasing V/F. For example, Lau et al. (62) saw a significant reduction in Vss/F in response to a single IV dose of rifampin inhibiting the hepatic uptake of atorvastatin. Zheng et al. (63) showed a similar effect of rifampin on the Vss/F for glyburide. Induction may increase the amount of drug getting into the liver, decrease the amount in plasma, and increase V/F. While the effects of altering function of hepatic apical efflux transporters may influence V/F, this effect isn’t always predictable. Decreasing hepatic basolateral efflux may keep drug within hepatocytes, possibly decreasing plasma levels and increasing V/F, while inducing basolateral efflux may lead to decreased V/F by causing there to be more drug in the plasma.
An extensive review of transporter effects on V has recently been published (170).
Conclusions
A wealth of knowledge exists regarding drug transporters, and increasingly their effects on pharmacokinetics are being characterized. Work in the field is far from complete though, and there are many areas in which there is a dearth of information.
While the substrate and inhibition profiles of transporters both new and old are being updated continuously (76–78), new transporters are still being discovered. For example, Engel et al. (79) recently cloned the proton-stimulated organic cation transporter (PMAT) expressed in the intestine, and Zhou et al. (80) determined that it may play a role in intestinal transport of metformin. The search goes on for appropriate probe substrates (81–83) and specific inhibitors (84–86) that will allow testing of single transporters without confounding effects from substrate overlap (12,87,88).
With widespread use of cell models to test substrate profiles and to determine permeabilities, care needs to be taken to ensure that reproducible results are obtained and to be aware of factors that can lead to discrepancies between different laboratories’ results (36,89,90).
The development of models that incorporate different transporters is a huge step forward in the field; however, these models need valid in vivo parameter values in order to accurately predict pharmacokinetics under a variety of conditions (32,91–95). Human and animal studies need to continue to generate data to develop complete and robust in silico models.
The understanding of transporter effects on oral pharmacokinetics has improved vastly over the last 20 years, yet it is still a vibrant and important area of research needing continued effort and advancement.
Acknowledgments
This work was supported in part by NIGMS grant GM75900, the American Foundation for Pharmaceutical Education (AFPE), and Amgen through a fellowship and an unrestricted grant.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta. 1976;455:152–162. [DOI] [PubMed]
- 2.Fromm MF. P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int. J. Clin. Pharmacol. Ther. 2000;38(2):69–74. [DOI] [PubMed]
- 3.Landowski CP, Sun D, Foster DR, Menon SS, Barnett JL, Welage LS, et al. Gene expression in the human intestine and correlation with oral valacyclovir pharmacokinetic parameters. J. Pharmacol. Exp. Ther. 2003;306(2):778–786. [DOI] [PubMed]
- 4.Mizuno N, Sugiyama Y. Drug transporters: their role and importance in the selection and development of new drugs. Drug Metab. Pharmacokinet. 2002;17(2):93–108. [DOI] [PubMed]
- 5.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 2005;22(1):11–23. [DOI] [PubMed]
- 6.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12(3):413–420. [DOI] [PubMed]
- 7.Srimaroeng C, Perry JL, Pritchard JB. Physiology, structure, and regulation of the cloned organic anion transporters. Xenobiotica. 2008;38(7–8):889–935. [DOI] [PMC free article] [PubMed]
- 8.Zaïr ZM, Eloranta JJ, Stieger B, Kullak-Ublick GA. Pharmacogenetics of OATP (SLC21/SLCO), OAT and OCT (SLC22) and PEPT (SLC15) transporters in the intestine, liver and kidney. Pharmacogenomics. 2008;9(5):597–624. [DOI] [PubMed]
- 9.Ciarimboli G. Organic cation transporters. Xenobiotica. 2008;38(7–8):936–971. [DOI] [PubMed]
- 10.Hagenbuch B, Gui C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica. 2008;38(7-8):778–801. [DOI] [PubMed]
- 11.Toyoda Y, Hagiya Y, Adachi T, Hoshijima K, Kuo MT, Ishikawa T. MRP class of human ATP binding cassette (ABC) transporters: historical background and new research directions. Xenobiotica. 2008;38(7–8):833–862. [DOI] [PubMed]
- 12.Zhou SF, Wang LL, Di YM, Xue CC, Duan W, Li CG, et al. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008;15(20):1981–2039. [DOI] [PubMed]
- 13.Kusuhara H, Sugiyama Y. ATP-binding cassette, subfamily G (ABCG family). Pflugers Arch. 2007;453(5):735–744. [DOI] [PubMed]
- 14.Choudhuri S, Klaassen CD. Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. Int. J. Toxicol. 2006;25(4):231–259. [DOI] [PubMed]
- 15.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007;47:566–578. [DOI] [PubMed]
- 16.Mottino AD, Catania VA. Hepatic drug transporters and nuclear receptors: regulation by therapeutic agents. World J. Gastroenterol. 2008;14(46):7068–7074. [DOI] [PMC free article] [PubMed]
- 17.Chinn LW, Kroetz DL. ABCB1 pharmacogenetics: progress, pitfalls, and promise. Clin. Pharmacol. Ther. 2007;81(2):265–269. [DOI] [PubMed]
- 18.Maeda K, Sugiyama Y. Impact of genetic polymorphisms of transporters on the pharmacokinetic, pharmacodynamic, and toxicological properties of anionic drugs. Drug Metab. Pharmacokinet. 2008;23(4):223–235. [DOI] [PubMed]
- 19.Nakamura T, Yamamori M, Sakaeda T. Pharmacogenetics of intestinal absorption. Curr. Drug Deliv. 2008;5(3):153–69. [DOI] [PubMed]
- 20.Food and Drug Administration. http://www.fda.gov/cder/drug/drugInteractions/tableSubstrates.htm#top (accessed 12/11/2008), part of U.S. Food and Drug Administration Center for Drug Evaluation and Research. http://www.fda.gov/Cder/ (accessed 12/11/2008).
