

Abstract
Salvinorin A is an unregulated potent hallucinogen isolated from the leaves of Salvia divinorum. It is the only known non-nitrogenous kappa-opioid selective agonist and rivals synthetic lysergic acid diethylamide (LSD) in potency. This objective of this study was to characterize the in vitro transport, in vitro metabolism, and pharmacokinetic properties of Salvinorin A. The transport characteristics of Salvinorin A were assessed using MDCK-MDR1 cell monolayers. The P-glycoprotein (P-gp) affinity status was assessed by the P-gp ATPase assay. In vitro metabolism studies were performed with various specific human CYP450 isoforms and UGT2B7 to assess the metabolic characteristics of Salvinorin A. Cohorts (n=3) of male Sprague Dawley rats were used to evaluate the pharmacokinetics and brain distribution of Salvinorin A (10 mg/kg, intraperitonal (i.p.) over a 240 min period. A validated UV-HPLC and LC/MS/MS method was used to quantify the hallucinogen concentrations obtained from the in vitro and in vivo studies, respectively. Salvinorin A displayed a high secretory transport in the MDCK-MDR1 cells (4.07±1.34 × 10-5 cm/s). Salvinorin A also stimulated the P-gp ATPase activity in a concentration (5-10 μm) dependent manner, suggesting that it may be a substrate of P-gp. A significant decrease in Salvinorin A concentration ranging from 14.7±0.80 % to 31.1±1.20 % was observed after incubation with CYP2D6, CYP1A1, CYP2C18, and CYP2E1, respectively. A significant decrease was also observed after incubation with UGT2B7. These results suggest that Salvinorin A may be a substrate of UGT2B7, CYP2D6, CYP1A1, CYP2E1 and CYP2C18. The in vivo pharmacokinetic study showed a relatively fast elimination with a half-life (t1/2) of 75 min and a clearance (Cl/F) of 26 L/h/kg. The distribution was extensive (Vd of 47.1 L/kg), however the brain to plasma ratio was 0.050. Accordingly, the brain half life was relatively short, 36 min. Salvinorin A is rapidly eliminated after i.p. dosing, in accordance with its fast onset and short duration of action. Further, it appears to be a substrate for various oxidative enzymes and multi-drug resistant protein, P-gp.
Keywords: Salvinorin A, Salvia divinorum, pharmacokinetics, metabolism, transport, blood brain barrier, hallucinogen
1. INTRODUCTION
Psychotropic natural products are widely available through a variety of commercial sources; however they represent a class of agents that are understudied, possibly toxic and possess pharmacologic properties consistent with drug abuse liability. Salvia divinorum and its active component, Salvinorin A (Figure 1), is a potent hallucinogen whose use is associated with “altered consciousness” and its status in this country and abroad has been under review. This substance has been banned in 5 states (Delaware, Louisiana, Missouri, Oklahoma, and Tennessee) and two states (New Jersey and New York) are currently formulating legislature on Salvia divinorum. Other countries like Australia, Denmark, Finland, Italy, and Sweeden have recently classified Salvia divinorum as a controlled substance [1-5]. Reports have predicted that its use will most likely reach the levels associated with similar hallucinogenic agents such as 3,4-methylenedioxy methamphetamine (MDMA), phencyclidine (PCP) and lysergic acid diethylamide (LSD) in the next 5 - 10 years [6,7]. This is evidenced by a sharp increase in its consumption by college students and young adults over the last few years [3, 6-10].
Fig. 1.
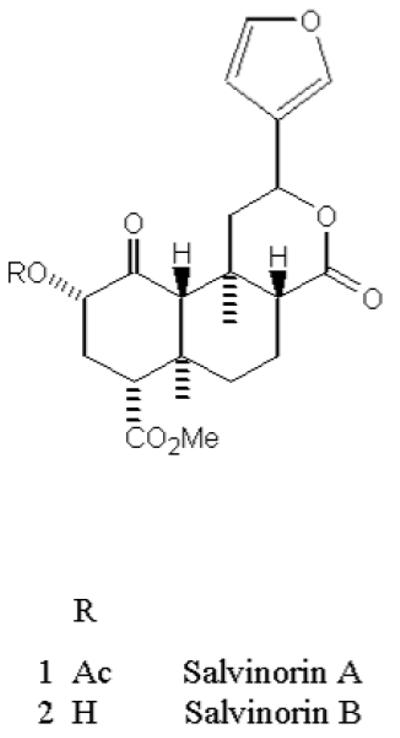
Chemical structure of Salvinorin A and B
Salvia divinorum is typically consumed by smoking a quantity of dried leaves, although buccal absorption by chewing dried leaves or ingesting a tincture is also used. The onset of action is relatively rapid, on the order of 30 sec for smoking and 5-10 min for buccal absorption after ingestion [2, 11]. Salvinorin A is an extremely potent naturally occurring hallucinogen, with an effective dose, when smoked, of 200 - 500 μg [2, 12]. It induces an intense, short-lived hallucinogenic experience in humans appearing in less than 1 min and lasting 15 min or less [13]. Its effect is reported to be qualitatively distinct from that induced by the classical hallucinogens such as LSD, psilocybin and mescaline. Salvinorin A is a neoclerodane diterpene (Figure 1) and chemically, it lacks a basic nitrogen structure which is an uncommon feature associated with psychoactive hallucinogenic agents, however, it does have a number of carbon atoms that enhance its lipopilicity. Pharmacologically, Salvinorin A does not act at the molecular target responsible for the actions of classic hallucinogens, the serotonin 5-HT2A receptor [14,15]. Salvinorin A is selective for the κ opioid receptor (KOR) [16,17], produces KOR-like discriminative effects in rhesus monkeys [18], low dopamine levels in the mouse caudate putamen [19], and rat nucleus acumbens [20] through activation of KORs. Presently, limited research has been conducted to characterize Salvinorin A. It is suggested that Salvinorin B (Fig. 1) is an inactive metabolite of Salvinorin A [21, 22] and that it possibly shares metabolic pathway(s) with cocaine, heroin, tetrahydrocannabinol (THC) and MDMA and is metabolized by esterase in the blood [23]. In addition, a pharmacokinetic study in rhesus monkeys found that its elimination half-life is rapid (56.6 ±24.8 min) [24] which corresponds with its short duration of action. Recently, positron emission tomography (PET) studies performed in baboons indicated extremely rapid brain uptake. [11C]-salvinorin A was distributed throughout the brain with the highest concentration in the cerebellum and a notable concentration the visiual cortex perhaps accounting for its physiological effects when smoked [25].
