

Abstract
Several studies have shown that catecholamines can inhibit the fibrillation of α-synuclein, a small presynaptic protein whose aggregation is believed to be a critical step in the etiology of Parkinson's disease and several other neurodegenerative disorders. However, the mechanism of this inhibition is uncertain. We show here that substoichiometric concentrations of DOPAC (3, 4-dihydroxyphenylacetic acid), a normal product of the metabolism of dopamine, can inhibit the fibrillation of α-synuclein (α-Syn), due to non-covalent binding of DOPAC to α-Syn monomer. Intriguingly, the presence of α-synuclein accelerates the spontaneous oxidation of DOPAC, and the oxidized form of DOPAC (the quinone) is responsible for the fibrillation inhibition. In addition, the presence of DOPAC leads to the oxidation of the methionine residues of α-Syn, probably due to the H2O2 production as a by-product of DOPAC oxidation. The lack of fibrillation results from the formation of stable oligomers, which are very similar to those observed transiently at early of the α-Syn fibrillation. A possible explanation for this phenomenon is that DOPAC stabilizes the normally transient oligomers and prevents them from subsequent fibril formation. The analysis of the α-synuclein Y39W variant suggests that DOPAC binds non-covalently to the same N-terminal region of α-Syn as lipid vesicles, probably in the vicinity of residue 39. In contrast to the compounds with 1,2-dihydroxyphenyl groups (DOPAC, catechol), their 1,4-dihydroxyphenyl isomers (hydroquinone, homogentisic acid) are able to modify α-Syn covalently, probably due to the less steric hindrance in the Michael addition.
Keywords: α-synuclein, DOPAC, DOPAC-stabilized oligomer, amyloid fibril, oxidative modification, methionine oxidation, dopamine
The abnormal movements associated with Parkinson's disease (PD) result from the death of the specific dopaminergic neurons in the substantia nigra. The oxidative instability of dopamine and its several metabolites create potentially greater predisposition for oxidative stress in the dopaminergic neurons, which is assumed to be associated with the particular vulnerability of these neurons. Several investigations have shown that catecholamines can inhibit the fibrillation of α-synuclein, a protein whose aggregation is believed to be a critical step in the etiology of PD and several related neurodegenerative diseases.
The diagnostic hallmark of PD is the presence of intracellular fibrillar inclusions, Lewy bodies and Lewy neuritis, in the surviving dopaminergic neurons 1-3. α-Synuclein (α-Syn), a 140-residue presynaptic protein of unknown function, is the primary component of these fibrillar inclusions 3. In vitro, far-UV CD, Fourier transform IR, fluorescence and NMR spectroscopies, as well as SAXS and several hydrodynamic technique show that α-Syn is intrinsically disordered 4-6. Intrinsic disorder refers to the lack of a fixed structure in proteins and many biologically active proteins were shown to remain unstructured, or incompletely structured, under physiological conditions 7-34. Intrinsic disorder has been reported both at a region as well as a whole protein level. There are several crucial differences between amino acid sequences of intrinsically disordered proteins and regions and structured globular proteins and domains 9;24;35. Intrinsically disordered proteins are highly abundant in nature and the overall amount of disorder in proteins increases from bacteria to archaea to eukaryota 36-39. These proteins carry out numerous biological functions, many of which obviously rely on high flexibility and lack of stable structure. These functions are diverse and complement those of ordered proteins and protein regions 27-29. Many intrinsically disordered proteins are associated with human diseases such as cancer 40, cardiovascular disease 41, amyloidoses 42, neurodegenerative diseases, diabetes and others 34. Based on these intriguing links among intrinsic disorder, cell signaling and human diseases, suggesting that protein conformational diseases may result not only from protein misfolding, but also from misidentification and missignaling, the “disorder in disorders” or D2 concept was recently introduced 34.
α-Syn assembles into filaments and fibrils after incubation under physiological conditions in vitro. Morphologies of these filaments and fibrils are similar to those extracted from the diseased brain 43-46. A variety of factors, such as low pH, high temperature, naturally occurring polyamines, and environmental PD risk factors (including heavy metals, pesticides and herbicides), accelerate fibrillation of α-Syn in vitro 5;47-51. Substantial evidence indicates that α-Syn aggregation is a critical step in the etiology of Parkinson's disease 51;52, as well as in a number of other neurodegenerative diseases, collectively known as synucleinopathies 51. However, the question on whether the mature fibrils, protofilaments, protofibrils, specific oligomers or some folding intermediates are the neurotoxic species responsible for the cell death in these diseases is still a subject of great controversy 53-58. Oligomers of α-Syn with globular, annular or chain-like conformations have been observed 59;60. Some of these oligomers were suggested to form pores that can permeabilize membranes 60-64. Since it is primarily dopaminergic neurons that appear to be affected in PD, there has been considerable speculation regarding the role of dopamine and its metabolites in the disease.
Although the normal function of α-Syn is still unknown this protein is believed to be involved in regulation of the dopamine neurotransmission via effects on vesicular dopamine storage and trafficking 65-69. Interestingly, dopamine and related compounds that have vicinal dihydroxy groups were shown to be the effective inhibitors of the α-Syn fibrillation 70-74. It was proposed that α-Syn is able to form α-Syn-dopamine-quinone (DAQ) adducts with oxidized dopamine, and that these adducts block the α-Syn fibrillation and stabilize the potentially most toxic α-Syn oligomers or protofibrils 70. However, the mechanism of fibrillation inhibition by α-Syn-DAQ adduct remains uncertain because the yield of adducts is very low as observed by the lack of characteristic signals from the dopamine adducts detectable by mass spectrometry 70.
A better understanding of the mechanism of inhibition of α-Syn fibrillation by catechols is of particular interest for a number of reasons, including an attempt to understand whether the resulting oligomers are toxic 75. It has been shown that the neurotoxicity of α-Syn is dopamine-dependent 76 and that α-Syn also facilitates the toxicity of oxidized catechol metabolites 77. SDS-PAGE analysis of α-Syn incubated with dopamine and other catechol compounds revealed a ladder of SDS-stable oligomers that suggested covalently cross-linked species. However, these effects were at concentrations of catechol compounds (0.18∼2 mM) far greater than those occurring in vivo 78. Recently it has been shown that intracellular catechols such as DA and DOPAC have the ability to modulate α-Syn aggregation in cultured human cells 79. In particular, an increase in cytosolic catechol levels was associated with a decrease in α-Syn-containing inclusions, possibly through the formation of catechol-induced oligomeric intermediates.
