

Abstract
Ca2+ / Calmodulin-dependent kinase II (CaMKII) plays a central role in long-term potentiation (LTP), which underlies some forms of learning and memory. Here we monitored the spatiotemporal dynamics of CaMKII activation in individual dendritic spines during LTP using 2-photon fluorescence lifetime imaging microscopy in combination with 2-photon glutamate uncaging. Induction of LTP and associated spine enlargement in single spines triggered transient (∼ 1 min) CaMKII activation restricted to the stimulated spines. CaMKII in spines was specifically activated by NMDA receptors and L-type voltage sensitive calcium channels, presumably via nanodomain Ca2+ near the channels, in response to glutamate uncaging and depolarization, respectively. The high degree of compartmentalization and channel specificity of CaMKII signalling allow stimuli-specific spatiotemporal patterns of CaMKII signalling and may be important for synapse-specificity of synaptic plasticity.
CaMKII is a serine/threonine protein kinase consisting of 12 subunits 1. Each subunit is activated by the association of Ca2+ bound Calmodulin (Ca2+/CaM). When a CaMKII subunit is autophosphorylated at site T286, its activity can be maintained after the dissociation of Ca2+/CaM 2. It has been suggested that CaMKII activation in postsynaptic density (PSD) may persist long-term (more than hours) to maintain LTP 2. CaMKII interacts with several ion channels including the NR2B subunit of NMDA receptors (NMDARs) 3 and voltage sensitive calcium channels (VSCCs) 4, suggesting that CaMKII activation may be initiated within the nanodomains of these channels to produce channel-specific signalling. However, at the level of single synapses it is unknown whether CaMKII activation is channel-specific, to what degree CaMKII activation is compartmentalized, and if it persists in the stimulated spine during LTP.
To measure CaMKII activation in spines, we developed a fluorescence resonance energy transfer (FRET)-based CaMKIIα sensor by modifying the previously reported CaMKIIα sensor, Camuiα, in which the N- and C-termini of CaMKIIα are labeled with donor and acceptor fluorophores 5. To optimize the sensitivity and brightness, we used the FRET pair of monomeric enhanced green fluorescent protein (mEGFP) 6 and REACh, a non-radiative YFP variant 7 (Green-Camuiα), and measured FRET using 2-photon fluorescence lifetime imaging microscopy (2pFLIM) 8-10. The activation of Green-Camuiα should change the conformation of CaMKIIα to the open state where its kinase domain is exposed 1, thereby decreasing FRET and increasing the fluorescence lifetime of mEGFP (Supplementary Fig. 1). We confirmed that Green-Camuiα is incorporated into a CaMKII dodecameric holoenzyme, and the fluorescence lifetime of Green-Camuiα reports CaMKII activation associated with T286 phosphorylation as well as Ca2+/CaM binding (Supplementary Note). We biolistically 11 transfected CA1 pyramidal neurons in hippocampal cultured slices with Geen-Camuiα and mCherry. The expression level of Green-Camuiα was estimated at 25 - 50 % of the level of endogenous CaMKIIα subunit, and the endogenous concentration of CaMKIIα subunit was estimated to be ∼10 - 100 μM (Supplementary Fig. 2, Supplementary Note).
CaMKII activation during structural plasticity of dendritic spines
Using this technique, we imaged CaMKII activation during structural plasticity of dendritic spines, which is considered to be associated with LTP12-14 (Fig. 1). To induce synapse-specific structural plasticity, we applied a low frequency train of 2-photon glutamate uncaging pulses (45 pulses at 0.5 Hz) to a single dendritic spine in zero extracellular Mg2+ 13, 14. The spine volume increased quickly following glutamate uncaging by 376 ± 68 % (Fig. 1c,e) and relaxed to an elevated level at 104 ± 21 % for more than 30 minutes (Fig. 1c,f)13, 14. The structural plasticity was associated with accumulation of Green-Camuiα in the stimulated spines roughly proportional to the volume change (Supplementary Fig. 3) 15. Structural plasticity of dendritic spines was abolished by blocking NMDARs (100 μM AP5) 13 (Fig. 1c,e,f). CaMKII inhibitor (10 μM KN62) partially inhibited the sustained structural plasticity (Fig. 1c,f), but not appreciably the transient phase (Fig. 1c, e), suggesting that the activation of CaMKII is required for the maintenance of the spine volume change 13. Overexpression of the T286A mutant of Green-Camuiα reduced the sustained spine enlargement (Fig. 1c, f, Supplementary Fig. 4), demonstrating that this mutant acts as dominant negative. Because the overexpression level of T286A mutant is relatively small (10 - 20 %; Supplementary Note), inhibiting autophosphorylation of T286 in relatively small fraction of CaMKII subunits in a holoenzyme may be sufficient for inhibiting structural plasticity. This result is consistent with previous studies reporting that LTP is attenuated in T286A heterozygous knockout mice 16.
Figure 1. Simultaneous measurements of CaMKII activation and structural plasticity in single spines using 2pFLIM combined with 2-photon glutamate uncaging.

a, Fluorescence lifetime images of Green-Camuiα during the induction of spine structural plasticity by 2-photon glutamate uncaging in the absence of extracellular Mg2+. Longer lifetimes imply increased activity. The white arrowhead indicate the location of uncaging laser spot.
b, Averaged time course of fluorescence lifetime change of Green-Camuiα in the stimulated spine (indicated as “Stim”) and adjacent spines (“Adj”, within 5 μm of stimulated spine). Data using pharmacological inhibitors and T286A mutant Green-Camuiα are also shown. The number of samples (spines / neurons) is 35 / 30 for stimulated spines, 29 / 28 for adjacent spines, 5 / 4 for AP5, 7 / 5 for KN62 and 18 / 11 T286A. Right panel: closer view of fluorescence lifetime change during uncaging.
c, Averaged time course of spine volume change in stimulated spines and adjacent spines. Right: closer view of the volume change.
d, CaMKII activation (averaged over 0 - 40 s or first 5 points). Stars denote statistical significance between control and others (p < 0.05).
e, Transient volume change (volume change averaged over 1.5 - 2 min subtracted by that averaged over 25 - 30 min) 13.
f, Sustained volume change (volume change averaged over 25 - 30 min).