To date, there are several investigations underway to evaluate Salvinorin A’s pharmacologic properties, however its blood brain barrier (BBB) transport, metabolism, and pharmacokinetics have not been well described. As such, the rate and extent of its distribution across the BBB into the central nervous system (CNS) responsible for producing hallucinogenic effects are unknown as are those dispositional properties that mediate its duration of action. To investigate these, the following three objectives were pursued: (1) assessment of the in vitro transport mechanism of Salvinorin A across MDCK-MDR1 cell monolayers, (2) characterization the in vitro metabolism of Salvinorin A using recombinant human CYP450 (InVitroSomes™) and UGT2B7 enzyme (Supersomes™ membrane fractions from insects cells expressing UDP-glucurosyltransferases (UGT) isoform), and (3) evaluation of the single-dose pharmacokinetics of Salvinorin A in male Sprague Dawley rats.
2.MATERIALS and METHODS
2.1.Materials
Salvinorin A was provided by Dr.Thomas Prisinzano (Iowa University, Iowa). 4-chloro benzotropine (BZT) was synthesized and provided by Dr.Amy H. Newman (NIH, Baltimore, MD). The purieties of salvinorin A and BZT were > 98%. All chemicals and solvents were American Chemical Society analytical grade or HPLC grade. InVitroSomes™, human recombinant cytochrome P450 enzymes were purchased from InVitro Technologies (Baltimore, MD). Human UGT2B7 Supersomes™ enzymes were purchased from BD Biosciences Discovery Labware (Woburn MA). MDCK-MDR1 cells were provided by Dr.Peter W. Swaan (University of Maryland). DMEM, phosphate buffered saline, non-essential amino acid, fetal bovine serum (FBS), L-glutamine, pencillin G-streptomycin sulfate antibiotic mixture and trypsin (0.25%)-EDTA (1mM) were purchased from Invitrogen Laboratories (Carlsbad, CA). Polymixin, amphotericin, heparin, and dextran were purchased from the Sigma Chemical Co. (St.Louis, MO). Twelve well transport plates (cell culture treated) were purchased from Corning Costar (Cambridge, MA).
2.2.SalvinorinA-stimulated P-gp ATPase activity
In order to assess whether or not Salvinorin A was a P-gp substrate, we determined its ability to stimulate ATPase activity. Salvinorin A stimulated P-gp ATPase activity was estimated by Pgp-GIO assay system (Promega, Madison, WI). This method relies on the ATP dependence of the light-generating reaction of firefly luciferase where ATP consumption is detected as a decrease in luminescence. In a 96 well plate recombinant human P-gp were incubated with P-gp-GIO assay buffer™ (20 μL), verapamil (200 μM), sodium orthovanidate (100 μM), Salvinorin A (2.5 to 100 μM). Each compound was loaded into four individual wells. Verapamil served as a positive control while sodium orthovanadate was used as a P-gp ATPase inhibitor. In the presence of sodium orthovanadate ATP consumption by P-gp is negligible and without sodium orthovanadate, P-gp consumes ATP to a greater or lesser extent than the control, dependent on the effect of the test compounds. The reaction was initiated by addition of MgATP (10 mM), stopped 40 min later by addition of 50 μl of firefly luciferase reaction mixture (ATP detection reagent) that initiated an ATP-dependent luminescence reaction. Signals were measured 100 min later by Lmax® luminometer (Molecular Devices Corporation, Sunnyvale, CA) and converted to ATP concentrations by interpolation from a luminescent ATP standard curve. The rate of ATP consumption (pmol/min/μg protein) was determined as the difference between the amount of ATP in absence and presence of sodium orthovanadate. Salvinorin A-stimulated P-gp ATPase activity was reported also as fold-stimulation relative to the basal P-gp ATPase activity in the absence of compound (control).
2.3. In vitro cell culture studies: MDCK-MDR1 cells
MDCK-MDR1 cells were cultured in Dulbecco’s modified eagle serum (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100U/mL of pencillin and streptomycin. The cells were plated onto 12 well Costar TranswellR inserts (0.4μM pore size, 1cm2 surface area) at a density of 425,000 cells/cm2. The cells were cultured and maintained in DMEM supplemented with 10% FBS, 2% L-glutamine, 1% non essential amino acid, 1% pencillin-streptomycin under standard conditions of 5% CO2, 37 ± 0.5 °C and 95% humidity until confluence was reached on day four. The medium was changed every day after seeding and confluent monolayers were used for transport studies outlined below. Monolayer integrity was checked by measuring the transepithelial resistance (TEER) and [14C]-mannitol permeability. P-gp functional confirmation was determined by [14C]paclitaxel efflux values.