Here, we take advantage of the good water solubility of DOPAC (3, 4-dihydroxyphenylacetic acid), a normal product of the dopamine metabolism, to show that: (a) lower concentrations of DOPAC than those used in previous studies are sufficient to inhibit fibrillation of α-Syn; (b) DOPAC binds to α-Syn non-covalently at low concentration but the covalent modifications of α-Syn occur at higher concentrations; (c) DOPAC can oxidize methionine groups of α-Syn; (d) in the absence of DOPAC, the formation of transient oligomers preceded the α-Syn fibrillation. Although in the presence of low DOPAC concentrations, the α-Syn oligomers were also formed, these oligomers did not assemble into fibrils.
Results
Unless otherwise noted, the conditions for α-synuclein incubation were 20 mM phosphate, pH 7.4, 100 mM NaCl, 37°C, with 70 μM α-Syn and agitation. Under these conditions, α-Syn formed fibrils with a lag phase of approx. 20 h, and fibrillation was complete by 35-40 h; the fibrillation kinetics were very sensitive to small changes in the rate of agitation.
Oxidation of DOPAC
Solutions of DOPAC at physiological pH and 37°C were spontaneously oxidized by dissolved oxygen to form DOPAC-quinone and other oxidized compounds. We will refer to this mixture as DOPAC-Q. This oxidation was accompanied by a color change and induction of fluorescence (excitation λmax 360 nm; emission λmax around 460 nm, Figure 1, 70). Sodium azide is frequently added to α-synuclein solutions to be incubated for long time periods in order to prevent the microbial contamination of the sample. Monitoring the fluorescence changes from oxidation of DOPAC during incubation showed that NaN3 increased the intensity of fluorescence from DOPAC-Q, indicating that NaN3 might accelerate the DOPAC oxidation. This could be explained as follows: since azide is a free radical scavenger, it could react with free radical products of DOPAC oxidation and thus could enhance the oxidation by mass action effects. Interestingly, the presence of 70 μM α-Syn greatly enhanced the fluorescence signal of DOPAC compared to 5 mM NaN3 (Figure 1). Although the fluorescence could come from formation of di-tyrosine cross-linked α-synuclein or an α-synuclein-DOPAC complex, incubation of DOPAC with a Tyr-minus mutant (the four tyrosines substituted with phenylalanines) of α-Syn showed no significant decrease in fluorescence compared to the wild-type α-Syn. This clearly indicated that the Tyr residues of α-Syn are not involved in the fluorescent species. Furthermore, incubation of α-Syn alone did not show this fluorescence signal. Therefore, these observations implied that α-Syn itself accelerated the oxidation of DOPAC or alternately that binding of oxidized DOPAC to α-Syn increased the fluorescence intensity of DOPAC-Q.
Figure 1.

Oxidation of DOPAC leads to formation of fluorescent species. A: Fluorescence (excited at360 nm) of freshly prepared DOPAC (short dash); after 4 days of incubation (long dash); and after 1 year of incubation (solid line). DOPAC concentration was 100 μM, excitation was 360 nm. B: Kinetics of DOPAC fluorescence (excitation at 360 nm, emission at 456 nm) on incubation with 70 μM α-synuclein (●); with 5 mM NaN3 (▼); and DOPAC alone (○).
DOPAC inhibits fibrillation of α-synuclein at micromolar concentrations
The effects of different concentrations of DOPAC on α-Syn fibrillation were analyzed at 37°C, pH 7.4 using the ThT fluorescence assay. DOPAC showed significant inhibitory effects on α-Syn fibrillation and the extent of inhibition was DOPAC concentration-dependent (Figure 2A). DOPAC, even at concentration of only one tenth of that of α-Syn, increased the elongation time and decreased the ThT fluorescence signal, which was roughly proportional to the amount of fibrils formed. The data indicated a stoichiometric inhibitory effect. At concentrations exceeding 1:1 molar ratio over α-Syn, DOPAC completely inhibited α-Syn fibrillation evidenced by ThT signal, EM image (which showed mostly oligomers) and nearly all α-Syn were soluble and could not be pelleted by centrifugation (Figure 2B) (α-Syn fibrils are insoluble and can be pelleted). Small amount of short fibrils and protofibrils in a form of small, thin, curvy fibrils, were also observed by EM image (see Figure 2Bb'). However, the amount of such fibrils and protofibrils was very small and they were not abundantly present in analyzed EM fields. These fibrils might originate from a subset of α-Syn molecules aggregating prior their interaction with DOPAC. Furthermore, no such fibrils were detected by AFM image (see below, Figure 3B), suggesting either these fibrils are not stable and dissociate due to the dilution during the AFM samples preparation or the chance to observe these aggregated species by AFM is small. The inhibitory effects of DOPAC on α-Syn fibrillation were greatly decreased in the presence of sodium thiosulfate (Na2S2O3), a reducing reagent. Freshly prepared DOPAC (in the presence of thiosulfate) showed little inhibition whereas pre-oxidized DOPAC still showed significant inhibition of α-Syn fibrillation even when Na2S2O3 was added to the solutions at the beginning of incubation (Figure 2C). Since Na2S2O3 does not reduce oxidized DOPAC, these observations indicate that it is the oxidized form of DOPAC that is responsible for the fibrillation inhibition. The possibility of α-Syn methionine oxidation by non-oxidized DOPAC (at low concentrations) was excluded by the failure to detect a change in the mass of α-Syn by MS.
Figure 2.