During structural plasticity, the activity of CaMKII, as measured by the fluorescence lifetime of Green-Camuiα, increased rapidly (Fig. 1a, b). The activity was restricted to the stimulated spines, and did not spread into dendrites nor adjacent spines (Fig. 1a,b,d). Activation of CaMKII preceded the enlargement of spine volume, suggesting that CaMKII triggers molecular processes that lead to the structural plasticity (Fig. 1a,b,c). The NMDAR blocker AP5 inhibited the fluorescence lifetime change, indicating that CaMKII is activated by Ca2+ from NMDARs (Fig. 1b,d). The CaMKII inhibitor KN62 decreased the fluorescence lifetime change by 40 % (Fig. 1b, d). The T286A and T305D mutant sensors reported less activation, suggesting that the binding of calmodulin and phosphorylation of T286 is required for the full activation of CaMKII in spines (Fig. 1b, d, Supplementary Fig. 4).
Mechanisms of spine specific CaMKII activation
In general, the degree of compartmentalization of CaMKII activation is determined by two factors: the inactivation time constant of CaMKII activity and the mobility of CaMKII 10, 17. Thus, we measured these parameters to study the mechanisms by which CaMKII activation is restricted. First, to measure the kinetics of CaMKII precisely, we imaged CaMKII activity with higher temporal resolution (2 s) in response to a brief train of glutamate uncaging pulses (0.5 Hz, 8 pulses in zero extracellular Mg2+). The activation of CaMKII reached a plateau within 6 s, and after the cessation of uncaging, it was inactivated with two time constants, 6 and 45 s (Fig. 2a). T286A Green-Camuiα was activated much less than wild type, and decayed within 2 s (Fig. 2a). These results indicate that T286 phosphorylation prolongs CaMKII activation and allows the accumulation of activated CaMKII during repetitive glutamate uncaging. To measure how the decay kinetics is regulated by CaMKII activation, we stimulated spines with different numbers of uncaging pulses from 1 to 45 (Fig. 2b). With 45 uncaging pulses, we observed enlargements of spines. Our data indicate that the decay time constants depend on neither the number of stimulations nor the level of the CaMKII activation (Fig. 2b).
Figure 2. Measurements of parameters that determine spine-specificity: inactivation kinetics and mobility of CaMKII.
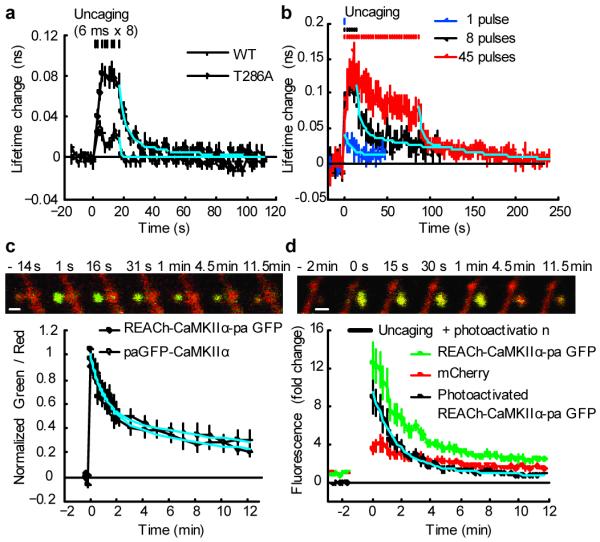
a, Measurements of CaMKII activation and inactivation following brief stimulation (8 pulses, 0.5 Hz) of spines using wild type and T286A Green Camuiα in zero extracellular Mg2+. These experiments were performed in an interleaved manner. The solid cyan curve indicates double exponential functions obtained by fitting to wild type data (time constants: 6 s [80%] and 45 s [20%]). The dashed cyan curve indicates single exponential function with 1 s time constant. The number of samples (spines / neurons) is 25 / 5 for wild type and 28 / 4 for T286A.
b. CaMKII inactivation kinetics following different number of uncaging pulses (1, 8 and 45 pulses, 0.5 Hz). Data were fitted with double exponential functions (cyan curves). The time constants were 9.3 s (71%) and > 100 s (29 %) for 1 pulse, 4.7 s (61 %) and 136.6 s (39 %) for 8 pulses, 4.5 s (61 %) and 102 s (39 %) for 45 pulses. The number of samples (spines / neurons) is 28 / 9 (1 pulses), 16 / 8 (8 pulses) and 13 / 7 (45 pulses).
c, Measurements of spine-dendrite coupling of CaMKII using photoactivatable GFP (paGFP). We used Green-Camuiα with mEGFP replaced by paGFP (REACh-CaMKIIα-paGFP) or paGFP-CaMKIIα. Top panel: Green and red fluorescence before and after photoactivation in a neuron expressing REACh-CaMKIIα-paGFP (green) and mCherry (red). Bar, 1 μm. Bottom panel: Averaged time course of the ratio of red and green. Signal was normalized to the peak fluorescence change. The first point is 1 s after the photoactivation. By fitting with double exponential functions, the decay time constants was obtained as 1.3 min (48 %) and 17.0 min (52 %) for REACh-CaMKIIα-paGFP (cyan solid curve), and 1.0 minutes (53 %) and 20.7 minutes (47 %) for paGFP-CaMKIIα (cyan dashed curve). The number of samples (spines / neurons) is 15 / 5 (REACh-CaMKIIα-paGFP) and 22 / 9 (paGFP-CaMKIIα).
d. Spine-dendrite coupling of Green-Camuiα during structural plasticity. During glutamate uncaging (720 nm, 6 ms, 45 pulses in Mg2+ free solution), REACh-CaMKIIα-paGFP can be photoactivated. We subtracted the normalized red fluorescence from the normalized green fluorescence, assuming that the small baseline fluorescence of REACh-CaMKIIα-paGFP behaves similarly to mCherry fluorescence change (see Supplementary Fig. 3). By fitting with double exponential functions (cyan curve), the decay time constants were obtained as 1.6 min (82 %) and 15.3 min (18 %). The number of samples (spines / neurons) is 10 / 7.