2.4. Transport study
MDCK-MDR1 transport studies were performed as previously described in our laboratory [26]. All transport experiments were performed at 37°C in phosphate buffered saline (PBS). Salvinorin A (5 μM), radiolabeled marker ([14C]mannitol, [14C]paclitaxel) or blank buffer were added to either the apical or basolateral side. Cell monolayers were continuously agitated on a plate shaker during transport experiments (60-70 rpm). For examination of transport in the apical to basolateral (A→B) direction, the transwell inserts were moved to wells with fresh PBS. At time t = 0, 0.5 mL of Salvinorin A solution was added to the apical side of the monolayer. Inserts were moved to new wells at times between 10 and 120 minutes. For examination in the basolateral to apical (B→A) direction, Salvinorin A (1.5 mL) was initially added to the basolateral side at time t = 0. At various time intervals between 10-120 min, samples were collected from the apical side and replaced with fresh, pre-warmed PBS. Samples were analyzed by a UV-HPLC method for Salvinorin A. Apparent permeability coefficients were determined for each transport study. Radioactive compounds were analyzed by scintillation counter (Beckman Coulter LS 6500).
2.5.In vitro metabolism screening with various human CYP450 isoforms and UGT2B7
A screening phenotyping approach was used to identify the isozyme(s) responsible for its metabolism using various cytochrome (CYP) isoforms including CYP2D6, CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2E1, CYP3A4 and CYP3A5. InVitroSomes™, used in this study, are single enzyme systems that express a specific CYP isoform (e.g., CYP2D6) and yeast CYP-reductase co-expressed in Saccharomyces cerevisiae. In VitroSomes™ are ideal for use in drug metabolism studies [27] particularly in specific CYP pathway identification (reaction phenotyping) and CYP inhibition screening. InVitroSomes™ expressing the aforementioned isoforms were incubated with Salvinorin A (0, 50 μm or 5 μm), and TE (500 mM Tris, 10 mM EDTA) reaction buffer. TSE (500 mM Tris, 2 M NaCl, 10 mM EDTA) reaction buffer was used for CYP2B6, CYP3A4, and CYP3A5 instead of TE buffer. This solution was incubated with shaking for 5 minutes at 37°C. At the end of this period, 20 μL of NADPH (5 mg/mL) was added to each sample to start the reaction. This was followed by one hour incubation at 37°C. At the end of this second incubation period, 100 μL of cold acetonitrile was added to stop the reaction. All samples were analyzed for Salvinorin A using our validated UV-HPLC method. Preliminary studies were performed to find the optimal incubation time which was used in subsequent studies. Salvinorin A (50 μM) was incubated with CYP2D6 at different incubation times (10, 30 and 60 min) at 37°C. Significant decrease was observed at 60 min, and this incubation time period was used for subsequent studies.
In vitro metabolism studies were also conducted to evaluate the role of UGT in Salvinorin A’s metabolism. Supersomes™ were used for possible glucuronidation activity. The reaction mixture (0.2 mL) containing 1 mg/mL protein (UGT2B7), 1mM uridine diphosphoglucuronic acid (UDPGA), 10 mM magnesium chloride, 0.025 mg/mL alamethicin and 0, 10 or 50 μM of Salvinorin A in 50 mM tris (pH 7.5) was incubated at 37°C for one hour. Two hour incubation time was used for 0 and 5 μM of Salvinorin A. After the incubation period, the reaction was stopped by the addition of 100 μL acetonitrile and centrifuged (10,000 rpm) for 15 minutes. All samples were analyzed using an UV-HPLC method.
2.6. Pharmacokinetic studies
2.6.1. Animals
Male Sprague Dawley rats weighing 275 to 300 g were purchased from Harlan Laboratories (Indianapolis, IN). The animals were housed in an AAALAC accredited facility run on a 12 hour light and dark cycle. The animals were allowed unrestricted access to food and water. The protocol for the animal studies was approved by the School of Pharmacy, University of Maryland IACUC.
2.6.2. Dosing and sampling
To evaluate the in vivo pharmacokinetic and brain distribution of Salvinorin A, a single dose study was performed in adult male Sprague Dawley rats. Salvinorin A was administered as a single i.p. dose of 10 mg/kg (in Cremophor EL: ethanol, 70%:30%). A destructive sampling study design was followed where cohorts of three animals were euthanized by CO2 asphyxiation at pre-dose and at the following time points post dosing: 5, 10, 15, 30, 60, 90, 120, and 240 minutes. Blood samples were collected by cardiac puncture using pre-heparinized syringes and immediately transferred into tubes containing acetonitrile to inhibit possible esterase metabolism. Blood samples were centrifuged at 3500 rpm for 10 minutes and plasma separated. Brain tissue was immediately excised, blotted dry and weighed. All samples were stored at -80°C until analyzed.
2.7. Quantification of Salvinorin A
2.7.1. UV-HPLC systems to quantifying Salvinorin A in in vitro samples
A UV-HPLC method was used to quantify the Salvinorin A concentrations in the transport and in vitro metabolism studies. The chromatographic conditions consisted of a Waters Symmetry (C18 5 μm, 150 × 4.6 mm) column plus Supelguard 5 μm LC-18, 2 cm guard column. The mobile phase (acetonitrile: water, 55:45 v/v) was filtered through a 0.45 μm nylon filter and degassed under ultrasound and vacuum for 15 min and pumped at a flow rate 1 mL/min over a 20 min period. The injection volume was 200 μL. Salvinorin A was quantified at a UV wavelength of 210 nm and its retention time was 12 min. The sensitivity limit for Salvinorin A using this method was 100 ng/mL.
2.7.2. LC/MS/MS analytical method to quantifying Salvinorin A in biological matrix
An LC/MS/MS analytical method was used to quantify Salvinorin A in plasma and brain. Four mL of hexane was added to 0.4 mL of plasma, vortexed (1 min) and centrifuged at 10,000 rpm for 10 min. The supernatant was evaporated to dryness at 40°C under a gentle stream of nitrogen and reconstituted with 100 μL of mobile phase. Brain tissue was homogenized, and diluted with an equal volume of PBS. Four mL of hexane was added to the brain homogenate (0.8 mL), vortexed for 2 minutes and centrifuged at 10,000 rpm for 10 min. The organic phase was transferred to a clean test tube, evaporated under nitrogen and reconstituted with 100 μL of mobile phase. The supernatant was transferred to a microvial and thirty μL were injected onto the LC/MS/MS system.