- 21.van Montfoort JE, Müller M, Groothuis GM, Meijer DK, Koepsell H, Meier PJ. Comparison of "type I" and "type II" organic cation transport by organic cation transporters and organic anion-transporting polypeptides. J. Pharmacol. Exp. Ther. 2001;298(1):110–115. [PubMed]
- 22.Okabe M, Szakács G, Reimers MA, Suzuki T, Hall MD, Abe T, et al. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008;7(9):3081–3091. [DOI] [PMC free article] [PubMed]
- 23.Li L, Meier PJ, Ballatori N. Oatp2 mediates bidirectional organic solute transport: a role for intracellular glutathione. Mol. Pharmacol. 2000;58(2):335–340. [DOI] [PubMed]
- 24.Satlin LM, Amin V, Wolkoff AW. Organic anion transporting polypeptide mediates organic anion/HCO3− exchange. J. Biol. Chem. 1997;272(42):26340–26345. [DOI] [PubMed]
- 25.Mahagita C, Grassl SM, Piyachaturawat P, Ballatori N. Human organic anion transporter 1B1 and 1B3 function as bidirectional carriers and do not mediate GSH-bile acid cotransport. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293(1):G271–278. [DOI] [PubMed]
- 26.Kosters A, Karpen SJ. Bile acid transporters in health and disease. Xenobiotica. 2008;38(7–8):1043–1071. [DOI] [PMC free article] [PubMed]
- 27.Food and Drug Administration. Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. www.fda.gov/cder/guidance/index.htm (accessed 12/17/2008), part of U.S. Food and Drug Administration Center for Drug Evaluation and Research. http://www.fda.gov/Cder/ (accessed 12/11/2008).
- 28.Takagi T, Ramachandran C, Mermejo M, Yamashita S, Yu LX, Amidon GL. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain and Japan. Mol. Pharmaceut. 2006;3:631–643. [DOI] [PubMed]
- 29.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Houssain AS, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharmaceut. 2004;1:85–96. [DOI] [PubMed]
- 30.Yang Y, Faustino PJ, Volpe DA, Ellison CD, Lyon RC, Yu LX. Biopharmaceutics classification of selected beta-blockers: solubility and permeability class membership. Mol. Pharmaceut. 2007;4(4):608–614. [DOI] [PubMed]
- 31.Benet LZ, Amidon GL, Barends DM, Lennernäs H, Polli JE, Shah VP, et al. The use of BDDCS in classifying the permeability of marketed drugs. Pharm. Res. 2008;25(3):483–488. [DOI] [PMC free article] [PubMed]
- 32.Lennernäs H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica. 2007;37(10–11):1015–1051. [DOI] [PubMed]
- 33.Li N, Zhang Y, Hua F, Lai Y. Absolute difference of hepatobiliary transporter MRP2/Mrp2 in liver tissues and isolated hepatocytes from rat, dog, monkey and human. Drug Metab. Dispos. 2009;37(1):66–73. [DOI] [PubMed]
- 34.Katoh M, Suzuyama N, Takeuchi T, Yoshitomi S, Asahi S, Yokoi T. Kinetic analyses for species differences in P-glycoprotein-mediated drug transport. J. Pharm. Sci. 2006;95(12):2673–2683. [DOI] [PubMed]
- 35.Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006;23(8):1675–1686. [DOI] [PubMed]
- 36.Hayeshi R, Hilgendorf C, Artursson P, Augustijns P, Brodin B, Dehertogh P, et al. Comparison of drug transporter gene expression and functionality in Caco-2 cells from 10 different laboratories. Eur. J. Pharm. Sci. 2008;35(5):383–396. [DOI] [PubMed]
- 37.Arthur JM. The MDCK cell line is made up of populations of cells with diverse resistive and transport properties. Tissue Cell. 2000;32(5):446–450. [DOI] [PubMed]
- 38.Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, et al. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm. Res. 2002;19(10):1400–1416. [DOI] [PubMed]
- 39.Chen ML, Yu L. The use of drug metabolism for prediction of intestinal permeability. Mol. Pharmaceutics. 2009;6(1):74–81. [DOI] [PubMed]
- 40.C.L. Larregieu and L.Z. Benet. The confounding FDA redefinition of intestinal permeability in terms of extent of absorption. AAPS 2009 Annual Meeting Abstract.
