

Abstract
Cytolytic T lymphocytes (CTL) undergo massive expansion upon appropriate antigenic stimulation. Homeostasis is maintained by a subsequent “contraction” of these cells. Activation-induced cell death (AICD) and programmed cell death, prevent the untoward side effects arising from excessive numbers and prolonged persistence of activated CTL that occur upon uncontrolled and/or continued expansion. However, effector cell persistence has been identified as a hallmark of successful T cell-mediated adoptive immunotherapy. Thus, prevention of AICD may be critical to achieve more successful clinical results. We have previously shown that treatment with c-jun N-terminal kinase (JNK)-inhibitor, SP600125, protects human melanoma epitope Mart-127-35 reactive CTL from apoptotic death upon their re-encounter with cognate antigen. However, inhibition of JNK also interferes with the functional capability of the CTL to secrete interferon (IFN)-γ. Here, we show that reactive oxygen species (ROS) inhibitors such as the superoxide dismutase mimetic, Mn (III) tetrakis (5, 10, 15, 20-benzoic acid) porphyrin (MnTBAP), efficiently protected Mart-127-35 reactive primary CTL from AICD without impairing their functional capability. MnTBAP prevented the increase in intracellular ROS, mitochondrial membrane collapse, and DNA fragmentation observed in control treated cells upon cognate antigen encounter. Furthermore, the mechanism of AICD prevention in primary CTL included blockade of JNK activation. Finally, tumor reactive in vitro expanded tumor infiltrating lymphocytes, which are used clinically in cancer immunotherapy, also benefit from MnTBAP mediated antioxidant treatment. Thus, modulation of the redox pathway might improve CTL persistence and lead to better clinical results for T cell-based immunotherapies.
Keywords: AICD, CTL, Oxidative stress, ROS, JNK
Introduction
Maintenance of T cell homeostasis is a complex process controlled by balancing the expansion and contraction of T cells. Programmed cell death (PCD) and activation-induced cell death (AICD) prevent untoward side effects of an uncontrolled and persistent T-cell effector response and maintain homeostasis (1, 2). However, AICD may be detrimental to immunotherapy, especially if activated CTL undergo apoptosis directly upon re-encountering their cognate antigen. Thus, prolonging T-cell survival by inhibiting AICD may enhance the therapeutic benefit of various immunotherapeutic strategies (3, 4).
Several experimental models of tumor immunotherapy demonstrate that T-cell persistence is important for therapeutic success. Long-term persistence of adoptively transferred TCR transgenic CTL was associated with improved tumor regression (5). Over-expression of the anti-apoptotic protein Bcl-2 in effectors directly inhibits apoptosis and thereby prolongs T-cell survival. Importantly, Bcl-2 over-expression in TCR transgenic CTL significantly enhanced adoptive immunotherapy of established murine melanomas (6). Cancer regression in patients receiving autologous tumor-infiltrating lymphocytes (TIL) was also significantly correlated with their persistence in peripheral blood (7). These studies suggest that AICD may limit T-cell survival in vivo and that inadequate T-cell persistence limits current adoptive immunotherapy protocols.
Death receptor (DR) ligation and activation of the caspase cascade has been considered the principal trigger for AICD. However, recent findings have established that some death signals originate internally and that not all types of cell death are caspase mediated (8). DNA damage, reactive oxygen species (ROS), nitric oxide, and excess mitochondrial Ca2+ may all promote AICD (9). Our previous studies have shown that cognate-antigen exposure induces AICD in primary human CTL (10). Furthermore, we found that the c-jun N-terminal kinase (JNK) inhibitor SP600125 rescued CD8+ T cells reactive to either a melanoma-associated epitope (Mart-127-35) or a influenza matrix protein epitope (MP58-66), from caspase independent AICD (11, 12). However, SP600125 concomitantly interfered with the ability of activated CTL to secrete interferon (IFN)-γ. A role for ROS in mitochondrial damage and caspase-independent death is documented in diverse models (13, 14). Interestingly, antioxidant MnTBAP was shown to block death of mouse CD4+ T cells, after exposure to strong polyclonal stimuli with the superantigen staphylococcal enterotoxin A (SEA) (15). Protection from cell death was attributed to blockade of ROS production, which is normally initiated upon T-cell activation and sensitizes T cells to apoptosis by decreasing Bcl-2 expression (16).
Here, we evaluated the effect of ROS inhibition on AICD following restimulation with the cognate epitope of Mart-127-35 antigen- reactive primary human CTL. Notably, MnTBAP could protect a large fraction of the activated CTL from undergoing AICD. Importantly, MnTBAP did not interfere with T-cell effector functions, including their ability to secrete cytokines. Further, clinically relevant effector types such as in vitro expanded TIL were also protected from AICD after MnTBAP pretreatment. Thus, strategies to modulate the redox pathway may improve T-cell survival (17, 18) without impairing effector cell functionality, thereby conferring therapeutic benefit to T-cell-based immunotherapies for various diseases (19, 20).