The presence of DOPAC inhibits alpha-synuclein fibrillation. A: ThT fluorescence assays of fibrillation of 70 μM α-Syn in the presence of 0 (○), 7 (▲), 14 (▽), 28 (●), 35 (□), 42 (▼), 49 (△), and 100 (◼) μM DOPAC in 20 mM pH 7.4 phosphate buffer and 100 mM NaCl at 37°C. B: Incubation of α-synuclein with DOPAC leads to formation of soluble oligomers. EM images and SDS gel of incubated α-Syn (70μM) without (a) or with DOPAC (100 μM) (b and b') in 20 mM phosphate buffer (pH 7.4) and 100 mM NaCl at 37°C, the scale bar is 200 nm. Panel b' represents a rear field where some protofibrils are seeing. The side panel shows the SDS gel of incubated and centrifuged solutions, S and M represent supernatant and non-centrifuged mixture respectively. C: Thiosulfate prevents DOPAC oxidation. ThT assay of fibrillation of 70 μM α-Syn and 5 mM Na2S2O3 in the presence of 0 (□), 100 μM fresh (◼) or pre-oxidized (◇) DOPAC in 20 mM phosphate buffer (pH 7.4), 100 mM NaCl at 37 °C.
Figure 3.


Formation of oligomers during the incubation of α-synuclein monitored by AFM. A: Time course of α-Syn (70 μM) incubated without DOPAC in 20 mM pH 7.4 phosphate buffer and 100 mM NaCl at 37°C, scale bar is 300 nm. The lower panels show the height distribution. B: Time course of incubated α-Syn solution (70 μM) with DOPAC (100 μM) in 20 mM pH 7.4 phosphate buffer and 100 mM NaCl at 37°C, scale bar is 300 nm. The lower panels show the heights distribution. Note: sometimes, due to the variance in tip's condition, particles on AFM images were stretched and looked “wider” than their real sizes. However, the heights of these particles are reliable.
DOPAC stabilizes transient oligomers of α-Syn
The fact that DOPAC (100 μM) completely inhibited fibrillation of α-Syn (70 μM) was supported by both the low ThT signals and EM images (Figure 2B). To better characterize the time-course of changes in the heights of these DOPAC-stabilized oligomers, AFM was used to study α-Syn incubated alone or with DOPAC. For α-Syn alone, AFM showed that oligomers (∼5 nm high) built up during the first ∼ 19 hours followed by fibrils (∼7 nm high) at ∼48 hours and the final mature fibrils (∼ 10 nm high) (Figure 3A). In the presence of DOPAC, increasingly larger oligomers formed (∼5 nm high at 20 hrs; ∼7 nm high at 44 hr and ∼10 nm at 72 hrs), and no fibrils were observed (Figure 3B). The heights of these DOPAC-induced α-Syn oligomers were comparable to heights of the oligomers or fibrils formed during the incubation of α-Syn alone at similar incubation time points. It appears that these oligomers were stabilized by DOPAC in such a way that they cannot enter the fibrillation pathway.
AFM data showed that the oligomers were built up during the first 20 hours of incubation with agitation (i.e., during the lag time) for both α-Syn incubated alone and in the presence of DOPAC. These oligomers were soluble and unlike fibrils were not pelletable by the centrifugation. Importantly, these oligomers dissociated easily during the gel-filtration experiments, as the SEC HPLC profiles contained predominantly the α-Syn monomeric peak, although small amount of oligomeric peak was also detected when incubating α-Syn with DOPAC (Figure 4). This suggests that oligomers were not stable and dissociated to monomers on SEC HPLC due to the sample dilution. The decrease of the monomeric peak intensity during incubation of α-Syn alone indicated the formation of insoluble fibrils, while the increase and the shift to right of the monomeric α-Syn peak after incubation with DOPAC suggested that DOPAC was bound to α-Syn and that these DOPAC-bound α-Syn species were slightly more compact than the non-incubated protein.
Figure 4.

Time course monitoring of incubating α-Syn (70 μM) alone (A) or with DOPAC (100 μM) (B) at pH 7.4, 37°C under fibrillation condition by size exclusion HPLC. Absorbance spectra were monitored at 280 nm. In these experiments, 15 μL of incubated solutions were injected on a TSK-GEL G2000SWXL size-exclusion column at a flow rate of 0.5 mL/min.
Taken together these data indicated that oligomers formed either during the incubation of α-Syn alone or in the presence of DOPAC were structurally similar. In fact, they shared similarity in morphology (heights) and in the stability on SEC HPLC. However, in contrast to the oligomers accumulated during the incubation of α-Syn alone, the oligomers “stabilized” by DOPAC did not transform into the fibrils.
At low concentrations, DOPAC binds to α-Syn non-covalently
Without agitation, α-Syn does not form fibrils being incubated for several weeks. To test whether DOPAC binds to monomeric α-Syn, SEC FPLC was used to analyze the mixture of α-Syn (70μM) with DOPAC (100 μM) during incubation without agitation (Figure 5). The area of the monomeric α-Syn peak increased and area of the DOPAC peak decreased with time. This suggested that DOPAC was bound to α-Syn slowly in a course of incubation. Finally, after the prolonged incubation, the DOPAC peak disappeared and there was a large increase in the α-Syn monomer peak. This indicated that most of the DOPAC was bound tightly to α-Syn, although a small amount of polymerized DOPAC was also observed at later stages. Reactive groups of α-Syn, such as Lys, Tyr and the N-terminal alpha-amino groups have been suggested to react with DOPAC-Q 70. However, no mass changes of α-Syn were observed from ESI-MS even when a 3 or 4-fold excess of DOPAC was used (see below, Figure 8). The possibility that a Schiff base, which is sensitive to acid hydrolysis, formed between α-Syn and DOPAC-Q is unlikely because the treatment of the incubated α-Syn and DOPAC solution with even 100-fold excess of NaBH4, which reduces Schiff bases, did not yield any new MS peaks corresponding to α-Syn-DOPAC-Q complex. Also, there was no obvious increase in the α-Syn peak area on reverse phase HPLC (data not shown), while SEC showed a big increase in α-Syn monomer peak area (Figure 5). These observations suggested that DOPAC at low concentration was bound to α-Syn non-covalently. This was also supported by the failure to detect crosslinked α-Syn on SDS-PAGE or by MS. Furthermore, no α-Syn-DOPAC-Q bands were observed after NBT red-ox staining of the SDS-PAGE gels at low concentrations of DOPAC (see below Figure 7). The DOPAC-bound monomeric α-Syn was separated by SEC FPLC, concentrated and incubated under normal fibrillation conditions. No fibrils were detected by ThT fluorescence assay and EM. This meant that the non-covalent binding of DOPAC to α-Syn monomer was sufficient for the complete inhibition of the α-Syn fibrillation.
Figure 5.