- 41.Shirasaka Y, Li Y, Shibue Y, Kuraoka E, Spahn-Langguth H, Kato Y, et al. Concentration-dependent effect of naringin on intestinal absorption of b1-adrenoceptor antagonist talinolol mediated by p-glycoprotein and organic anion transporting polypeptide (Oatp). Pharm. Res. 2009;26(3):560–567. [DOI] [PubMed]
- 42.Fagerholm U. The role of permeability in drug ADME/PK, interactions and toxicity-presentation of a permeability-based classification system (PCS) for prediction of ADME/PK in humans. Pharm. Res. 2008;25(3):625–638. [DOI] [PubMed]
- 43.Wu CY, Benet LZ, Hebert MF, Gupta SK, Rowland M, Gomez DY, et al. Differentiation of absorption and first-pass gut and hepatic metabolism in humans: studies with cyclosporine. Clin. Pharmacol. Ther. 1995;58(5):492–497. [DOI] [PubMed]
- 44.Cao X, Yu LX, Barbaciru C, Landowski CP, Shin HC, Gibbs S, et al. Permeability dominates in vivo intestinal absorption of P-gp substrate with high solubility and high permeability. Mol. Pharmaceut. 2005;2(4):329–340. [DOI] [PubMed]
- 45.Cummins CL, Jacobsen W, Benet LZ. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002;300(3):1036–1045. [DOI] [PubMed]
- 46.Cummins CL, Salphati L, Reid MJ, Benet LZ. In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model. J. Pharmacol. Exp. Ther. 2003;305(1):306–314. [DOI] [PubMed]
- 47.Schwarz UI, Hanso H, Oertel R, Miehlke S, Kuhlisch E, Glaeser H, et al. Induction of intestinal P-glycoprotein by St John’s wort reduces the oral bioavailability of talinolol. Clin. Pharmacol. Ther. 2007;81(5):669–678. [DOI] [PubMed]
- 48.Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, et al. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin. Pharmacol. Ther. 2000;68(4):345–355. [DOI] [PubMed]
- 49.Lee JS, Obach RS, Fisher MB. Drug Metabolizing Enzymes: Cytochrome P450 and Other Enzymes in Drug Discovery and Development. New York: Marcel Dekker; 2003.
- 50.Sesink AL, Arts IC, de Boer VC, Breedveld P, Schellens JH, Hollman PC, et al. Breast cancer resistance protein (Bcrp1/Abcg2) limits net intestinal uptake of quercetin in rats by facilitating apical efflux of glucuronides. Mol. Pharmacol. 2005;67(6):1999–2006. [DOI] [PubMed]
- 51.Su Y, Hu P, Lee SH, Sinko PJ. Using novobiocin as a specific inhibitor of breast cancer resistant protein to assess the role of transporter in the absorption and disposition of topotecan. J. Pharm. Sci. 2007;10(4):519–536. [DOI] [PubMed]
- 52.Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin. Pharmacol. Ther. 2007;81(3):362–370. [DOI] [PubMed]
- 53.Y. Kato, T. Miyazaki, T. Kano, T. Sugiura, Y. Kubo, and A. Tsuji. Involvement of influx and efflux transport systems in gastrointestinal absorption of celiprolol. J. Pharm. Sci. 2009;98(7):2529–2539. [DOI] [PubMed]
- 54.Koitabashi Y, Kumai T, Matsumoto N, Watanabe M, Sekine S, Yanagida Y, et al. Orange juice increased the bioavailability of pravastatin, 3-hydroxy-3-methylglutaryl CoA reductase inhibitor, in rats and healthy human subjects. Life Sci. 2006;78(24):2852–2859. [DOI] [PubMed]
- 55.Dahan A, Amidon GL. Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: the role of efflux transport in the oral absorption of BCS Class III drugs. Mol. Pharmaceut. 2009;6(1):19–28. [DOI] [PubMed]
- 56.Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999;104(2):147–53. Erratum in: J. Clin. Invest. 110(4):571 (2002). [DOI] [PMC free article] [PubMed]
- 57.Food and Drug Administration. Guidance for Industry: Food- Effect Bioavailability and Fed Bioequivalence Studies. http://www.fda.gov/cder/guidance/index.htm (accessed 12/17/2008).