Next, we measured the mobility of CaMKII, another determinant of the compartmentalization of CaMKII, using photoactivatable GFP (paGFP; Fig. 2c, d). We used either a Green-Camuiα variant with EGFP replaced by photoactivatable GFP (REACh-CaMKII-paGFP) or paGFP tagged CaMKIIα (paGFP-CaMKII). Following photoactivation of paGFP, green fluorescence decayed due to the diffusion of CaMKIIα out of the spine with two time constants, ∼1 and ∼20 minutes (Fig. 2c). Fluorescence recovery after photobleaching (FRAP) of Green-Camuiα also showed similar recovery time constants (Supplementary Fig. 5b-c). We also measured the mobility of CaMKII during structural plasticity by monitoring the decay of REACh-CaMKIIα-paGFP which is photoactivated by glutamate uncaging protocol (6 ms pulse, 45 times). The photoactivated REACh-CaMKIIα-paGFP decayed with the time constants similar to the resting condition (Fig. 2d). Consistent with this spine-dendrite coupling time constant, accumulation of CaMKIIα into spines during the transient phase of structural plasticity was smaller compared to the spine volume change (Supplementary Fig. 3). Since the inactivation of CaMKII is faster than its diffusion out of the spine, CaMKII activation must be restricted to the stimulated spine, which agrees with our measurements of CaMKII activity during structural plasticity (Fig. 1).
CaMKII activation during paring-induced LTP
In the presence of extracellular Mg2+, LTP induction in hippocampal CA1 synapses requires coincident postsynaptic depolarization and synaptic activation to release Mg2+ block of NMDARs 18. To study the dynamics of CaMKII activation under such condition, we imaged CaMKII activation during LTP induced by pairing postsynaptic depolarization with 2-photon glutamate uncaging in the presence of extracellular Mg2+ (1 mM) 13 (Fig. 3). We performed whole-cell patch clamp recordings on neurons transfected with Green-Camuiα and mCherry, and measured the postsynaptic current evoked by 2-photon glutamate uncaging (uEPSC) at -65 mV. LTP was induced by pairing uncaging pulses (30 pulses at 0.5 Hz) with postsynaptic depolarization (0 mV). This LTP protocol induced a sustained increase in both uEPSC and spine volume by ∼150%, which was maintained for more than 60 minutes (Fig. 3c, d). In the same LTP protocol, depolarization produced large calcium elevation in spines (2.6 ± 0.5 μM) and in dendritic shafts (2.6 ± 0.5 μM) presumably due to activation of VSCCs 19 (Supplementary Fig. 6a-c). Although the depolarization was held for more than 1 min, [Ca2+] decayed rapidly within 2 s 20 (Supplementary Fig. 6b, c). On top of small residual Ca2+ (0.21 ± 0.07 μM), subsequent glutamate uncaging produced calcium transients between 1.8 ± 0.3 μM (first uncaging) and 0.8 ± 0.2 μM (30th uncaging), largely restricted to the heads of the stimulated spines 21, 22 (Supplementary Fig. 6b, c).
Figure 3. Imaging CaMKII activation during LTP induction by 2-photon glutamate uncaging paired with postsynaptic depolarization.

a, Fluorescence lifetime images of Green-Camuiα during the induction of LTP by 2-photon glutamate uncaging paired with postsynaptic depolarization.
b, Averaged time course of fluorescence lifetime change of Green-Camuiα in the stimulated spine, adjacent spines (within 5 μm of stimulated spine), and dendritic shaft close to the stimulated spine. Right: closer view of fluorescence lifetime change during uncaging. Average of 8 stimulated spines, 13 adjacent spines and 8 dendrites from 8 neurons. Plot of stimulated spines is noisier due to smaller number of samples. Plots of stimulated spines in b - d are from the same spines.
c, Averaged time course of changes in spine volume and the amplitude of uncaging-evoked EPSC at -65 mV (uEPSC) in the stimulated spine and adjacent spines (within 5 μm). For uEPSC, each point is average of four trials at 5 s intervals. Uncaging spot was relocated to the tip of spines before each four trial measurement.
d, uEPSC at single spines before and 60 minutes after LTP induction (average of 4 trials, filtered with 2 ms window).
In response to depolarization, CaMKII activity increased in dendritic shafts (lifetime change = 0.079 ± 0.018 ns), but less so in spines (0.028 ± 0.010 ns) (Fig. 3a,b). Glutamate uncaging pulses caused additional CaMKII activation in the stimulated spines (0.097 ± 0.014 ns), but not in the adjacent spines (Fig. 3a,b). CaMKII activation in spines and dendritic shafts rapidly decayed within ∼ 2 min. The large gradient of CaMKII activation between spines and dendritic shafts during LTP induction demonstrates the high degree of compartmentalization of CaMKII activation in individual spines (Fig. 3a,b).
Channel specific activation of CaMKII
It has been reported that Ca2+ through VSCCs is required for LTP induced by several protocols 19, 23-26. Therefore, we further characterized the depolarization-induced CaMKII activation with higher temporal resolution (2 s; Fig. 4). We found that CaMKII activation in dendritic shafts increased within 2 s after depolarization, was sustained during the depolarization (16 s), and then decayed rapidly following repolarization (Fig. 4a). Activation of CaMKII in spines (0.034 ± 0.004 ns) was smaller than in dendrites (0.076 ± 0.007 ns). [Ca2+] during the depolarization decayed within 2 s, and little extra Ca2+ remained for more than 10 s (0.21 ± 0.07 μM) (Supplementary Fig. 6b). Thus, these results suggest that small sustained increases in [Ca2+] are sufficient to sustain CaMKII activation during the depolarization for more than ∼10 s. The T286A mutant was inactivated much more rapidly (in less than 4 s) than the wild type during the depolarization (Fig. 4b), indicating that the prolonged activation of CaMKII requires autophosphorylation at T286, and the decay of CaMKII activity after repolarization is due to the dephosphorylation of T286. Bursts of back-propagating action potentials (83 Hz, 0.5 s, 8 times, 2 s interval) also produced similar CaMKII activation in spines and dendrites (Fig. 4d).