The LC/MS/MS system consisted of a quattro micro triple quadrapole mass spectrometer (Micromass-Waters, Millford, Boston) operated in the positive ion mode with an ESI-probe. The mass spectrometer was operated in the multiple reaction monitoring (MRM) mode. The source was operated at 140 degree and nitrogen was used as the nebulizer gas and argon was used as the collision gas set at 10 psi. The cone voltage was 45 V, capillary voltage was 3.5 V, and the entrance and exit voltages were -5 and 1 respectively. The HPLC system consisted of a Waters 2695 quaternary system, Xterra MS C18 (2.5 μm, 2.1 × 50 mm,) column and the mobile phase was composed of acetonitrile and water (55:45, v/v). The mobile phase was pumped at flow rate of 0.2 mL/min and the injection volume was 30 μL. Following HPLC separation, the Salvinorin A peak area corresponding to 433.5-373 parent-daughter transition and the internal standard peak (BZT) peak area corresponding to 342.5-201.1 parent daughter transition were quantitated. The retention time for Salvinorin A was 2.8 min and 1.6 min for the internal standard. The total run time was five minutes. The plasma and brain calibration curves were linear in the range of 7.5 to 500 ng/mL (r2 ≥ 0.999) and 7.5 to 200 ng/mL (r2 ≥ 0.997) for plasma and brain, respectively.
2.8. Data analysis
2.8.1. ATPase assay
Basal P-gp activity, test compound stimulated P-gp activity and fold stimulation by a test compound were calculated according to the following equations:
| Eq.(1) |
| Eq.(2) |
Where ATPvanadate is the number of non consumed (total) pmoles of ATP in the presence of sodium orthovanadate. ATPcontrol is the number of non-consumed pmoles of ATP in presence of the assay buffer. ATPcompound is the number of non consumed pmoles of ATP in presence of a test compound.
| Eq.(3) |
2.8.2 Permeability calculations and cell culture data analysis
The calculation of apparent permeability (Papp), for transport studies across cell monolayers was determined from the following equation:
| Eq.(4) |
where Papp is the permeability, Vr is the receiver compartment volume, dCr is the receiver compartment concentration at the end of the interval, dt is the time of the interval, A is the area of the filter, and Cd is the donor compartment concentration at the start of the interval. All experiments were performed in triplicate and data from the transport experiments are presented as mean ± standard deviation (SD). The efflux ratio (R) was calculated according to the following equation:
| Eq.(5) |
2.8.3. In vitro metabolism data analysis
In vitro metabolism data were converted to the % remaining of Salvinorin A as below:
| Eq.(6) |
where Ctreated is enzyme treated Salvinorin A concentration, Cinitial is initial Salvinorin A concentration. All metabolism experiments for each enzyme were done in triplicate and data was presented as mean ± SD. Data were statistically compared by Student’s t-test and significance set at p<0.05.
2.8.4. Pharmacokinetic analysis
Non-compartmental modeling was used to estimate Salvinorin A pharmacokinetics parameters after single dose administration. The Salvinorin A plasma or brain concentration-time data were evaluated using the nonlinear regression program WinNonlin™ (Pharsight Corp., Mountainview, CA; version 4.1). Pharmacokinetic parameters Cmax (maximum Salvinorin A concentration), tmax (the time that Cmax occurred), AUC0-240 (area under the plasma concentration time curve from 0 up to 240 min), AUC0-inf (area under the plasma concentration time curve from 0 up to infinity), λz (elimination rate constant), t1/2 (half-life), Vd/F (volume of distribution), and Cl/F (clearance) were determined using non-compartmental analysis. Brain distribution of Salvinorin A was determined by calculating brain to plasma partition coefficient of Salvinorin A (Ri). The AUC0-inf for both plasma and brain were determined by noncompartmental methods utilizing the linear trapezoidal rule. RiAUC was calculated according to the following equation using the brain and plasma AUC0-inf values:
| Eq.(7) |
It should be noted that this was a destructive sampling study design and hence variability associated with the pharmacokinetic parameters could not be determined.
3. RESULTS
3.1. Salvinorin A - stimulated P-gp ATPase activity
Various concentrations of Salvinorin A (2, 5, 10 μM) concentrations were examined for their effects n P-gp ATPase activity. Each Salvinorin A concentration together with a known excess of ATP was incubated with recombinant human P-gp for 100 minutes. ATP consumption was detected as a decrease in luminescence i.e. the higher the stimulation of the P-gp ATPase activity, the lower the luminescence signal. As seen in Table 1, concentrations of Salvinorin A of 5 μM, 10 μM, the rate of ATP consumption (47.48 ± 3.36 and 41.14 ± 5.86 pmol/μg P-gp/min) were significantly different (p <0.05) from the control (21 ± 4.21 pmol/μg P-gp/min). The known P-gp substrate, verapamil (200 μM), stimulated the rate of ATP consumption by 103.01 ± 5.86 pmol/μg P-gp/min (p <0.05). The P-gp ATPase assay indicated that the tested Salvinorin A concentrations stimulated P-gp ATPase activity in a concentration dependant manner (5 μM, 10 μM ) indicating that the hallucinogen is most likely a P-gp substrate.
Table 1.