Monitoring the interaction of α-synuclein with DOPAC by SEC FPLC. UV absorbance (280 nm) traces of incubated solutions (no agitation) of α-Syn (70μM) with DOPAC (100μM) at pH 7.4, 37°C by SEC FPLC. In these experiments, 300 μL of incubated solutions were injected on a SUPERDEX™ 200 size-exclusion column at a flow rate of 2.0 mL/min.
Figure 8.

Formation of covalent adducts during incubation of α-synuclein with hydroquinone or homogentisic acid. Mass spectra of: a) α-Syn alone; b) α-Syn incubated with 3 molar equivalent of DOPAC or dopamine for 1 day; c) α-Syn incubated with 3 molar equivalent of HGA for 1 day and d:) α-Syn incubated with 3 molar equivalent of HQ for 1 day.
Figure 7.
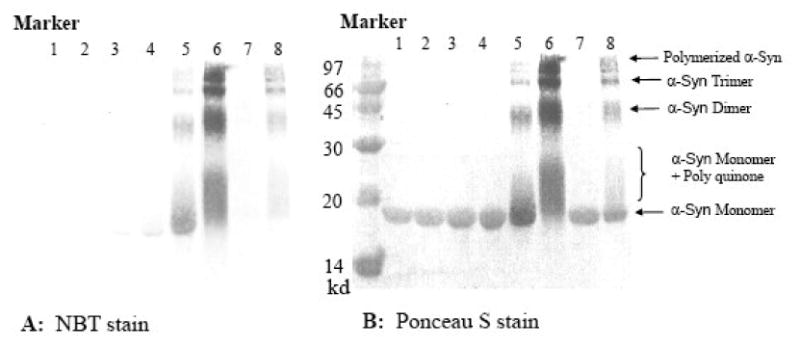
Covalent modification of α-synuclein by the quinones of DOPAC and dopamine. SDS PAGE of incubated α-Syn (70μM; for lane 6, 140 μM was used) with different concentrations of DOPAC (lane 1-6) or dopamine (lane 7-8): Lane 1 (0 μM); 2 (10 μM); 3 (50 μM); 4 (100 μM); 5 (1000 μM); 6 (5000 μM); 7 (100 μM); 8 (1000 μM). Panel A shows the results with the NBT stain, which only detects quinone-modified proteins. Panel B shows all proteins present on the same gel using Ponceau S stain after NBT staining.
What is the binding site for DOPAC on α-Syn?
It is interesting to know where DOPAC (at low concentrations) binds non-covalently to α-Syn since the physiological concentration of dopamine-like chemicals is quite low (≤ nano-molar range). Taking the advantage that the WT α-Syn has no Trp residue, three mutants of α-syn with Tyr to Trp substitutions at positions 39, 125, 136 were created and incubated with equivalent molar ratios of DOPAC. The idea here was that if DOPAC-Q (excitation λmax = 360 nm) binds to α-Syn at a position close to Trp (excitation λmax = 290 nm, emission λmax ∼ 350 nm), it might quench the fluorescence of Trp by fluorescence resonance energy transfer (FRET). Y39W α-Syn showed a much faster decrease in Trp fluorescence than the other two mutants (Figure 6A). Unfortunately, no fluorescence of DOPAC from FRET was detected when solutions were excited at 290 nm. This could be due to the low intensity of DOPAC-Q fluorescence (only 2∼5 % of Trp fluorescence intensity). It is also possible that DOPAC-Q quenches Trp fluorescence through direct interactions rather than through FRET. Monitoring the time-dependence of DOPAC-Q fluorescence showed that WT α-Syn and its Trp mutants had similar effects on DOPAC oxidation (data not shown). This means that fast Trp fluorescence decrease of Y39W in the presence of DOPAC was not due to the fast oxidation of DOPAC.
Figure 6.

Changes in Trp fluorescence of Trp-containing mutants of α-synuclein (Y39W, Y125W, and Y136W) on incubation with DOPAC. Note, the wild type α-synuclein has no tryptophan residues. A: the kinetics of Trp fluorescence emission change for Y39W (○), Y125W (△) and Y136W (□) α-Syn (70 μM) in the presence of one molar equivalent of DOPAC (pH 7.4, 37°C). B: Trp Fluorescence of α-Syn (70 μM) mutants before and after incubation with one molar equivalent of DOPAC: Y39W before (black solid) and after incubation (red solid); Y125W before (black long dash) and after (red long dash); Y136W before (black short dash) and after (red short dash). C: Tryptophan fluorescence of α-synuclein mutants: Y39W in fibril (blue solid) and native monomer (black solid); Y125W in fibril (dash dot) and in native form (dash); Y136W in fibril (dotted) and in native form (dash dot dot). Fluorescence intensities were normalized.
The Trp fluorescence of the Y39W, Y125W, and Y136W mutants showed that the environment of Trp in Y39W was more hydrophobic (emission λmax = 350 nm) than the Trp environments in Y125W and Y136W mutants (emission λmax = 357 nm). We propose that DOPAC-Q binds to the hydrophobic region of α-Syn. There was no obvious Trp fluorescence peak shift after incubating the Y125W or Y136W mutants with DOPAC, and there was a small (2∼3 nm) red shift for Y39W. This implied that the residues at positions 39, 125, 136 were still, or even more, solvent exposed in the transient α-Syn oligomers (Figure 6B). In contrast, the Trp emission λmax was blue-shifted ∼7 nm (from 350 to 342 nm) after fibrillation of the Y39W mutant while no peak shifts were observed for Y125W and Y136W α-Syn fibrils (Figure 6C). These observations suggest that the residue 39 is buried inside the fibrils but is still solvent exposed in the DOPAC-stabilized oligomers.