Figure 4. Differential activation of CaMKII in spines and dendrites by postsynaptic depolarization.
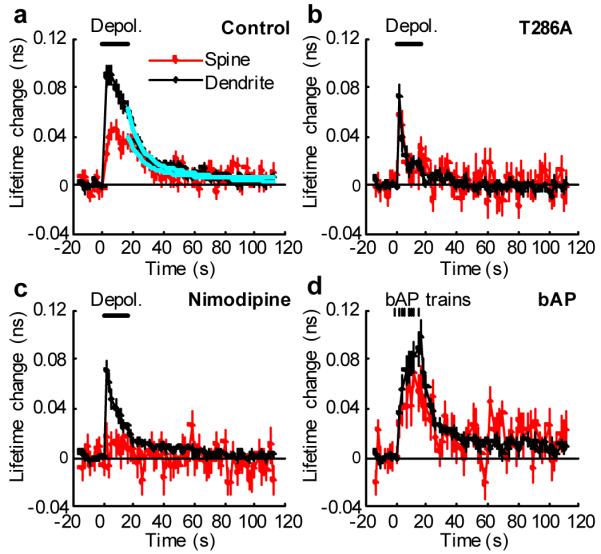
a, High temporal resolution measurements of the fluorescence lifetime change of Green-Camuiα in spines and dendrites in response to postsynaptic depolarization (0 mV, 16 s, indicated by the bar) in spines (red) and dendritic shafts (black). Average of 53 spines and 30 dendrites from 12 neurons. The cyan lines are a double exponential function obtained by fitting. The time constants are 11.3 s (84 %) and > 300 s (16%) for spines, and 6.8 s (60 %) and 43.7 s (40 %) for dendrites.
b, Fluorescence lifetime change of T286A mutant of Green-Camuiα. Average of 19 spines and 14 dendrites from 8 neurons.
c, Fluorescence lifetime change of Green-Camuiα in the presence of L-type VSCC inhibitor (20 μM Nimodipine). Average of 22 spines and 15 dendrites from 7 neurons.
d, Bursts of back-propagating action potentials (bAP) induced CaMKII activation at proximal dendrites (< 100 μm). We applied 8 bAP bursts, each 83 Hz for 0.5 s, at 2 s intervals. Average of 22 spines and 12 dendrites from 7 neurons).
Nimodipine, a blocker of L-type VSCCs, abolished CaMKII activation in dendritic spines during depolarization (Fig. 4c). The peak of CaMKII activation in dendritic shafts was not affected by the blockade of L-type VSCCs, but the activity decayed more rapidly (Fig. 4c). Because L-type VSCCs produce only a small fraction of Ca2+ in spines and dendritic shafts during cell depolarization (Supplementary Fig. 6d), L-type VSCC-dependent CaMKII activation presumably occurs in the nanodomain at the mouth of L-type VSCCs 4, 27, 28. Also, global Ca2+ elevation by opening of non-L-type VSCCs did not activate CaMKII in spines (Fig. 4c), although it is as large as uncaging evoked Ca2+ elevation (Supplementary Fig. 6). This result suggests that global Ca2+ elevation alone is insufficient to activate CaMKII in spines.
To further test the hypothesis of the nanodomain activation of CaMKII, we performed whole-cell patch clamp using a pipette including Ca2+ chelators: EGTA or BAPTA. BAPTA and EGTA have similar dissociation constants to Ca2+, but the binding rate of BAPTA is ∼100 times higher 29. Thus, EGTA inhibits global Ca2+ over nanodomain Ca2+ more selectively than BAPTA does 29. The activation of CaMKII induced by depolarization was inhibited by BAPTA much more effectively compared to the same concentration of EGTA (Fig. 5a-f), suggesting that nanodomain Ca2+ near calcium channels is sufficient to activate CaMKII both in spines and dendrites. With EGTA loaded in neurons, depolarization induced larger CaMKII activation in spines, suggesting that there is global Ca2+-dependent CaMKII inactivation (Fig. 5b, c). Slower CaMKII activation kinetics in the presence of EGTA (Fig. 5b, c) are presumably caused by smaller Ca2+-dependent inactivation combined with smaller activation per unit time due to reduced Ca2+ elevation. Unlike depolarization-induced CaMKII activation, EGTA and BAPTA both similarly reduced glutamate-uncaging induced CaMKII activation (Fig. 5g-i), suggesting that nanodomain Ca2+ alone is not sufficient for uncaging-induced CaMKII activation.
Figure 5. Effects of Ca2+ chelators on CaMKII activation.

a-e, Fluorescence lifetime change of Green-Camuiα in spines and dendrites in response to postsynaptic depolarization (0 mV, 0 - 16 s, indicated by the black bar) in spines (red) and dendritic shafts (black) in neurons patch-clamped with electrodes containing no Ca2+ chelator (a), 5 mM EGTA (b), 20 mM EGTA (c), 5 mM BAPTA (d) or 20 mM BAPTA (e). Number of samples (spines / dendrites / neurons) is 18 / 11 / 6 for control (a), 29 / 14 / 7 for 5 mM EGTA (b), 15 / 7 / 4 for 20 mM EGTA (c), 21 / 7 / 5 for 5 mM BAPTA (d) and 18 / 7 / 4 for 20 mM BAPTA (e). Imaging was performed more than 15 min after establishing whole-cell patch.
f, Dependence of fluorescence lifetime change (averaged over 0 - 16 s) by depolarization on Ca2+ chelator concentration (EGTA: closed circle, BAPTA: open circle, red: spine, black: dendrite). Fluorescence lifetime was normalized to control.
g-h, Fluorescence lifetime change of Green-Camuiα in response to glutamate uncaging (6 ms, 45 stimuli in zero extracellular Mg2+) in neurons patch-clamped with electrodes containing 1 - 20 mM EGTA (g), 1 - 5 mM BAPTA (h) in a current-clamp mode. Controls are measured in the same neurons before patch-clamp. Patch-clamping with internal solution without Ca2+ chelator did not alter fluorescence lifetime change (Supplementary Fig. 7). Number of samples (spines / neurons) is 22 / 9 for control in g, 12 / 3 for 1 mM EGTA, 15 / 3 for 5 mM EGTA, 17 / 3 for 20 mM EGTA, 16 / 7 for control in h, 17 / 4 for 1 mM BAPTA, 14 / 3 for 5 mM BAPTA.
i, Dependence of fluorescence lifetime change (averaged over 0 - 40 s) induced by glutamate uncaging on Ca2+ chelator concentration (EGTA: open circle, BAPTA: closed circle). Fluorescence lifetime was normalized to control.