Salvinorin A enhancement of P-gp ATPase activity in drug-treated membranes compared with non treated membranes
| Drug | no. of wells | pmol/μg P-gp/min | Fold Stimulation |
|---|---|---|---|
| Non treated | 4 | 21.31± 4.21 | 1.0 |
| Veramapil (200 μM) | 4 | 103.01 ± 1.99a | 4.83 |
| Salvinorin A (2.5 μM) | 3 | 50.94 ± 17.87 | 2.39 |
| Salvinorin A (5 μM) | 3 | 47.78 ± 3.36a | 2.24 |
| Salvinorin A (10 μM) | 3 | 41.14 ± 5.86b | 1.93 |
| Salvinorin A (25 μM) | 3 | 31.08 ± 21.18 | 1.46 |
indicates significant difference from the non treated at p < 0.01
indicates significant difference from the non treated at p < 0.05
3.2. Salvinorin A transport across MDCK-MDR1 cell line
The objective of this study was to determine if this hallucinogen is highly permeable across cell lines expressing P-gp. In MDCK-MDR1 cells, the secretory transport (4.07 ± 1.34 × 10-5 cm/s) of Salvinorin A was higher than the absorptive transport which could not be determined. It should be noted that the limit of detection of the analytical method was 100 ng/ml for the UV-HPLC method and 7.5 ng/ml for the LC/MS/MS method. In MDCK-MDR1 cells TEER values above 900 Ω cm2 were used in the study. Mannitol permeability was 3.23±1.16 × 10-6 cm/s and the paclitaxel efflux was 15.2.
3.3. In vitro metabolism of Salvinorin A
Salvinorin A’s chemical structure suggests that it may be a substrate of CYP450 (oxidative metabolism), UGT (hydrolysis) or carboxylesterases (ChEs) (hydrolysis). A recent study has provided evidence for Salvinorin A’s metabolism by blood ChEs [23]. However, CYP450 and UGT enzymes that contribute to its metabolism have not been elucidated. Salvinorin A displays a short duration of action after ingestion, suggesting that it is cleared rapidly and that metabolism most likely plays a significant role in the dissipation of its effect. Metabolism studies using a series of specific human CYP450 isoforms and UGT2B7 enzyme were performed. After 1 h, there was a significant decrease (p<0.05) of 10(± 1.2) %, 5.3(±2) %, 6(±1.2) %, and 6.4(±1.6) %, when Salvinorin A (50 μM) was incubated with CYP2D6, CYP1A1, CYP2C18, and CYP2E1, respectively, as seen in Fig. 2(a). Further, when Salvinorin A was incubated at a concentration of 5 μM with CYP2D6, CYP1A1, CYP2C18, and CYP2E1, there was also statistically significance (p <0.01) reduction of 14.7(± 0.80) %, 31(±1.20) %, 20.6(±1.00) %, and 22(±0.80) %, respectively (Fig. 2b). Salvinorin A (50 μM) percent reductions for CYP1A1, CPY2C18 and CYP2E1 when compared to the 5 μM concentrations were found to be lower. These results suggest that the metabolism of Salvinorin A in these CYP450 isoforms follows Michaelis Menten kinetics.
Fig. 2.

Salvinorin A in vitro metabolism screening with various CYP 450 isoforms. Salvinorin was incubated with CYP450 enzymes at a concentration of 50 μM (a), or 5 μM (b) as indicated. The values show the % reduction on initial concentration (mean ± SD, n=3). Different from initial concentration *p< 0.05 and **p< 0.01.
In vitro metabolism studies were also conducted to evaluate the role of UGT in Salvinorin A’s metabolism. Based on its structure, it was postulated that it would also undergo glucuronidation. Among the UGTs identified in humans, UGT2B7 is the major enzyme involved in glucuronidation of most drugs [28-30]. Because of this UGT2B7 was used to examine the possible subsequent glucuronidation of Salvinorin A using a baculovirus expression system (Supersomes™). UGT2B7 was incubated with Salvinorin A at 0, 5, 10 and 50 uM. A decrease of 7(±5.60) % (p <0.05), 18.1(± 5.20) % (p <0.05) and 51(± 4.00) % (p <0.01), was observed for the 50, 10 and 5 μM Salvinorin A concentration vs the control as illustrated in Fig. 3. In addition, as observed for CYP1A1, CPY2C18 and CYP2E1, Salvinorin A metabolism via UGTB27 appears to be saturable at higher concentrations.
Fig. 3.

Salvinorin A in vitro metabolism screening with UGT2B7 (n=3). Salvinorin was incubated with UGT2B7 at a concentration of 50, 10 and 5 μM as indicated. The values show the % reduction on initial concentration (mean ± SD, n=3). Different from initial concentration *p< 0.05 and **p< 0.01.
3.4. Pharmacokinetic evaluation
The mean plasma and brain concentration versus time profiles for Salvinorin A after a single i.p. dose of Salvinorin A are presented in Fig. 4 (a) and (b), respectively. Brain levels were detected over 1 h. It should be noted that the limit of detection of LS/MS/MS method was 7.5 ng/mL and recovery was >92%. The pharmacokinetic parameters were calculated using non-compartmental analysis method using WinNonlin™ and pharmacokinetic parameters are summarized in Table 2. Due to the study design (destructive sampling), the variability associated with the pharmacokinetic parameters could not be determined. The plasma profile and brain uptake of Salvinorin A was found to be rapid with an apparent tmax occurring at 10-15 min after i.p. administration. AUC0-inf in plasma and brain were as 410 μg h/L and 20.6 μg h/L, respectively. The elimination of Salvinorin A was relatively fast with a t1/2 of 75 min and a clearance (Cl/F) of 26 L/h/kg. The distribution was extensive (Vd of 47.1 L/kg); however the brain to plasma ratio was very low ranging from 0.092 to 0.074 over a 60 min period (Fig. 4). Accordingly, the brain half life was relatively short, 36 minutes and the brain/plasma partitioning was 0.050.
Fig. 4.

Salvinorin A (a) plasma concentration versus time profile (b) brain concentration versus time profile. After a 10 mg/kg i.p. dose to Sprague Dawley rats (n =3/pt), (mean ± SD).
Table 2.