Covalent modifications of α-Syn by DOPAC occur at high DOPAC concentrations
At higher concentrations of DOPAC (1 mM), dimers, trimers and higher order oligomers of α-Syn were observed on SDS-PAGE. NBT red-ox stain (which only detects quinone modified protein) showed that all monomeric and oligomeric α-Syn were associated with DOPAC quinones and most α-Syn was still in its monomeric form (Figure 7). The smear above the monomeric α-Syn band may indicate that the polymerized DOPAC-Q bind to α-Syn monomer. This smear was most noticeable when higher concentrations of DOPAC (5 mM) were used. Interestingly, for dopamine, SDS gel electrophoresis analysis after NBT staining showed only dopamine-Q-associated oligomeric α-Syn and the extended α-Syn monomer bands, but no dopamine-Q associated α-Syn monomer band. Therefore, it appears that dopamine-Q preferentially modifies oligomeric α-Syn and only polymerized dopamine quinones can modify monomeric α-Syn. This could be due to faster polymerization of dopamine than DOPAC. The cross-links between α-Syn and DOPAC or dopamine quinones might occur after oligomerization of α-Syn, which could result in the side-chains involved in cross-linking being brought together.
Regio-isomers of DOPAC and homogentisic acid or catechol and hydroquinone react with α-Syn differently
It is interesting that there were no covalent modifications of α-Syn induced by low concentrations of DOPAC or dopamine. It has been reported that hydroquinone (HQ) but not catechol modified α-Syn by Michael addition 75. This observation suggested that compounds with the 1,2-dihydroxyphenyl groups did not covalently modify α-Syn, whereas their para-isomers; i.e. 1,4-dihydroxyphenyl derivatives, can modify α-Syn covalently. We chose catechol and DOPAC (Figure 8) as representatives of the former, and HQ and homogentisic acid (HGA) as representatives of the latter. All four compounds showed substantial inhibition of α-Syn fibrillation by ThT assay (data not shown).
These four compounds were incubated with α-Syn at 3:1 molar ratio. Based on ESI MS analysis, catechol and DOPAC did not form covalent α-Syn adducts, although a small amount of methionine oxidized α-Syn was formed after the prolonged incubation in the presence of NaN3. However, covalent modifications of α-Syn, through Michael additions of the quinones, were observed with HQ and HGA by ESI-MS already after one day of incubation, and longer times of incubation gave more covalent α-Syn adducts (Figure 8). Methionine oxidation of α-Syn was also seen by MS after the incubation for two days. This implies that, if covalent modification of α-Syn occurs by catechol-related quinones, it occurs before methionine oxidation of α-Syn. The Michael addition of HGA (mass 168) or HQ (mass 110) quinones to α-Syn, resulted in mass changes of 162 or 104 daltons respectively on α-Syn, which correspond to the adducts with mass (chemical + α-Syn − 6) daltons. The mass loss of 6 daltons comes from the three times of oxidation of the phenols to the quinones after Michael addition of two amino groups from α-Syn to one quinone molecule 75. Interestingly, no mass loss of 4 daltons, corresponding to one amino group adding to one quinone, was detected by ESI-MS. This indicated that the addition of two amino groups of α-Syn to the quinones was a rather cooperative reaction.
For HGA and HQ, amino groups from α-Syn can be added to the quinones from opposite sides of the phenyl ring. But for DOPAC and catechol, amino groups have to attack quinones from the same side on two adjacent carbon atoms, which suggests an unfavorable steric effect. This could be the reason why no Michael addition occurred for DOPAC and catechol. For HGA and HQ, which did modified α-Syn through Michael addition, at a 1:1 molar ratio, only small amounts of α-Syn were modified (only mono-adduct) after many days of incubation and no modification occurred for the first 2 days (data not shown). However, at a 3:1 ratio, substantial amounts of mono-adduct, and some di-adduct were observed on ESI-MS after only one day of incubation. Even after incubation for many days, tri-adducts were never observed. This indicated that some α-Syn sites were involved in the non-covalent binding of HGA and HQ. These α-Syn sites were likely incompatible with the Michael addition taking place.
DOPAC can oxidize the methionine groups of α-Syn
Dopaminergic neurons are likely to be subjected to higher levels of ROS than most cells due to the oxidative instability of dopamine. Previous studies have shown that the four methionine groups of α-Syn are vulnerable to oxidation by H2O2 leading to formation of soluble oligomers and inhibition of fibrillation 80-83. When 70 μM α-Syn was incubated with 100 μM or higher concentration of DOPAC for 3 days at 37°C, ESI MS showed that α-Syn was not oxidized, although DOPAC underwent oxidation. However, under the same conditions but in the presence of 5 mM NaN3 the methionine groups of α-Syn were oxidized by DOPAC (data not shown). The excess of methionine oxidation depended on DOPAC concentration: the more DOPAC added, the greater the level of α-Syn oxidation observed. In contrast, the addition of higher concentrations of NaN3 (e. g. 100 mM), caused no more methionine oxidation of α-Syn than oxidation induced by 5 mM NaN3. Co-incubation of α-Syn (70 μM) with 5 mM NaN3 did not lead to the methionine oxidation. The possible catalytic effects of trace amounts of iron from the buffer were excluded by the addition of the chelator DETAPAC (200 μM) during incubation. Furthermore, the methionine oxidation of α-Syn by DOPAC was inhibited by the reducing reagent Na2S2O3. This implied that DOPAC was responsible for the methionine oxidation, and NaN3 acted as an activator, probably by interacting with certain ROS from the oxidation of DOPAC and leading to oxidation pathways that generate more peroxide.
Since methionine oxidation is also known to inhibit α-Syn fibrillation 80, it was not clear what was the main cause of the α-Syn fibrillation inhibition when DOPAC was present, DOPAC binding or methionine oxidation. ESI-MS was used to monitor methionine oxidation of α-Syn (70 μM) by DOPAC (100 μM) under the fibrillation condition. The noticeable methionine oxidation of α-Syn by DOPAC was only detected after at least 30 hours of incubation (a time point where some fibrils were already formed for intact α-Syn). More specifically, ESI-MS revealed that 1 or 2 Met residues per α-Syn were oxidized under these conditions whereas there are four oxidizable methionine residues in α-Syn. Figure 5 shows that more than 60% DOPAC is bound to α-Syn during the same time period. Furthermore, partial methionine oxidation of α-Syn (i.e., oxidation of only two Met residues) did not completely inhibited α-Syn fibrillation 83. This suggested the binding of DOPAC to α-Syn preceded methionine oxidation of α-S and that this binding dominated in the inhibition of α-Syn fibrillation.