Discussion
In this study, we visualized CaMKII activation in single dendritic spines during LTP. Using this method, we demonstrated that CaMKII activation is largely restricted to the stimulated spine during LTP induced by 2-photon glutamate uncaging (Figs. 1, 3). This suggests that CaMKII activation is important for the synapse specificity of LTP. This finding is in contrast to the activation of Ras, which is another molecule essential for LTP induction 30. In response to LTP-inducing stimuli, Ras is activated in stimulated spines, and then activated Ras spreads over ∼ 10 μm of dendritic length and invades nearby spines by diffusion 10. The restricted CaMKII activation can be explained by the rapid CaMKII inactivation in spines and dendrites (∼ 5 s and ∼ 1 min: Figs. 2a, b, 4a) and slow spine-dendrite diffusion coupling (∼ 1 and ∼ 20 min: Fig. 2c, d) 31: activated CaMKII at the stimulated spine gets inactivated before it diffuses out of the spine. In contrast, Ras inactivation is much slower (∼ 5 min) and spine-dendrite coupling time constant is smaller (∼ 5 s) 10. This allows activated Ras to spread and invade nearby spines before it gets inactivated. Because CaMKII and Ras act in parallel pathways to induce LTP 10, the spine specificity of LTP may be produced by CaMKII, while Ras may be permissive for LTP and responsible for dendritic level signalling such as the regulation of threshold for LTP induction 10, 14.
Rapid inactivation of CaMKII during LTP induction (Figs. 1, 3) is consistent with previous studies showing that persistent activation of CaMKII is not required for LTP maintenance 32, 33. However, this appears inconsistent with biochemical studies demonstrating that T286 phosphorylation is sustained after LTP induction 34, 35 . This discrepancy may be due to the difference in sample preparation (acute slices vs. cultured slices), in the population of CaMKII measured (whole lysate including soma vs. single synapses) or in stimulation paradigms: most of the previous biochemical studies used strong stimulations to induce LTP in a large number of synapses, whereas we induced LTP in a single synapse. Alternatively, only the subpopulation of CaMKII subunits that bind to NR2B subunit of NMDARs may maintain their activity 3, 36. Given that there is more CaMKII than NMDARs in the PSD 37, the subpopulation of CaMKII that undergoes persistent activation may be below the detection threshold of our method.
We found that T286 phosphorylation is required for high CaMKII activation in response to repetitive 2-photon uncaging (Figs. 1b, 2a). [Ca2+] transients last only ∼0.2 s after each uncaging (Supplementary Fig. 6a-c), due to strong Ca2+ extrusion in spines and dendrites 17. Thus, Ca2+ / CaM should dissociate from CaMKII rapidly after these stimulations as indicated in the activation time course of T286A Green-Camuiα (Figs. 2a, 4b). The phosphorylation of T286 prolongs the activation of CaMKII by ∼ 5 s (Figs. 2a, 4a), and allows for the accumulation of activated CaMKII during repetitive uncaging (Fig. 2a).
Our imaging results indicate that CaMKII activation in dendritic spines and dendritic shafts are regulated by different Ca2+ sources in response to cell depolarization: CaMKII in dendritic spines is activated specifically by L-type VSCCs, while CaMKII in dendritic shafts is activated mostly by non-L-type channels in addition to a small contribution from L-type VSCCs (Fig. 4). Because L-type VSCCs do not contribute to the global Ca2+ in spines and in dendritic shafts (Supplementary Fig. 6d), the L-type VSCC dependent activation of CaMKII must be produced in the nanodomain at the mouth of L-type VSCCs 4, 27, 28. Experiments using Ca2+ chelators with different binding rate (Fig. 5a-f) also suggest that nanodomain Ca2+ at calcium channels is sufficient for CaMKII activation in response to cell depolarization both in spines and dendrites. Because L-type VSCCs are required for hippocampal LTP induced by several protocols 19, 23-26, CaMKII activation by L-type VSCCs, although smaller than that by NMDARs (Figs. 3, 4), is likely to be important for the modulation of LTP.
Unlike CaMKII activation in response to depolarization, nanodomain Ca2+ alone was insufficient for NMDAR-mediated CaMKII activation (Fig. 5g-i). Because global Ca2+ elevation through non-L-type VSCCs, although it is as high as and longer-lasting than uncaging-evoked [Ca2+] elevation (Supplementary Fig. 6d), was not sufficient to activate CaMKII in spines (Fig. 4c), NMDAR-mediated CaMKII activation presumably requires both global and nanodomain Ca2+. Thus, CaMKII activation mechanisms depend on channels from which Ca2+ influx occurs. The difference may be due to different CaM localization near channels. Also, the dynamics of Ca2+-dependent protein phosphatase activity are likely to be important in shaping the spatiotemporal dynamics of CaMKII (Fig. 5a-f).
Most LTP induction protocols involve postsynaptic depolarization 18, which alone can produce large Ca2+ in spines (Supplementary Fig. 6) 19. However, it had remained a mystery why postsynaptic depolarization alone cannot induce synapse non-specific LTP 18, 19. Our results demonstrate that depolarization activates only a subset of CaMKII that interact with L-type VSCCs, which presumably is insufficient for LTP induction 38. Thus, local Ca2+-mediated, channel specific CaMKII activation in spines play an important role in the induction of synapse-specific LTP.