Non-compartmental pharmacokinetic parameters for Salvinorin A after single i.p dose of 10 mg/kg to male Sprague Dawley rats
| Pharmacokinetic Parameter | Plasma | Brain |
|---|---|---|
| AUC0-240 (μg h /L) | 348 | 13.0 |
| AUC0-inf (μg h/L) | 410 | 20.6 |
| Cmax (ng/mL) | 345 | 23.9 |
| tmax (min) | 15 | 10 |
| λz (h-1) | 0.552 | 0.115 |
| t1/2 (min) | 75.4 | 36.1 |
| Vd/F (L/kg) | 47.1 | |
| Cl/F (L/h/kg) | 26.0 |
4. DISCUSSION
Salvinorin A displays a fast onset of its pharmacological action with a relatively short duration of action. After ingestion through smoking, it is transported across the BBB with relative ease and accumulates into the CNS. To better understand the psychotropic activity of Salvinorin A from a pharmacokinetic perspective we investigated its transport and pharmacokinetic characteristics, potential metabolism pathways, plasma to brain distribution, and the rate and extent of its distribution across the BBB.
The P-gp affinity status of Salvinorin A was evaluated using efflux studies and assessing its ATPase activity. P-gp is an ATP-dependent efflux pump that is important in multi-drug resistance and certain drug-drug interactions. The MDCK-MDR1 cell line has been used as model of transepithelial transport due to its formation of highly confluent monolayer and the expression of the multi drug resistance gene (MDR1) encoding high levels of P-gp [31]. In addition, these cell monolayers have been used as an in vitro model to predict brain uptake potential and the brain uptake potential of compounds that interact with P-gp [32]. We observed a secretory transport for Salvinorin A (4.07 ±1.34 × 10-5 cm/s) which was higher than the absorptive transport (not detected), suggesting P-gp affinity status for this hallucinogen.
The ATPase activity of Salvinorin A was evaluated by assessing P-gp dependent decrease in luminescence, an indication of ATP consumption by P-gp. The results obtain suggest that Salvinorin A (5 and 10 μM) is a substrate of P-gp based on comparison to non-treated cells. We observed a trend toward a decrease in P-gp stimulation with increasing Salvinorin A concentrations, however the fold stimulation range observed of 1.74 -2.39 (Table 1) is within the range which indicates P-gp stimulation and ATP consumption. The value of 21.31 pmol/μg/P-gp/min for the untreated group is well below the range (37.08 - 57.08 pmol/μg/P-gp/min ) observed for Salvinorin A concentrations (2.5 - 25 μM) evaluated, suggesting that it is not a P-gp inhibitor. In addition, statistical differences were observed for the Salvinorin A concentrations (5 and 10 μM) vs. the non-treated group, but no statistical differences were observed among the P-gp stimulation for the Salvinorin A concentrations observed.
Even though these results suggest that Salvinorin A is a substrate of P-gp, the implication of these results have to be evaluated within the context of its known in vivo potency and brain uptake. Salvinorin A is very lipophilic (XlogP = 2.3), displays an extremely rapid onset of activity in vivo, and is extremely potent (dose of 200-500 μg) [2,3,10,13,18,21,24,]. Indicated in our pharmacokinetic studies and a recent PET imaging study, Salvinorin A crosses the BBB rapidly after administration [25]. These findings would suggest that even though Salvinorin A appears to be a P-gp substrate, its high lipophilicity, potency and passive permeability offset the impact of P-gp mediated efflux on the CNS levels achieved.
The chemical structure of Salvinorin A suggests that it may be a substrate of CYP450, UGTs or ChEs. As illustrated in Figure 1, Salvinorin A has an ester group which serves as a potential site for hydrolytic metabolism by ChEs or conjugative (glucuronidation) metabolism by UGTs enzymes. To determine potential biotransformation pathways of Salvinorin A, we performed in vitro metabolism studies by screening with various human CYP450 isoforms and human UGT2B7. We observed significant decreases of 14.7(±0.80), 31.1(±1.20), 20.6(±1.00) and 22(±0.80) % (p<0.01) in the concentration of Salvinorin A (5 μM) upon incubation with CYP2D6, CYP1A1, CYP2C18 and CYP2E1, respectively (Figure 2). These enzymes therefore may be involved in the metabolism of Salvinorin A in vivo. We also observed a significant decrease of 51.0(±4.00) % (p<0.01) in Salvinorin A (5 μM) concentration when incubated with UGT2B7 (Figure 3). These results suggest that Salvinorin A is a substrate of UGT2B7. It should be noted that there were lower rates of metabolism for CYP1A1, CPY2C18 CYP2E1 and UGT2B7 at the 50μM vs. 5μM, suggesting saturable metabolism.
Sharing metabolic pathways as well as having the potential to be used concurrently, clearly suggests the potential for drug interactions with certain abused drugs and Salvinorin A [33]. It appears that Salvinorin A may share pathways of metabolism with drugs of abuse and this may result in drug-drug interactions. It should be noted that a number of “drugs of abuse” [heroin, codeine and morphine (UGT2B7), oxycodone (CYP2D6, CYP3A4), MDMA (CYP2D6, CYP3A4, CYP1A2), THC (CYP3A4), ketamine (CYP2B6), cocaine (ChEs)] share one or more of the aforementioned metabolic pathways with Salvinorin A [28, 29, 34-37].