Discussion
Our data showed that even substoichiometric concentrations of DOPAC can inhibit the fibrillation of α-Syn, and that the non-covalent binding of DOPAC to α-Syn monomer was sufficient to inhibit the fibrillation process. Determining the molecular mechanism of DOPAC-induced inhibition of α-synuclein fibrillation was complicated by the oxidative instability of the compound. The results of the experiments using sodium thiosulfate which prevented the oxidation of DOPAC but did not reduce the oxidized forms indicated that the oxidized form of DOPAC (the quinone) was mostly responsible for the inhibition. The presence of α-synuclein accelerated the spontaneous oxidation of DOPAC, possibly due to the presence of trace metal ions associated with α-synuclein 84, or due to interactions with potentially catalytic side chains of the protein in the DOPAC-α-synuclein complex. We also found that the presence of DOPAC led to the oxidation of the methionine residues in α-synuclein. This probably reflects the production of H2O2 as a by-product of DOPAC oxidation, since peroxide is well known to oxidize the methionines of α-synuclein to the sulfoxide.
At concentrations of DOPAC equivalent to, or less than that of α-synuclein, DOPAC bound to the protein in a non-covalent manner. Various observations demonstrated that the non-covalent interaction of DOPAC with α-Syn induced the formation of α-Syn oligomers, which did not form fibrils.
Non-covalently bound DOPAC appeared to bind to the same N-terminal region of α-synuclein as lipid vesicles, probably at a location close to residue 39, based on the analysis of the Y39W variant of α-synuclein. It is interesting to note that AFM imaging indicated that the oligomers induced by non-covalently bound DOPAC were very similar to those observed transiently during the incubation of α-Syn alone and their heights were similar to those of fibrils. One possibility was that the oxidized DOPAC stabilized the normally transient oligomers and prevented them from entering the fibril formation pathway, perhaps by altering or blocking the oligomer interface involved in fibrillation. Regardless of the exact mechanism, it was clear that the oxidized DOPAC stabilized species, oligomers or protofibrils, prior to fibril formation.
In contrast, at higher DOPAC concentrations (≥3:1 stoichiometry) α-Syn was covalently cross-linked by DOPAC quinones, and Michael addition products were formed. Interestingly, formation of these species involved dimerization, indicating that two Michael additions occurred. This suggested that appropriately located Lys side-chains of two adjacent α-synuclein molecules in an oligomer were optimally oriented for such a reaction. The possible products of the α-Syn interaction with DOPAC are summarized in Figure 9.
Figure 9.

Potential reactions between DOPAC and α-Syn: i) at low concentration, DOPAC quinone monomer binds α-Syn non-covalently (Mα-S + DOPAC); ii) at high concentration, monomeric DOPAC quinone can react with α-Syn monomer to form covalent products (Mα-S-DOPAC); iii) at high concentration, oxidized DOPAC can polymerize first. Polymerized DOPAC can react covalently either with one α-Syn molecule forming poly-DOPAC modified α-Syn monomer (Mα-S-Poly-DOPAC) or with several α-Syn molecules to get polymerized α-Syn (Poly-α-S-DOPAC).
It is notable that there were no covalent modifications of α-Syn by low concentrations of DOPAC or dopamine quinones, but it has been reported that the structurally related compound, hydroquinone, modified α-Syn by Michael addition 75. We confirmed that HQ did indeed form covalent adducts via Michael addition when present at low concentrations, and showed that HGA, which also has a 1,4-dihydroxyphenyl group, also formed such adducts, whereas catechol, with its 1,2-dihydroxyphenyl structure, did not. These observations indicated that the outcome was determined by whether the polyphenol had 1,2- or 1,4-dihydroxy (or quinone) groups. Here, compounds with 1,2-dihydroxyphenyl groups did not covalently modify α-Syn, whereas their 1,4-dihydroxyphenyl isomers made covalent modifications of α-Syn. We attributed this difference to a steric hindrance effect for Michael addition to the quinones from the 1,2-dihydroxyphenyl compounds. Since both the 1,2- and 1,4-dihydroxyphenyl compounds examined were strong inhibitors of α-synuclein fibrillation it appeared that the covalent modification was not directly related to the prevention of fibril formation.
With HQ and HGA, the MS data indicated that the Michael addition occurred between the two nucleophilic groups of α-Syn (presumably lysine side-chains) and only one HGA or HQ molecule. This observation, along with the stoichiometry of the reaction, suggested that the dihydroxyphenyl compounds bound at a specific site at the interface of two α-synuclein molecules in an oligomer.
DOPAC, HGA, catechol and HQ were able to oxidize the methionine groups of α-Syn to the methionine sulfoxide in the presence of sodium azide. This suggested that more hydrogen peroxide was produced during oxidation of these chemicals in the presence of azide, presumably due to its radical scavenging activity leading to a shift in the distribution of oxidation products.
In summary, at physiologically relevant concentrations, DOPAC binds non-covalently to α-Syn and results in the formation of specific α-Syn oligomers that do not form fibrils. Spontaneous oxidation of DOPAC leads to the formation of the quinone, which at high concentrations, can result in covalent modification of α-synuclein via a Michael addition.
Materials and Methods
Materials
Recombinant human α-Syn and its mutants Y39W, Y125W, Y136W were expressed in Escherichia coli and purified as described previously 85. Lyophilized protein was freshly prepared before use by dissolving protein in 2 mM phosphate buffer at pH 7.4 and centrifuged at 95000 rpm with a Beckmann Airfuge. 3, 4-dihydroxyphenylacetic acid (DOPAC), dopamine, catechol, hydroquinone, homogentisic acid (HGA) and diethylenetriaminepentaacetic acid (DETAPAC) were obtained from Aldrich Chem. Co. All other chemicals were analytical grade and purchased from Fisher chemicals or EM science.
Methionine oxidation of α-Syn by DOPAC
α-Syn (70 μM) was incubated with 100 μM or higher concentration of DOPAC at 37°C in 20 mM phosphate buffer pH 7.4 with or without 5 mM or 100 mM NaN3 for 1-3 days. To prevent methionine oxidation of α-Syn, 5 mM Na2S2O3 was added to the above solutions. 200μM DETAPAC was used to remove trace amounts of metal ion in buffer solutions. The methionine oxidation of α-Syn was detected by ESI-MS.
Covalent modification of α-Syn by Catechol, DOPAC, Hydroquinone and Homogentisic acid
70 μM α-Syn was incubated with these compounds at the indicated molar ratios at 37°C, pH 7.4 for 1-3 days. Modifications were detected by ESI-MS and SDS-PAGE.