- 58.Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv. Drug Deliv. Rev. 2008;60(6):717–733. [DOI] [PMC free article] [PubMed]
- 59.Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J. Pharmacol. Exp. Ther. 2003;304(1):223–228. [DOI] [PubMed]
- 60.Su Y, Zhang X, Sinko PJ. Human organic anion-transporting polypeptide OATP-A (SLC21A3) acts in concert with P-glycoprotein and multidrug resistance protein 2 in the vectorial transport of saquinavir in Hep G2 cells. Mol. Pharmaceut. 2004;1(1):49–56. [DOI] [PubMed]
- 61.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 2009;328(2):652–662. [DOI] [PubMed]
- 62.Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin. Pharmacol. Ther. 2007;81(2):194–204. [DOI] [PubMed]
- 63.Zheng HX, Huang Y, Frassetto L, Benet LZ. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin. Pharmacol. Ther. 2009;85(1):78–85. [DOI] [PMC free article] [PubMed]
- 64.Deng JW, Song IS, Shin HJ, Yeo CW, Cho DY, Shon JH, et al. The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet. Genomics. 2008;18(5):424–433. [DOI] [PubMed]
- 65.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 2008;83(2):273–280. [DOI] [PMC free article] [PubMed]
- 66.Tian X, Swift B, Zamek-Gliszczynski MJ, Belinsky MG, Kruh GD, Brouwer KL. Impact of basolateral multidrug resistance-associated protein (Mrp) 3 and Mrp4 on the hepatobiliary disposition of fexofenadine in perfused mouse livers. Drug Metab. Dispos. 2008;36(5):911–915. [DOI] [PMC free article] [PubMed]
- 67.Zelcer N, van de Wetering K, Hillebrand M, Sarton E, Kuil A, Wielinga PR, et al. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proc. Natl. Acad. Sci. U S A. 2005;102(20):7274–7279. [DOI] [PMC free article] [PubMed]
- 68.Parker RB, Yates CR, Soberman JE, Laizure SC. Effects of grapefruit juice on intestinal P-glycoprotein: evaluation using digoxin in humans. Pharmacotherapy. 2003;23(8):979–987. [DOI] [PubMed]
- 69.Benet LZ, Izumi T, Zhang Y, Silverman JA, Wacher VJ. Intestinal MDR transport proteins and P-450 enzymes as barriers to oral drug delivery. J. Control. Rel. 1999;62(1–2):25–31. [DOI] [PubMed]
- 70.Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, et al. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gasotroenterology. 2001;120:525–533. [DOI] [PubMed]
- 71.Solas C, Simon N, Drogoul MP, Quaranta S, Frixon-Marin V, Bourgarel-Rey V, et al. Minimal effect of MDR1 and CYP3A5 genetic polymorphisms on the pharmacokinetics of indinavir in HIV-infected patients. Br. J. Clin. Pharmacol. 2007;64(3):353–362. [DOI] [PMC free article] [PubMed]
- 72.Zhou S, Chan E, Pan SQ, Huang M, Lee EJ. Pharmacokinetic interactions of drugs with St John’s wort. J. Psychopharmacol. 2004;18(2):262–276. [DOI] [PubMed]
- 73.Peng SX, Ritchie DM, Cousineau M, Danser E, Dewire R, Floden J. Altered oral bioavailability and pharmacokinetics of P-glycoprotein substrates by coadministration of biochanin A. J. Pharm. Sci. 2006;95(9):1984–1993. [DOI] [PubMed]
- 74.Tannergren C, Knutson T, Knutson L, Lennernäs H. The effect of ketoconazole on the in vivo intestinal permeability of fexofenadine using a regional perfusion technique. Br. J. Clin. Pharmacol. 2003;55(2):182–190. [DOI] [PMC free article] [PubMed]
- 75.Sahin S, Benet LZ. The operational multiple dosing half-life: a key to defining drug accumulation in patients and to designing extended release dosage forms. Pharm. Res. 2008;25(12):2869–2877. [DOI] [PMC free article] [PubMed]
- 76.Kato Y, Takahara S, Kato S, Kubo Y, Sai Y, Tamai I, et al. Involvement of multidrug resistance-associated protein 2 (Abcc2) in molecular weight-dependent biliary excretion of beta-lactam antibiotics. Drug Metab. Dispos. 2008;36(6):1088–1096. [DOI] [PubMed]
- 77.Luna- Tortós C, Fedrowitz M, Löscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55(8):1364–1375. [DOI] [PubMed]
- 78.Kis E, Nagy T, Jani M, Molnár E, Jánossy J, Ujhelly O, et al. Leflunomide and its metabolite A771726, are high affinity substrates of BCRP: implications for drug resistance. Ann. Rheum. Dis. 2009;68(7):1201–1207. [DOI] [PubMed]
- 79.Engel K, Wang J. Interaction of organic cations with a newly identified plasma membrane monoamine transporter. Mol. Pharmacol. 2005;68(5):1397–1407. [DOI] [PubMed]