Methods summary
Green-Camuiα was made from Camuiα 5 by replacing ECFP with mEGFP and Venus with REACh (VenusA206K,T145W 7, 39). Hippocampal cultured slices were prepared from postnatal day 6-8 rats as described 40. Neurons were sparsely transfected with Green-Camuiα and mCherry using ballistic gene transfer 11 at days in vitro 8-14, and imaged 2-5 days after transfection. CaMKII activity was measured using 2pFLIM and spine volume change was monitored by measuring the fluorescence intensity of mCherry (red) in spines using regular 2-photon microscopy 9, 10. Statistical tests were performed using ANOVA followed by the least significant difference post-hoc tests. All error bars indicate s.e.m.
Supplementary Material
Acknowledgements
We thank H. Murakoshi for purified mEGFP and technical advice, Y. Hayashi for cDNAs and shRNA, A.Wang for cultured slices, T. Zimmerman and D. Kloetzer for laboratory management, M. Ehlers, C. Harvey, J. Lisman, M. Patterson, S. Raghavachari, K. Svoboda, R. Weinberg, and H. Zhong for comments on the manuscript. This study was funded by Burroughs Wellcome Fund, Dana Foundation, Alfred P. Sloan Foundation, Autism Speaks, National Alliance of Autism Research, Whitehall Foundation, Alzheimner’s Association, National Institute of Mental Health (R01MH08004), National Science Foundation (0642000), Duke University Undergraduate Research Support Grants (S.-J.R.L), Howard Hughes Neuroscience Forum Fellowship (S.-J.R.L), and Ruth K. Broad foundation (E.M.S.).
Appendix
Methods
Constructs
Camuiα (Venus-CaMKIIα-CFP) and short-hairpin (sh-) RNA against CaMKIIα was kindly provided by Dr. Y. Hayashi 5. Green-Camuiα was made by exchanging Venus with REACh (Venus 41 with monomeric mutation, A206K 6 and dark mutation, T145W 7, 39, 42) and CFP with mEGFP (EGFPA206K) 39.
Preparation
Hippocampal slice cultures were prepared from postnatal day 6 or 7 rats, as described 40. After 9-14 days in culture, cells were transfected with ballistic gene transfer 11 using gold beads (9-11 mg) coated with plasmids containing cDNA of Green-Camui (35 μg) and mCherry (16 μg). We used one neuron per one slice for most of our experiments. Samples were prepared in accordance with the animal care and use guidelines of Duke University Medical Center.
Imaging
Details of FRET imaging using 2pFLIM have been described previously 9, 39. Green-Camuiα and mCherry were simultaneously excited with a Ti:Sapphire laser tuned at 900 or 920 nm 39. Both epi- and trans- fluorescence were used for the detection. The fluorescence was divided with a dichroic mirror (565 nm) and detected with PMTs placed after wavelength filters (Chroma, HQ510/70-2p for green and HQ620/90-2p for red). For fluorescence lifetime imaging in the green channel, PMT with low transfer time spread (H7422-40; Hamamatsu) placed at epi-fluorescence site. For intensity imaging, wide aperture PMTs were used (R3896; Hamamatsu). Fluorescence lifetime image was produced on a PCI board (SPC-730; Becker-Hickl) controlled with custom software written using Matlab 7.0.
Fluorescence lifetime image analysis
Fluorescence lifetime of Green-Camuiα should have at least three components including open conformation, close conformation, and donor with unfolded acceptor 9. Thus, it is difficult to make a complete model and perform curve fitting. Instead, the mean fluorescence lifetime averaged over multiple populations τm was measured from the mean photon arrival time <t> as follows 9:
where F(t) is the fluorescence lifetime decay curve (Supplementary Fig. 1a), t is time, and t0 is the offset. We estimated the offset t0 before each experiment by fitting fluorescence lifetime decay curve of Green-Camuiα expressed in a neuron with double exponential function and comparing <t> with the fluorescence lifetime averaged over two populations as follows:
where P1 and P2 are the fraction and τ1 and τ2 are fluorescence lifetime of the two populations obtained by fitting. Change in fluorescence lifetime is independent of t0, therefore it depends on neither model nor curve fitting.
The fluorescence lifetime of Green-Camuiα in spines under resting condition was 1.82 ± 0.02 ns for wildtype (samples in Fig. 1 and Supplementary Fig. 4 pooled together), 1.84 ± 0.02 ns for T286A (Fig. 1 and Supplementary Fig. 4) and 1.81 ± 0.03 ns for T305D (Supplementary Fig. 4). The fluorescence lifetime under resting condition in the presence of pharmacological drugs was 1.77± 0.06 ns for AP5 (Fig. 1), 1.67 ± 0.03 ns for KN62 (Fig. 1), 1.81 ± 0.02 ns for Nimodipine (Fig. 4), 1.80 ± 0.01 ns for 1 - 20 mM EGTA (Fig. 5), and 1.83 ± 0.02 ns for 1 - 20 mM BAPTA (Fig. 5). Thus, most of the drugs we used in this study did not change the fluorescence lifetime, except that KN62 decreased the fluorescence lifetime significantly (p < 0.05, comparison between control and KN62 for samples in Fig. 1).