Oxidation of DOPAC by air
100 μM DOPAC was incubated in 20 mM phosphate buffer (pH 7.4) with or without 5 mM NaN3 at 37°C for 1-3 days. In some experiments, 100 mM NaN3 was used, as indicated in the text. “Complete” and “incomplete” pre-oxidized DOPAC were obtained by incubating 10 mM DOPAC for periods of several months or by incubating 200 μM DOPAC for 2 weeks with 5 mM NaN3 at 37°C. The oxidation of DOPAC was confirmed by color change from colorless to brown or dark red depending on the extent of oxidation and increase in fluorescence (excitation: 360 nm; emission: 400-520 nm) 70. Oxidized DOPAC was a mixture of the quinone and many other compounds and the total mixture was named DOPAC-Q herein.
Fibril formation and ThT assay
Fibril formation was monitored by ThT (thioflavine T) fluorescence using a Fluoroskan Ascent CF plate reader (Labsystems, Inc.). Protein solutions with 70 μM α-Syn and 20 μM ThT were incubated in 100 mM NaCl and 20 mM phosphate buffer (pH 7.4) in a 96-well plate, each well containing a 1/8 inch diameter Teflon spherical bead. The plate was incubated at 37°C with shaking at 120 rpm, 20 mm diameter or 600 rpm, 2 mm diameter. The fluorescence was measured at 30 min intervals with excitation at 450 nm and emission at 485 nm. Data from six wells were averaged and plotted as a function of time. Lag times were calculated based on the equation described previously 86.
Size-exclusion HPLC and FPLC measurements
Aliquots of solutions were removed during incubation and centrifuged for 20 min at 14000 rpm to remove any insoluble materials before running SEC chromatography. For HPLC, 15 μL of incubated solutions were injected on a TSK-GEL G2000SWXL size-exclusion column (7.8 mm ID × 30 cm) at a flow rate of 0.5 mL/min and for FPLC, 300 μL of incubated solutions were injected on a SUPERDEX™ 200 size-exclusion column at a flow rate of 2.0 mL/min in 10 mM phosphate buffer (pH 7.0) and 100 mM Na2SO4. HPLC was performed using a Waters 2695 separations module with a Waters 996 Photodiode Array (PDA) detector, and data were collected and analyzed by Millennium software. Absorbance of the eluant was monitored over the wavelength range from 220 to 450 nm with bandwidth of 1.2 nm. For FPLC (AKTA instrument), spectra were monitored at 280 nm.
SDS-PAGE gel electrophoresis measurements
Samples for sodium dodecyl sulfate PAGE (SDS-PAGE) gel electrophoresis were boiled in a water bath for 5 to 10 min in 4% SDS prior to electrophoretic separation. A PhastSystem with 8-25% gradient gels (Amersham Biosciences) was used and experiments were run with SDS buffer strips containing 0.20 M Tricine, 0.20 M Tris and 0.5% SDS, pH 7.5 for 30 min at 250 V. Gels were stained with Coomassie blue. For NBT redox cycling stain, SDS-PAGE was run on a 15% polyacrylamide gel, proteins was transferred to nitrocellulose membrane and stained with 0.24 mM NBT (NitroBlue Tetrazolium chloride) in 2 M potassium glycinate at pH 10 in the dark for 1 hour. The same membrane was stained with Ponceau S for all proteins after NBT staining 87.
Mass spectrometry
Samples for MS analysis were desalted with C8 RP column and eluted with 4:1 acetonitrile:H2O (pH adjusted to 2.0 by TFA) solution. The desalted solution was directly loaded onto the mass spectrometer. Mass spectra were obtained using a MicroMass Quattro II electrospray mass spectrometer operating in positive ion mode. Injection was carried out using a syringe pump (Harvard Apparatus, Holliston, MA) at a flow-rate of 20 μL/min. The source temperature was set to 80 °C and the capillary voltage was 3.25 kV. Protein molecular weight was determined from mass-to-charge ratios by MassLynx software.
Electron Microscopy Measurements
Samples for electron microscopy (EM) were deposited on carbon-coated pioloform 300 mesh copper grids and negatively stained with 1% (w/v) aqueous uranyl acetate. EM images were obtained on a JEOL JEM-100B transmission electron microscope operated at 80 kV.
Atomic force microscopy (AFM) measurements
AFM images were collected with a PicoScan LE system (Molecular Imaging, Phoenix, AZ) equipped with Acoustic AC mode (Tapping mode) for ex-situ experiment. Triangular cantilevers with 160 kHz resonance frequency and 2 N/m spring constant, a V-shaped cantilever NSC16/AIBS (MikroMasch), were used in Tapping mode imaging. The imaging was carried out at a scan rate of 1 line/s with 512 data points per line, at a drive current of 10 ± 4 Å. Aliquots of 10 μL of sample containing 100 mM NaCl were placed on a freshly cleaved mica substrate. After incubation at least 60 min, the substrate was rinsed with water several times to remove salt and loosely bound protein and dried with high-purity Nitrogen.
Heights ranging from 0.1 to 100 nm were estimated by section analysis, and lateral sizes were calibrated with standard calibration grid. At least four regions of the mica surface were examined to verify that similar structures existed through the sample. No filter treatment was used to modify the images. SPIP 4.0 (Image Metrology) was used to analyze the height, area and volume distribution of the α-Syn aggregations.
Acknowledgments
This research was supported in part by grants R01 NS39985 (A.L.F.), R01 LM007688-01A1 (V.N.U.) and GM071714-01A2 (V.N.U.) from the National Institutes of Health, and from the Program of the Russian Academy of Sciences «Molecular and Cellular Biology» (V.N.U.). We gratefully acknowledge the support of the IUPUI Signature Centers Initiative.