- 80.Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. 2007;35(10):1956–1962. [DOI] [PMC free article] [PubMed]
- 81.Urquhart BL, Ware JA, Tirona RG, Ho RH, Leake BF, Schwarz UI, et al. Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet. Genomics. 2008;18(5):439–448. [DOI] [PMC free article] [PubMed]
- 82.Magnusson MO, Dahl ML, Cederberg J, Karlsson MO, Sandström R. Pharmacodynamics of carbamazepine-mediated induction of CYP3A4, CYP1A2, and Pgp as assessed by probe substrates midazolam, caffeine, and digoxin. Clin. Pharmacol. Ther. 2008;84(1):52–62. [DOI] [PubMed]
- 83.Ishiguro N, Maeda K, Saito A, Kishimoto W, Matsushima S, Ebner T, et al. Establishment of a set of double transfectants coexpressing organic anion transporting polypeptide 1B3 and hepatic efflux transporters for the characterization of the hepatobiliary transport of telmisartan acylglucuronide. Drug Metab. Dispos. 2008;36(4):796–805. [DOI] [PubMed]
- 84.Wang Q, Strab R, Kardos P, Ferguson C, Li J, Owen A, et al. Application and limitation of inhibitors in drug-transporter interactions studies. Int. J. Pharmaceut. 2008;356(1–2):12–18. [DOI] [PubMed]
- 85.Bailey DG, Dresser GK, Leake BF, Kim RB. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin. Pharmacol. Ther. 2007;81(4):495–502. [DOI] [PubMed]
- 86.Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36(1):164–172. [DOI] [PubMed]
- 87.Muenster U, Grieshop B, Ickenroth K, Gnoth MJ. Characterization of substrates and inhibitors for the in vitro assessment of Bcrp mediated drug-drug interactions. Pharm. Res. 2008;25(10):2320–2326. [DOI] [PubMed]
- 88.Takenaka K, Morgan JA, Scheffer GL, Adachi M, Stewart CF, Sun D, et al. Substrate overlap between Mrp4 and Abcg2/Bcrp affects purine analogue drug cytotoxicity and tissue distribution. Cancer Res. 2007;67(14):6965–6972. [DOI] [PubMed]
- 89.Volpe DA. Variability in Caco-2 and MDCK cell-based intestinal permeability assays. J. Pharm. Sci. 2008;97(2):712–725. [DOI] [PubMed]
- 90.Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell AL, Karlsson J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007;35(8):1333–1340. [DOI] [PubMed]
- 91.Lohitnavy M, Lu Y, Lohitnavy O, Chubb LS, Hirono S, Yang RS. A possible role of multidrug resistance-associated protein 2 (Mrp2) in hepatic excretion of PCB126, an environmental contaminant: PBPK/PD modeling. Toxicol. Sci. 2008;104(1):27–39. [DOI] [PubMed]
- 92.Lavé T, Parrott N, Grimm HP, Fleury A, Reddy M. Challenges and opportunities with modelling and simulation in drug discovery and drug development. Xenobiotica. 2007;37(10–11):1295–1310. [DOI] [PubMed]
- 93.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispos. 2007;35(10):1766–1780. [DOI] [PubMed]
- 94.Wang Z, Yan A, Yuan Q, Gasteiger J. Explorations into modeling human oral bioavailability. Eur. J. Med. Chem. 2008;43(11):2442–2452. [DOI] [PubMed]
- 95.Parrott N, Lave T. Applications of physiologically based absorption models in drug discovery and development. Mol. Pharmaceut. 2008;5(5):760–775. [DOI] [PubMed]
- 96.Bonen A, Heynen M, Hatta H. Distribution of monocarboxylate transporters MCT1-MCT8 in rat tissues and human skeletal muscle. Appl. Physiol. Nutr. Metab. 2006;31(1):31–39. [DOI] [PubMed]
- 97.Food and Drug Administration. Drug Information: Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers http://www.fda.gov/Cder/drug/drugInteractions/tableSubstrates.htm#(accessed 12/17/2008), part of U.S. Food and Drug Administration Center for Drug Evaluation and Research. http://www.fda.gov/Cder/ (accessed 12/11/2008).
- 98.Grube M, Kock K, Oswald S, Draber K, Meissner K, Eckel L, et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006;80(6):607–620. [DOI] [PubMed]
- 99.Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, et al. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005;280(10):9610–9617. [DOI] [PubMed]
- 100.Bijvelds MJ, Jorna H, Verkade HJ, Bot AG, Hofmann F, Agellon LB, et al. Activation of CFTR by ASBT-mediated bile salt absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289(5):G870–879. [DOI] [PubMed]