Non-compartmental modeling was used to estimate Salvinorin A pharmacokinetics parameters after single i.p. dose administration. Salvinorin A has been reported to have a rapid onset of action and a short duration [1,2]. Accordingly, the absorption and brain uptake of Salvinorin A was found to be rapid with an apparent tmax occurring at 10-15 min after i.p. administration. These data support the immediate effect of Salvinorin A reported in a recent PET imaging study using female baboons [25]. This study observed extremely rapid brain uptake of Salvinorin A reaching a peak 40 s after dosing. In our study the maximum plasma concentration was found to be 345 ng/mL. Salvinorin A was cleared quickly from the plasma with a Cl/F of 26 L/hr/kg and a t1/2 of 75.4 min (Table 2). A pharmacokinetic analysis of Salvinorin A was performed after a single dose in non-human primates (n=4) and plasma t1/2 was reported to be 56.6 min [24]. Salvinorin A enters the CNS after reaching the systemic circulation via buccal absorption, oral ingestion or smoking. Recent findings have revealed exceptionally rapid brain uptake for Salvinorin A when smoked [25]. The major determinants of permeation of drugs across the BBB have long been thought to be lipophilicity and molecular weight [38]. Salvinorin A has a molecular weight of 432 Da and is highly lipophilic which may explain its rapid uptake in the CNS and it would appear to penetrate the BBB easily. However, our in vivo brain uptake results show that although it does rapidly enter the brain the levels achieved are extremely low as compared to plasma concentrations. Also its short duration of action suggests that its clearance from the systemic circulation is relatively high.
5. CONCLUSION
In conclusion, the BBB transport, metabolism and pharmacokinetics of Salvinorin A contribute significantly to its rapid onset and short duration of action. Salvinorin A is likely a P-gp substrate, however this does not appear to significantly minimize the rate in which Salvinorin A enters the brain. Further, its major metabolic pathway is glucuronidation, and as such it may share pathways with abused agents producing botanical/drug interactions. Studies are underway to better understand the fundamental pharmacologic properties of Salvinorin A and the studies performed herein provide an insight to those BBB transport, pharmacokinetic and metabolic properties of this agent that drive its hallucinogen effect.
REFERENCES
- [1].Giroud C, Felber F, Augsburger M, Horisberger B, Rivier L, Mangin P. Salvia divinorum: An hallucinogenic mint which might become a new recreational drug in Switzerland. Forensic Sci. Int. 2000;112:143–150. doi: 10.1016/s0379-0738(00)00180-8. [DOI] [PubMed] [Google Scholar]
- [2].Valdes LJ., 3rd Salvia divinorum and the unique diterpene hallucinogen, Salvinorin A (divinorin) J. Psychoactive Drugs. 1994;26:277–283. doi: 10.1080/02791072.1994.10472441. [DOI] [PubMed] [Google Scholar]
- [3].Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sci. 2005;78:527–531. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- [4].Vortherms TA, Roth BL. Salvinorin A: From natural product to human theraputics. Molecular Interventions. 2006;6:257–265. doi: 10.1124/mi.6.5.7. [DOI] [PubMed] [Google Scholar]
- [5].Grudmann O, Phipps SM, Zadezensky I, Butterweck V. Salvia divinorum and Salvinorin A: An update on pharmacology and analytical methodology. Planta Med. 2007;73:1039–1046. doi: 10.1055/s-2007-981566. [DOI] [PubMed] [Google Scholar]
- [6].Bagott MJ, Erowid E, Wrowid F, Mendelson JE. Use of salvia divinorum, an unscheduled hallucinogenic plant: a web-based survey of 500 users. College of Problems of Drug Dependence; San Juan Puerto Rico, CPDD: 2004. [Google Scholar]
- [7].Sumnall HR, Wagstaff GF, Cole JC. Self-reported psychopathology in polydrug users. J. Psychopharm. 2004;18:75–82. doi: 10.1177/0269881104040239. [DOI] [PubMed] [Google Scholar]
- [8].Halpern JH. Hallucinogens and dissociative agents naturally growing in the United States. Pharmacol. Ther. 2004;102:131–138. doi: 10.1016/j.pharmthera.2004.03.003. [DOI] [PubMed] [Google Scholar]
- [9].Dennehy CE, Tsourounis C, Miller AE. Evaluation of herbal dietary supplements marketed on the internet for recreational use. Annals Pharmacother. 2005;39:1634–1639. doi: 10.1345/aph.1G185. [DOI] [PubMed] [Google Scholar]
- [10].Babu KM, McCurdy CR, Boyer EW. Opioid receptors and legal highs: Salvia divinorum and Kratom. Clinical Toxicology. 2008;46:146–152. doi: 10.1080/15563650701241795. [DOI] [PubMed] [Google Scholar]
- [11].Siebert DJ. Salvia divinorum and salvinorin A: new pharmacologic findings. J. Ethnopharmacol. 1994;43:53–56. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- [12].Hanes KR. Antidepressant effects of the herb Salvia divinorum: A case report. J. Clin. Pharmacol. 2001;21:634–635. doi: 10.1097/00004714-200112000-00025. [DOI] [PubMed] [Google Scholar]
- [13].Gonzalez D, Riba J, Bouso JC, Gomez-Jarabo G, Barbanoj MJ. Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend. 2006;85:157–162. doi: 10.1016/j.drugalcdep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [14].Sheffler DJ, Roth BL. Salvinorin A: the “magic mint” hallucinogen finds a molecular target in the KOR. Trends Pharmacol. Sci. 2003;24:107–109. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- [15].Yan F, Roth BL. Salvinorin A: a novel and highly selective kappa-opioid receptor agonist. Life Sci. 2004;75:2615–2619. doi: 10.1016/j.lfs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- [16].Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steiberg S, Ernberger P, Rothman RB. Salvinorin A: A potent naturally occurring non-nitrogenous kappa opioid selective agonists. Proc. Natl. Acad. Sci. USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen B, Roth BL. Salvinorin A, an active component of the hallucinogenic sage salvia divinorum is a highly efficacious κ-opioid receptor agonist: Structural and functional considerations. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- [18].Butelman ER, Harris TJ, Kreek MJ. The plant-derived hallucinogen, salvinorin A, produces kappa-opioid agonist-like discriminative effects in rhesus monkeys. Psychopharmacology (Berl) 2004;172:220–224. doi: 10.1007/s00213-003-1638-0. [DOI] [PubMed] [Google Scholar]