Footnotes
Footnote: Prof. Anthony L. Fink has passed away on March 2, 2008
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dunnett SB, Bjorklund A. Prospects for new restorative and neuroprotective treatments in Parkinson's disease. Nature. 1999;399:A32–9. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 2.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 5.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 6.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 7.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 8.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 9.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–484. [PubMed] [Google Scholar]
- 10.Dunker AK, Obradovic Z. The protein trinity--linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 11.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 12.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of Protein Folding. Wiley-VCH, Verlag GmbH & Co. KGaA; Weinheim, Germany: 2005. pp. 271–353. [Google Scholar]
- 13.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 14.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–84. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 15.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9 1:S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 17.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–54. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 18.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 19.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 20.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 22.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–1871. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in the Protein Data Bank. J Biomol Struct Dyn. 2007;24:325–42. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- 24.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–56. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35:D786–93. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uversky VN, Radivojac P, Iakoucheva LM, Obradovic Z, Dunker AK. Prediction of intrinsic disorder and its use in functional proteomics. Methods Mol Biol. 2007;408:69–92. doi: 10.1007/978-1-59745-547-3_5. [DOI] [PubMed] [Google Scholar]
- 27.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–32. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–98. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, Vacic V, Obradovic Z, Uversky VN. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9 2:S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008 doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Dunker AK, Uversky VN. Signal transduction via unstructured protein conduits. Nat Chem Biol. 2008;4:229–30. doi: 10.1038/nchembio0408-229. [DOI] [PubMed] [Google Scholar]
- 33.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, Asturias FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol. 2008;4:728–37. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 35.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 36.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, Dunker AK. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput. 1998:437–48. [PubMed] [Google Scholar]
- 37.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- 38.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 39.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 41.Cheng Y, LeGall T, Oldfield CJ, Dunker AK, Uversky VN. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry. 2006;45:10448–60. doi: 10.1021/bi060981d. [DOI] [PubMed] [Google Scholar]
- 42.Uversky VN. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res. 2008;5:260–87. doi: 10.2174/156720508784533312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 44.Crowther RA, Daniel SE, Goedert M. Characterisation of isolated alpha-synuclein filaments from substantia nigra of Parkinson's disease brain. Neurosci Lett. 2000;292:128–130. doi: 10.1016/s0304-3940(00)01440-3. [DOI] [PubMed] [Google Scholar]
- 45.El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson's disease. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 46.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM. NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 2004;23:2039–46. doi: 10.1038/sj.emboj.7600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 49.Fink AL. The aggregation and fibrillation of alpha-synuclein. Acc Chem Res. 2006;39:628–34. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 50.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 51.Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 52.Trojanowski JQ, Lee VM. Parkinson's disease and related alpha-synucleinopathies are brain amyloidoses. Ann N Y Acad Sci. 2003;991:107–110. doi: 10.1111/j.1749-6632.2003.tb07468.x. [DOI] [PubMed] [Google Scholar]
- 53.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 54.Giasson BI, Lee VM. Are ubiquitination pathways central to Parkinson's disease? Cell. 2003;114:1–8. doi: 10.1016/s0092-8674(03)00509-9. [DOI] [PubMed] [Google Scholar]
- 55.Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 56.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 57.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 58.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–7. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 59.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 61.Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- 62.Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- 63.Ding TT, Lee SJ, Rochet JC, Lansbury PT., Jr Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–17. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 64.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 65.Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–42. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 66.Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2002;11:2395–407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 67.Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884–94. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- 68.Tao-Cheng JH. Activity-related redistribution of presynaptic proteins at the active zone. Neuroscience. 2006;141:1217–24. doi: 10.1016/j.neuroscience.2006.04.061. [DOI] [PubMed] [Google Scholar]
- 69.Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–21. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 71.Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 2005;280:21212–9. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL. Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease. Faseb J. 2004;18:962–964. doi: 10.1096/fj.03-0770fje. [DOI] [PubMed] [Google Scholar]
- 73.Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J Biol Chem. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 74.Li J, Zhu M, Rajamani S, Uversky VN, Fink AL. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol. 2004;11:1513–1521. doi: 10.1016/j.chembiol.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 75.Li HT, Lin DH, Luo XY, Zhang F, Ji LN, Du HN, Song GQ, Hu J, Zhou JW, Hu HY. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J. 2005;272:3661–72. doi: 10.1111/j.1742-4658.2005.04792.x. [DOI] [PubMed] [Google Scholar]
- 76.Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- 77.Hasegawa T, Matsuzaki-Kobayashi M, Takeda A, Sugeno N, Kikuchi A, Furukawa K, Perry G, Smith MA, Itoyama Y. Alpha-synuclein facilitates the toxicity of oxidized catechol metabolites: implications for selective neurodegeneration in Parkinson's disease. FEBS Lett. 2006;580:2147–52. doi: 10.1016/j.febslet.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 78.Sulzer D. alpha-synuclein and cytosolic dopamine: stabilizing a bad situation. Nat Med. 2001;7:1280–2. doi: 10.1038/nm1201-1280. [DOI] [PubMed] [Google Scholar]
- 79.Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 2006;26:10068–78. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002;517:239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 81.Yamin G, Glaser CB, Uversky VN, Fink AL. Certain metals trigger fibrillation of methionine-oxidized alpha-synuclein. J Biol Chem. 2003;278:27630–27635. doi: 10.1074/jbc.M303302200. [DOI] [PubMed] [Google Scholar]
- 82.Glaser CB, Yamin G, Uversky VN, Fink AL. Methionine oxidation, alpha-synuclein and Parkinson's disease. Biochim Biophys Acta. 2005;1703:157–169. doi: 10.1016/j.bbapap.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 83.Hokenson MJ, Uversky VN, Goers J, Yamin G, Munishkina LA, Fink AL. Role of individual methionines in the fibrillation of methionine-oxidized alpha-synuclein. Biochemistry. 2004;43:4621–4633. doi: 10.1021/bi049979h. [DOI] [PubMed] [Google Scholar]
- 84.Zhu M, Qin ZJ, Hu D, Munishkina LA, Fink AL. Alpha-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry. 2006;45:8135–42. doi: 10.1021/bi052584t. [DOI] [PubMed] [Google Scholar]
- 85.Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J Mol Biol. 2005;353:357–72. doi: 10.1016/j.jmb.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 86.Nielsen L, Khurana R, Coats A, Frokjaer S, Brange J, Vyas S, Uversky VN, Fink AL. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–46. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 87.Paz MA, Fluckiger R, Boak A, Kagan HM, Gallop PM. Specific detection of quinoproteins by redox-cycling staining. J Biol Chem. 1991;266:689–92. [PubMed] [Google Scholar]