- 101.Gray JH, Mangravite LM, Owen RP, Urban TJ, Chan W, Carlson EJ, et al. Functional and genetic diversity in the concentrative nucleoside transporter, CNT1, in human populations. Mol. Pharmacol. 2004;65:512–519. [DOI] [PubMed]
- 102.Owen RP, Gray JH, Taylor TR, Carlson EJ, Huang CC, Kawamoto M, et al. Genetic analysis and functional characterization of polymorphisms in the human concentrative nucleoside transporter, CNT2. Pharmacogenet. Genomics. 2005;15(2):83–90. [DOI] [PubMed]
- 103.Bodo A, Barkos E, Szeri F, Varadi A, Sarkadi B. Differential modulation of the human liver conjugate transporters MRP2 and MRP3 by bile acids and organic anions. J. Biol. Chem. 2003;278(26):23529–23537. [DOI] [PubMed]
- 104.Tian Q, Zhang J, Chan SY, Tan TM, Duan W, Huang M, et al. Topotecan: topotecan is a substrate for multidrug resistance associated protein 4. Curr. Drug Metab. 2006;7(1):105–118. [DOI] [PubMed]
- 105.Ci L, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Involvement of MRP4 (ABCC4) in the luminal efflux of ceftoxime and cefazolin in the kidney. Mol. Pharmacol. 2007;71(6):1591–1597. [DOI] [PubMed]
- 106.Huber RD, Gao B, Pfandler M-AS, Zhang-Fu W, Leuthold S, Hagenbuch B, et al. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Cell Physiol. 2007;292:C795–C806. [DOI] [PubMed]
- 107.Gao B, Huber RD, Wenzel A, Vavricka SR, Ismair MG, Reme C, et al. Localization of organic anion transporting polypeptides in the rat and human ciliary body epithelium. Exper. Eye Res. 2005;80(1):61–72. [DOI] [PubMed]
- 108.Jedlitschky G, Burchell B, Keppler D. cGMP: the multidrug resistance protein 5 (MRP5) functions as an ATP-dependent export pump for cyclic nucleotides. J. Biol. Chem. 2000;275(39):30069–30074. [DOI] [PubMed]
- 109.Farias M, San Martin R, Puebla C, Pearson JD, Casado JF, Pastor-Anglada M, et al. Nitric oxide reduces adenosin transporter ENT1 gene (SLC29A1) promoter activity in human fetal endothelium from gestational diabetes. J. Cell Physiol. 2006;208(2):451–460. [DOI] [PubMed]
- 110.Yao SY, Ng AM, Sundaram M, Cass CE, Baldwin SA, Young JD. Transport of antiviral 3′-deoxy-nucleoside drugs by recombinant human and rat equilibrative, nitrobenzylthioinosine (NBMPR)-insensitive (ENT2) nucleoside transporter proteins produced in Xenopus oocytes. Mol. Membr. Biol. 2001;18(2):161–167. [DOI] [PubMed]
- 111.Kirat D, Masuoka J, Hayashi H, Iwano H, Yokota H, Taniyama H, et al. Monocarboxylate transporter (MCT1) plays a direct role in short-chain fatty acids absorption in caprine rumen. J. Physiol. 2006;576(Pt. 2):635–647. [DOI] [PMC free article] [PubMed]
- 112.Meredith D, Boyd CAR, Bronk JR, Bailey PD, Morgan KM, Collier ID, et al. PEPT-1: 4-aminomethylbenzoic acid is a non-translocated competitive inhibitor of the epithelial peptide transporter PepT1. J. Physiol. 1998;512(3):629–634. [DOI] [PMC free article] [PubMed]
- 113.Knutter I, Theis S, Hartrodt B, Born I, Brandsch M, Daniel H, et al. A novel inhibitor of the mammalian peptide transporter PEPT1. Biochemistry. 2001;40:4454–4458. [DOI] [PubMed]
- 114.Groneberg DA, Eynott PR, Doring F, Dinh QT, Oates T, Barnes PJ, et al. Distribution and function of the peptide transporter PEPT2 in normal and cystic fibrosis human lung. Thorax. 2002;57(1):55–60. [DOI] [PMC free article] [PubMed]
- 115.Hayer-Zillgen M, Bruss M, Bonisch H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2, and hOCT3. Br. J. Pharmacol. 2002;136:829–836. [DOI] [PMC free article] [PubMed]
- 116.Sakamoto S, Kusuhara H, Miyata K, Shimaoka H, Kanazu T, Matsuo Y, et al. Glucuronidation converting methyl 1-(3, 4-dimethoxyphenyl)-3-(3-ethylvaleryl)-4-hydroxy-6, 7, 8-trimethoxy-2-naphthoate (S-8921) to a potent apical sodium-dependent bile acid transporter inhibitor, resulting in a hypocholesterolemic action. J. Pharmacol. Exp. Ther. 2007;322(2):610–618. [DOI] [PubMed]
- 117.Li H, Chen F, Shang Q, Pan L, Shneider BL, Chiang JYL, et al. FXR-activating ligands inhibit rabbit ASBT expression via FXR-SHP-FTF cascade. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;288:G60–G66. [DOI] [PubMed]
- 118.Gorraitz E, Sanmartin C, Errasti-Murugarren E, Casado FJ, Pastor-Anglada M, Lostao MP. Interaction of nucleoside-derivatives with the human Na+/nucleoside cotransporters CNT1 and CNT3. FASEB J. 2008;22: lb133.