2-photon glutamate uncaging
A laser tuned at 720 nm was used to uncage MNI-caged glutamate (2 mM) in extracellular solution. The same galvano-scanning mirrors were used to steer both imaging and uncaging lasers. The laser spot was steered to the tip of a spine image (Supplementary Fig. 8), and 4-6 ms pulses at 4 mW were applied to stimulate glutamate receptors on the spine. This produced 2-20 pA postsynaptic current (Fig. 3d, Supplementary Fig. 8). The resolution of glutamate uncaging was ∼ 1 μm (Supplementary Fig. 8). The evoked current weakly depends on the distance between the tip of spine and the centre of uncaging spot (Supplementary Fig. 8). The intensity of the laser was modulated by a pockels cell (Con-Optics). For LTP experiment (Fig. 3), the uncaging spot was relocated before each uncaging session (each session includes 4 trials of glutamate uncaging at 0.2 Hz. The session was repeated every 10 minutes, Fig. 3c). Series and input resistance was continuously monitored, and if they change more than 20 %, the measurements were rejected from further analysis. To induce structural plasticity of spines (Figs. 1, 2b, d, 5g-i), we used ACSF (130 mM NaCl, 2.5 mM KCl, 2 mM NaHCO3, 1.25 mM NaH2PO4 and 25 mM glucose) containing 4 mM CaCl2, 1μM TTX and 2 mM MNI-caged-L-glutamate at 25 - 27 °C. To induce LTP with the pairing protocol, we added 1 mM MgCl2.
Fluorescence recovery after photo-bleaching (FRAP) and photoactivation
For FRAP (Supplementary Fig. 4b-c), spine fluroescence was photo-bleached by parking the imaging laser (tuned at 920 nm) for 1 - 8 s at a spine of interest. To activate photoactivatable GFP (paGFP; Fig. 2c, d), the uncaging laser (tuned at 800 nm) was parked for 20 ms at a spine of interest.
Electrophysiology
Whole-cell patch clamp was performed with patch pipettes (4-9 MΩ). We used cells with series resistance lower than 25 MΩ for experiments. To induce LTP with the pairing protocol (Fig. 3) or to induce cell depolarization (Figs. 4a-d, 5a-f), we performed experiments in a voltage-clamp mode using Cs+ internal solution (130 mM CsMeSO3, 10 mM Na-phosphocreatine, 4 mM MgCl2, 4 mM Na2-ATP, 0.4 mM Na2-GTP, 10 mM Cs-HEPES [pH 7.3]). To induce back-propagating action potentials (Fig. 4d), we injected 4 nA, 4 ms current pulses in a current-clamp mode using K+ based internal solution (Cs+ was substituted with K+) and ACSF with 4 mM CaCl2, 4 mM MgCl2, 100 μM APV and 10 μM NBQX. For experiments using BAPTA or EGTA (Fig. 5), we started imaging ∼ 15 min after establishing the whole-cell patch clamp with pipette including BAPTA or EGTA. Depolarization (Fig. 5a-f) and glutamate uncaging (Fig. 5g-i) were performed in voltage and current clamp modes, respectively. To prepare internal solution with 1, 5 and 20 mM EGTA or BAPTA (Fig. 5), we prepared 100 mM K-EGTA and K-BAPTA (pH 7.3), and mixed 1%, 5% and 20% volume of this solution and 99 %, 95 % and 80 % volume of Cs+- (Fig. 5a-f) or K+- (Fig. 5g-i) based internal solution, respectively. Then, we re-adjusted the osmolarity to 290-300 and pH to 7.3.
Calcium imaging
Ca2+ imaging (Supplementary Fig. 6) was performed as described previously 43. We performed whole-cell patch clamp to a neuron in a cultured hippocampal slice using an electrode containing Ca2+-sensitive green dye (500 μM Fluo-4FF) and Ca2+-insensitive red dye (300 μM Alexa-594) in Cs+ internal solution. Images were acquired every 32 ms (Supplementary Fig. 6a-c) or 256 ms (Supplementary Fig. 6d). The ratio of the fluorescence change of green normalized to red signal average over an imaging session (ΔG/R) was used as a measure of the [Ca2+] transient. Both fluorophores were simultaneously excited with a Ti:Sapphire laser tuned at 920 nm. The change in [Ca2+] (Δ[Ca2+]) was measured as:
where (G/R)sat is the ratio of green and red signals at saturating Ca2+ (10 mM) measured in a pipette, and KD is 10.4 μM for Fluo-4FF 43.
Measurements of the concentration of Green-Camuiα in cells
To measure the concentration of mEGFP-CaMKII or Green-Camuiα in neurons, we measured the fluorescence intensity of mEGFP at thick dendrites of neurons under 2-photon microscopy relative to the brightness of purified mEGFP (1 μM) measured under the same imaging setup. When measuring objects larger than 2-photon excitation volume (∼0.9 femtoliter), the brightness should be proportional to the concentration of fluorophores. We overexpressed mEGFP tagged with poly-histidine tag (His6) (pRSET-His6-mEGFP) in Escherichia coli and purified them using a Nickel-NTI affinity column. The concentration of the purified protein was measured by the absorbance of mEGFP (A489 = 56,000 cm-1M-1) 44. For Green-Camuiα, because FRET reduces its green fluorescence, we corrected for this effect by multiplying τmEGFP / τGreen-Camuiα ∼1.4, where τmEGFP and τGreen-Camuiα is the fluorescence lifetime of mEGFP (2.6 ns) and Green-Camuiα (1.8 ns), respectively.
In vitro fluorescence lifetime assay
To measure fluorescence lifetime of Green-Camuiα in cell lysates (Supplementary Fig. 1), we transfected HEK-293 cells with Green-Camuiα using Lipofectamine 2000. Cells were lysed with a solution containing 40mM HEPES (pH 8.0), 0.1mM EGTA, 5mM Magnesium Acetate, 0.01% Tween 20, and concentrated with a centrifugal filter (Microcon, Millipore) to remove CaM and ATP. The lysate was diluted to 15 times in 136.5 mM potassium gluconate, 17.5 mM KCl, 9 mM NaCl, 1 mM MgCl2, 0.2 mM EGTA, 10 mM HEPES-KOH (pH 7.2). We measured the fluorescence lifetime in response to increased free [Ca2+] by adding 0.5 mM CaCl2 and then reduced free [Ca2+] by adding 2 mM EGTA.