- [19].Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: Agonist actions at kappa opioid receptors. Psychopharmacology. 2005;179:551–558. doi: 10.1007/s00213-004-2087-0. [DOI] [PubMed] [Google Scholar]
- [20].Carlezon WA, Beguin C, Jr., Dinieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Depressive-Like effects of the {kappa}-opioid receptor agonist Salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- [21].Valdes LJ, 3rd, Chang HM, Visger DC, Koreeda M. Salvinorin C, a new neoclerodane diterpene from a bioactive fraction of the hallucinogenic Mexican mint Salvia divinorum. Org. Lett. 2001;3:3935–3937. doi: 10.1021/ol016820d. [DOI] [PubMed] [Google Scholar]
- [22].Roth LB, Lopez E, Beischel S, Westkaemper RB, Evans JM. Screening the receptorome to discover the molecular targets for plant-derved psychoactive compounds: A novel approach for CNS drug discovery. Pharm&Therap. 2004;102:99–110. doi: 10.1016/j.pharmthera.2004.03.004. [DOI] [PubMed] [Google Scholar]
- [23].Schmidt MS, Prisinzano TE, Tidgewell K, Harding W, Butelman ER, Kreek MJ, Murry DJ. Determination of Salvinorin A in body fluids by high performance liquid chromatography-atmospheric pressure chemical ionization. J Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2005;818:221–225. doi: 10.1016/j.jchromb.2004.12.041. [DOI] [PubMed] [Google Scholar]
- [24].Schmidt MD, Schmidt MS, Butelman ER, Harding WW, Tidgewell K, Murry DJ, Kreek MJ, Prisinzano TE. Pharmacokinetics of the plant-derived kappa-opioid hallucinogen salvinorin A in nonhuman primates. Synapse. 2005;58:208–210. doi: 10.1002/syn.20191. [DOI] [PubMed] [Google Scholar]
- [25].Hooker JM, Xu Y, Schiffer Wynne, Shea C, Carter P, Fowler JS. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. NeuroImage. 2008;41:1044–1050. doi: 10.1016/j.neuroimage.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cox DS, Scott KR, Gao H, Raje S, Eddington ND. Influence of MDR proteins at the blood-brain barrier on the transport and brain distribution of enaminone anticonvulsants. J Pharm Sci. 2001;90:1540–1552. doi: 10.1002/jps.1104. [DOI] [PubMed] [Google Scholar]
- [27].Truan G, Cullin C, Reisdorf P, Urban P, Pompon D. Enhanced in vivo monooxygenase activities in mammalian P450s in engineered yeast cells producing high levels of NADPH-P450 reductase and human cytochrome b5. Gene. 1993;125:49–55. doi: 10.1016/0378-1119(93)90744-n. [DOI] [PubMed] [Google Scholar]
- [28].Coffman BL, Rios GR, King CD, Trphly TR. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos. 1997;25:1–4. [PubMed] [Google Scholar]
- [29].Coffman BL. The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268) Drug Metab Dispos. 1998;26:73–77. [PubMed] [Google Scholar]
- [30].King CD, Green MD, Rios GR, Coffman BL, Owens IS, Bishop WP, Tephly TR. The glucuronidation of exogenous and endogenous compounds by stably expressed rat and human UDP-glucuronosyltransferase. Arch. Biochem. Biophys. 1996;332:92–100. doi: 10.1006/abbi.1996.0320. [DOI] [PubMed] [Google Scholar]
- [31].Cho MJ, Thompson DP, Cramer CT, Vidmar TJ, Scieszka JF. The Madin Darby canine kidney (MDCK) epithelial cell monolayer as model cellular transport barrier. Pharm Res. 1989;6:71–77. doi: 10.1023/a:1015807904558. [DOI] [PubMed] [Google Scholar]
- [32].Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int. J. Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- [33].Griffiths P, Vingoe L. The use of amphetamines, ecstacy and LSD in the Eurpoean Community: a review of data on consumption patters and current epidemiological literature. The National Addiction Centre; London: 1997. [Google Scholar]
- [34].Guengerich FP, Miller GP, Hanna IH, Sato H, Martin MV. Oxidation of methoxyphenethylamines by cytochrome P450 2D6. Analysis of rate-limiting steps. J. Biol. Chem. 2002;277:33711–33719. doi: 10.1074/jbc.M205146200. [DOI] [PubMed] [Google Scholar]
- [35].Kreth K, Kovar K, Schwab M, Zanger UM. Identification of the human cytochromes P450 involved in the oxidative metabolism of “Ecstasy”-related designer drugs. Biochem. Pharmacol. 2000;59:1563–1571. doi: 10.1016/s0006-2952(00)00284-7. [DOI] [PubMed] [Google Scholar]
- [36].Pindel EV, Kedishvili NY, Abrahams TL, Brzezinski MR, Zhang J, Dean RA, Bosron WF. Purification and cloning of broad substrate sepcifity human liver carboxylesterase that catalyzes the hydrolysis of cocaine and heroin. The J. Biolog. Chem. 1997;272:14769–14775. doi: 10.1074/jbc.272.23.14769. [DOI] [PubMed] [Google Scholar]
- [37].Tyndale RF, Sunahara R, Inaba T, Kalow W, Gonzalez FJ, Niznik HB. Neuronal cytochrome P450IID1 (debrisoquine/sparteine-type): potent inhibition of activity by (-)-cocaine and nucleotide sequence identity to human hepatic P450 gene CYP2D6. Mol. Pharmacol. 1991;40:63–68. [PubMed] [Google Scholar]
- [38].Pardridge WM. Peptide Drug Delivery to the Brain. Vol. 3. Raven Press; New York: 1991. Overview of blood-brain barrier transport biology and experimental methodologies; pp. 52–98. [Google Scholar]