- 119.Li JY, Boado RJ, Pardridge WM. Differential kinetics of transport of 2’, 3’-dideoxyinosine and adenosine via concentrative Na + nucledoside transporter CNT2 cloned from rat blood-brain barrier. J. Pharmacol. Exp. Ther. 2001;299(2):735–740. [PubMed]
- 120.Yabuuchi H, Tamai I, Nezu JI, Sakamoto K, Oku A, Shimane M, et al. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J. Pharmacol. Exp. Ther. 1999;289(2):768–773. [PubMed]
- 121.Elimrani I, Lahjouji K, Seidman E, Roy M-J, Mitchell GA, Qureshi I. Expression and localization of organic cation/carnitine transporter OCTN2 in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;284:G863–871. [DOI] [PubMed]
- 122.Aye ILMH, Paxton JW, Evseenko DA, Keelan JA. Expression, localisation and activity of ATP binding cassette (ABC) family of drug transporters in human amnion membranes. Placenta. 2007;28(8–9):868–877. [DOI] [PubMed]
- 123.Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002;1:417–425. [PubMed]
- 124.Janneh O, Hartkoorn RC, Jones E, Owen A, Ward SA, Davey R, et al. Cultured CD4T cells and primary human lymphocytes express hOATPs: intracellular accumulation of saquinavir and lopinavir. Br. J. Pharmacol. 2008;155(6):875–883. [DOI] [PMC free article] [PubMed]
- 125.Fujiwara K, Adachi H, Nishio T, Unno M, Tokui T, Okabe M, et al. Identification of thyroid hormone transporters in humans: different molecules are involved in a tissue-specific manner. Endocrinology. 2001;142(5):2005–2012. [DOI] [PubMed]
- 126.Potschka H, Baltes S, Löscher W. Inhibition of multidrug transporters by verapamil or probenecid does not alter blood-brain barrier penetration of levetiracetam in rats. Epilep. Res. 2004;58(3):85–91. [DOI] [PubMed]
- 127.Pollex E, Lubetsky A, Koren G. The role of placental breast cancer resistance protein in the efflux of glyburide across the human placenta. Placenta. 2008;29(8):743–747. [DOI] [PubMed]
- 128.Sampath J, Adachi M, Hatse S, Naesens L, Balzarini J, Flatley R, Matherly L, Schuetz J. Role of MRP4 and MRP5 in biology and chemotherapy. AAPS PharmSci. 2002;4(3): article 14. [DOI] [PMC free article] [PubMed]
- 129.Boraldi F, Quuaglino D, Croce MA, Garcia Fernandez MI, Tiozzo R, Gheduzzi D, et al. Multidrug resistance protein-6 (MRP6) in human dermal fibroblasts. Comparison between cells from normal subjects and from Pseudoxanthoma elasticum patients. Matrix Biol. 2003;22(6):491–500. [DOI] [PubMed]
- 130.Lin W, Buolamwini JK. Synthesis and flow cytometric evaluation, and identification of highly potent dipyridamole analogues as equilibrative nucleoside transporter 1 inhibitors. J. Med. Chem. 2007;50:3906–3920. [DOI] [PMC free article] [PubMed]
- 131.Robillard KR, Bone DBJ, Hammond JR. Hypoxanthine uptake and release by equilibrative nucleoside transporter 2 (ENT2) of rat microvascular endothelial cells. Microvasc. Res. 2008;75(3):351–357. [DOI] [PubMed]
- 132.Smith AJ, van Helvoort A, van Meer G, Szabo K, Welker E, Szakacs G, et al. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J. Biol. Chem. 2000;275(31):23530–23539. [DOI] [PubMed]
- 133.Hirano M, Maeda K, Hayashi H, Kusuhara H, Sugiyama Y. Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J. Pharmacol. Exp. Ther. 2005;314(2):876–882. [DOI] [PubMed]
- 134.Jigorel E, Le Vee M, Boursier-Neyret C, Bertrand M, Fardel O. Functional expression of sinusoidal drug transporters in primary human and rat hepatocytes. Drug Metab. Dispos. 2005;33(10):1418–1422. [DOI] [PubMed]
- 135.Leslie EM, Watkins PB, Kim RB, Brouwer KLR. Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SCL10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J. Pharmacol. Exp. Ther. 2007;321(3):1170–1178. [DOI] [PubMed]
- 136.Kim RB, Leake B, Cvetkovic M, Roden MM, Nadeau J, Walubo A, et al. Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J. Pharmacol. Exp. Ther. 1999;291(3):1204–1209. [PubMed]
- 137.Khamdung S, Takeda M, Shimoda M, Noshiro R, Narikawa S, Huang XL, et al. Interactions of human- and rat-organic anion transporters with pravastatin and cimetidine. J. Pharmacol. Sci. 2004;94(2):197–202. [DOI] [PubMed]
- 138.Halwachs S, Kneuer C, Honscha W. Endogenous expression of liver-specific drug transporters for organic anions in rat hepatocytoma fusion cell line HPCT-1E3. Eur. J. Cell Biol. 2005;84(7):677–686. [DOI] [PubMed]
- 139.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001;276:14581–14587. [DOI] [PubMed]
- 140.Burk O, Arnold KA, Nussler AK, Schaeffeler E, Efimova E, Avery BA, et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 2005;67:1954–1965. [DOI] [PubMed]
- 141.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002;277:2908–2915. [DOI] [PubMed]
- 142.Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab. Dispos. 2001;29:1467–1472. [PubMed]
- 143.Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, et al. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J. Biol. Chem. 2004;279:22250–22257. [DOI] [PubMed]