Immunofluorescence imaging
Slices were transfected with Green-Camuiα (or mEGFP-CaMKIIα) and empty vector, which substitutes mCherry. In parallel experiments shRNA against CaMKIIα (35 μg) and mEGFP (16 μg) were used to verify the specificity of immunostaining. Slices were fixed in 4 % paraformaldehyde plus 4 % sucrose (in 0.1 M phosphate buffer) for 15 minutes followed by incubation in 30 % sucrose for 15 minutes at room temperature. Slices were then re-sectioned using a vibratome at a thickness of 25 μm. The sections were permeabilized and blocked overnight at 4° C in 1% Triton-X100 plus 5% normal goat serum, followed by 30 minutes blocking in 10% goat serum (in 0.1 M phosphate buffer). Immunostaining was performed for 24 hours at 4° C using as primary antibody mouse anti-CaMKIIα (GeneTex Inc., San Antonio, TX) diluted 1:400 in 5% goat serum in PBS-T (phosphate buffer saline plus 0.1% Triton-X100). After 3 washings (20 minutes each) in PBS-T, sections were incubated with a Texas-red conjugated goat anti-mouse secondary antibody (Invitrogen/Molecular Probes) diluted 1:400 in 10% normal goat serum in PBS-T. The immunofluorescence was quantified under 2-photon microscopy in cell body excluding nucleus (Supplementary Fig. 2). The value was subtracted by the dark noise of photomultiplier. There was a few percent of bleed-through of mEGFP fluorescence to the red channel. To correct this effect, we measured the ratio (r) between the red and green fluorescence of mEGFP fluorescence using a purified mEGFP. Then, the bleed-through was corrected as:
where IRed and ITexas-red are the immunofluorescence before and after the correction, and IGreen is mEGFP fluorescence in the green channel.
Immunoprecipitation
HEK-293 cells were transfected with Green-Camuiα and/or CaMKIIα using Lipofectamine 2000. Cells were lysed in a buffer containing 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 0.5% deoxycholic acid, 25 mM beta-glycerophosphate, 1 mM sodium orthovanadate, 2 mM sodium-pyrophosphate, 2 mM EDTA, 1 mM sodium fluoride, 0.5 mM DTT, EDTA-free protease inhibitor cocktail (Roche) and incubated for 30 min at 4°C. The supernatants were collected after a 30 min microfuge centrifugation at 13,000 rpm at 4°C. After precleaning with protein A-Sepharose (Sigma, St. Louis, MO), samples were incubated with 5 μg/sample of mouse monoclonal anti-Green Fluorescent Protein antibody (MBL, Nagoya, Japan) or mouse non-immune IgG (Jackson Laboratories, ME) at 4°C overnight. The immunocomplexes were precipitated with protein G-Sepharose beads (Sigma, St. Louis, MO) for 1 h at 4°C and analyzed by Western blotting (Supplementary Fig. 9).
Fluctuation Correlation Spectroscopy (FCS)
FCS was performed under a 2-photon microscope with a Ti:Sa laser tuned at 900 nm at 25-27°C 45. Fluorescence signal was detected with a photomultiplier (PMT; H7422-40, Hamamtsu), and acquired with a PCI board (SPC-140; Becker-Hickl). HEK293 cells were transfected by Lipofectamine 2000, allowed to express for 2 days, lysed in a solution containing 40mM HEPES (pH 8.0), 0.1mM EGTA, 5mM Magnesium Acetate, 0.01% Tween 20, and diluted to 10-100 times in a solution containing 10 mM Na-HEPES (pH 7.4), 150 mM NaCl and 0.1 mM EGTA.
References
- 1.Rosenberg OS, et al. Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. Febs J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- 2.Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 3.Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Hudmon A, et al. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takao K, et al. Visualization of synaptic Ca2+ /calmodulin-dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 7.Ganesan S, Ameer-Beg SM, Ng TT, Vojnovic B, Wouters FS. A dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc Natl Acad Sci U S A. 2006;103:4089–4094. doi: 10.1073/pnas.0509922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yasuda R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr. Opin. Neurobiol. 2006;16:551–561. doi: 10.1016/j.conb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda R, et al. Super-sensitive Ras activation in dendrites and spines revealed by 2-photon fluorescence lifetime imaging. Nat. Neurosci. 2006;9:283–291. doi: 10.1038/nn1635. [DOI] [PubMed] [Google Scholar]
- 10.Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAllister AK. Biolistic transfection of neurons. Sci. STKE. 2000;2000:PL1. doi: 10.1126/stke.2000.51.pl1. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzaki M, et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otmakhov N, et al. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno M, Frankland PW, Silva AJ. A pharmacogenetic inducible approach to the study of NMDA/alphaCaMKII signaling in synaptic plasticity. Curr Biol. 2002;12:654–656. doi: 10.1016/s0960-9822(02)00767-4. [DOI] [PubMed] [Google Scholar]
- 17.Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- 18.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Conti R, Lisman J. A large sustained Ca2+ elevation occurs in unstimulated spines during the LTP pairing protocol but does not change synaptic strength. Hippocampus. 2002;12:667–679. doi: 10.1002/hipo.10096. [DOI] [PubMed] [Google Scholar]
- 20.Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995;487(Pt 1):67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noguchi J, Matsuzaki M, Ellis-Davies GC, Kasai H. Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron. 2005;46:609–622. doi: 10.1016/j.neuron.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 25.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 26.Remy S, Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proc Natl Acad Sci U S A. 2007;104:17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat. Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- 28.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 33.Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- 34.Fukunaga K, Muller D, Miyamoto E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long-term potentiation. J Biol Chem. 1995;270:6119–6124. doi: 10.1074/jbc.270.11.6119. [DOI] [PubMed] [Google Scholar]
- 35.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 36.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 37.Cheng D, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics. 2006;5:1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron. 1992;9:1175–1183. doi: 10.1016/0896-6273(92)90075-o. [DOI] [PubMed] [Google Scholar]
- 39.Murakoshi H, Lee S-J, Yasuda R. Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative YFP. Brain Cell Biol. 2008;36:31–42. doi: 10.1007/s11068-008-9024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 41.Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 42.Kwok S, et al. Genetically encoded probe for fluorescence lifetime imaging of CaMKII activity. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda R, et al. Imaging calcium concentration dynamics in small neuronal compartments. Sci. STKE. 2004;2004:l5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- 44.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 45.Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.