- 144.Szatmari I, Vámosi G, Brazda P, Balint BL, Benko S, Széles L, et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J. Biol. Chem. 2006;281:23812–23823. [DOI] [PubMed]
- 145.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001;276:28857–28865. [DOI] [PubMed]
- 146.Eloranta JJ, Jung D, Kullak-Ublick GA. The human Na + -taurocholate cotransporting polypeptide gene is activated by glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma coactivator-1alpha, and suppressed by bile acids via a small heterodimer partner-depende. Mol. Endocrinol. 2006;20:65–79. [DOI] [PubMed]
- 147.Miki Y, Suzuki T, Kitada K, Yabuki N, Shibuya R, Moriya T, et al. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res. 2006;66:535–542. [DOI] [PubMed]
- 148.Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002;62:638–646. [DOI] [PubMed]
- 149.Jung D, Podvinec M, Meyer UA, Mangelsdorf DJ, Fried M, Meier PJ, et al. Human organic anion transporting polypeptide 8 promoter is transactivated by the farnesoid X receptor/bile acid receptor. Gastroenterology. 2002;122:1954–1966. [DOI] [PubMed]
- 150.Morita Y, Sakaeda T, Horinouchi M, Nakamura T, Kuroda K, Miki I, et al. MDR1 genotype-related duodenal absorption rate of digoxin in healthy Japanese subjects. Pharm. Res. 2003;20:552–556. [DOI] [PubMed]
- 151.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA. 2000;97(7):3473–3478. [DOI] [PMC free article] [PubMed]
- 152.Kurata Y, Ieiri I, Kimura M, Morita T, Irie S, Urae A, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin. Pharmacol. Ther. 2002;72(2):209–219. [DOI] [PubMed]
- 153.Yi SY, Hong KS, Lim HS, Chung JY, Oh DS, Kim JR, et al. A variant 2677A allele of the MDR1 gene affects fexofenadine disposition. Clin. Pharmacol. Ther. 2004;76:418–427. [DOI] [PubMed]
- 154.de Jong FA, Scott-Horton TJ, Kroetz DL, McLeod HL, Friberg LE, Mathijssen RH, et al. Irinotecan-induced diarrhea: functional significance of the polymorphic ABCC2 transporter protein. Clin. Pharmacol. Ther. 2007;81:42–49. [DOI] [PubMed]
- 155.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin. Pharmacol. Ther. 2004;76:38–44. [DOI] [PubMed]
- 156.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, et al. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol. Ther. 2005;4:650–658. [DOI] [PubMed]
- 157.Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, et al. Role of BCRP 421C > A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin. Chim. Acta. 2006;373:99–103. [DOI] [PubMed]
- 158.Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin. Pharmacol. Ther. 2003;73:554–564. [DOI] [PubMed]
- 159.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin. Pharmacol. Ther. 2004;75:415–421. [DOI] [PubMed]
- 160.Niemi M, Kivistö KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1). Br. J. Clin. Pharmacol. 2005;59:602–604. [DOI] [PMC free article] [PubMed]
- 161.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, et al. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin. Pharmacol. Ther. 2005;77:468–478. [DOI] [PubMed]
- 162.Ieiri I, Suwannakul S, Maeda K, Uchimaru H, Hashimoto K, Kimura M, et al. SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin. Pharmacol. Ther. 2007;82:541–547. [DOI] [PubMed]
- 163.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007;82:726–733. [DOI] [PubMed]
- 164.Zhang W, He YJ, Han CT, Liu ZQ, Li Q, Fan L, et al. Effect of SLCO1B1 genetic polymorphism on the pharmacokinetics of nateglinide. Br. J. Clin. Pharmacol. 2006;62:567–572. [DOI] [PMC free article] [PubMed]
- 165.Katz DA, Carr R, Grimm DR, Xiong H, Holley-Shanks R, Mueller T, et al. Organic anion transporting polypeptide 1B1 activity classified by SLCO1B1 genotype influences atrasentan pharmacokinetics. Clin. Pharmacol. Ther. 2006;79:186–196. [DOI] [PubMed]
- 166.Xiang X, Jada SR, Li HH, Fan L, Tham LS, Wong CI, et al. Pharmacogenetics of SLCO1B1 gene and the impact of *1b and *15 haplotypes on irinotecan disposition in Asian cancer patients. Pharmacogenet. Genomics. 2006;16:683–691. [DOI] [PubMed]
- 167.Oswald S, Scheuch E, Cascorbi I, Siegmund W. A LC-MS/MS method to quantify the novel cholesterol lowering drug ezetimibe in human serum, urine and feces in healthy subjects genotyped for SLCO1B1. J. Chromatogr. B. 2006;830:143–150. [DOI] [PubMed]
- 168.Vormfelde SV, Toliat MR, Schirmer M, Meineke I, Nürnberg P, Brockmöller J. The polymorphisms Asn130Asp and Val174Ala in OATP1B1 and the CYP2C9 allele *3 independently affect torsemide pharmacokinetics and pharmacodynamics. Clin. Pharamacol. Ther. 2008;83:815–817. [DOI] [PubMed]
- 169.Miura M, Satoh S, Inoue K, Kagaya H, Saito M, Inoue T, et al. Influence of SLCO1B1, 1B3, 2B1 and ABCC2 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Eur. J. Clin. Pharmacol. 2007;63:1161–1169. [DOI] [PubMed]
- 170.Grover A, Benet LZ. Effects of drug transporters on volume of distribution. AAPS J. 2009;11:250–261. [DOI] [PMC free article] [PubMed]