Materials and Methods
Cells
Peripheral blood mononuclear cells (PBMC) from HLA-A2-positive healthy donors were obtained with informed consent. TIL1235 (reactive to the human Mart-127-35 antigen), HLA-A2+ human melanoma MEL624 and its HLA-A2- variant MEL624-28, were obtained from surgical specimens of patients undergoing experimental immunotherapies at the Surgery Branch, NCI (21). T2 cells are transporter-associated protein-deficient and its empty surface HLA-A2 molecules were used for direct presentation of epitopes to the antigen-reactive CTL.
Culture medium and reagents
Mart-127-35 peptide (AAGIGILTV) and MP58-66 peptide (GILGFVFTL) were purchsed from MP Systems (San Diego, CA). Culture medium was Iscove’s Modified Dulbecco’s Medium (GIBCO BRL, Grand Island, NY) supplemented with 10 % fetal bovine serum (Gemini Bioproducts Inc., Calabasas, CA). Media for TIL1235 was supplemented with 6000 IU/ml interleukin (IL)-2 (Chiron, Emeryville, CA). Ficoll-Paque was obtained from Amersham Bioscience (Piscataway, NJ). Recombinant cytokines were purchased from R & D Systems (Minneapolis, MN). Major Histocompatibility Complex (MHC) class I tetramers and pentamers were purchased from Beckman Coulter (Fullerton, CA) and ProImmune (Oxford, UK), respectively. These reagents bind directly to TCR of a particular specificity, determined by the MHC allele and peptide combination and thus can be used to detect and separate antigen-specific CD8+ T cell populations. Antibody to detect single-stranded (ss) DNA was obtained from Alexis Biochemicals (San Diego, CA) while all other fluorochrome-labeled monoclonal antibodies and Annexin V were purchased from BD Biosciences (San Jose, CA). 7AAD was purchased from Calbiochem (La Jolla, CA) and Hydroethidium (HE) was from Sigma Chemicals (St. Louis, MO). Inhibitors as SP600125 for JNK, PD098059 for ERK and antioxidants MnTBAP, L-NAC were purchased from EMD Biosciences (La Jolla, CA).
Generation of dendritic cells from peripheral blood monocytes
The procedure for generating dendritic cells (DC) from peripheral blood monocytes has been published (10). Briefly, circulating monocytes were enriched by adherence of Ficoll-Hypaque density gradient-isolated PBMC. The adherent cells were cultured in complete media (CM) containing 1000 IU/ml GM-CSF and 500 IU/ml IL-4 for 3-5 days to obtain a population of immature DCs (iDCs). Maturation of iDCs was performed by first adding IFN-γ (1000 U/ml) for 2 hours followed by 100 ng/ml lipopolysaccharide (LPS) overnight.
Activation of CD8+ T cells by DC-based presentation of epitopes in vitro
The procedure for peptide-loaded DC-based in vitro activation and expansion of epitope reactive CD8+ T cells has been described (10, 11). Briefly, Ficoll-Hypaque gradient-separated PBMC were purified for CD8+ T cells (≥ 90% purity) by Dynal magnetic bead isolation kit (Invitrogen, Grand Island, NY) and co-cultured with autologous irradiated (3000 rad) DCs pulsed with relevant peptides (100 μg/ml) and 5 μg/ml of β2 microglobulin at a CD8+ T cell-to-DC ratio of 100:1. Activated CTL were maintained in media containing IL-15 (10 ng/ml).
AICD induction and evaluation
CTL were preincubated with inhibitors at predetermined optimal concentration for 30 min at 37 °C and then exposed to cognate or non cognate antigens in the form of either MHC class I pentamer reagents, peptide-loaded T2 cells or tumor cell lines for induction of antigen specific apoptosis (11, 12). Apoptosis was determined by flow cytometry with three or four color staining (e.g. CD8, HLA-A2 pentamer, 7AAD and Annexin V) and acquired on a FACSCalibur (Becton Dickinson, Mountain View, CA) or an Accuri C6 (Accuri Cytometers, Ann Arbor, MI) flow cytometer and data analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Measurement of mitochondrial membrane potential (Δψ)
Δψ was estimated by staining with 20 nM DiOC6 (Molecular Probes, Carlsbad, CA), a cationic lipophilic dye, for 15 min at 37 °C in the dark before analysis by flow cytometry. Fluorescence of DiOC6 is oxidation-independent and correlates with Δψ (22).
Cytokine release assay
Cytokine release by the effector cells was determined by co-culturing 1×104 to 1×105 effector cells in a 1:1 ratio with melanoma tumor cells or peptide-pulsed T2 cells as described previously (10). After 16 to 24 hours, culture supernatants were harvested and cytokine concentrations measured by sandwich ELISA per the manufacturer’s protocol (R & D Systems) using a spectrophotometer (BioTek, Winooski, VT).
CD107a degranulation assay
Cell surface expression of CD107a was used as a surrogate marker for degranulation (23). 1×105 CTL were cocultured with melanoma tumor cells in a 1:1 ratio over night before stained with an anti-CD107a mAb and analysed by FACS.
Western blot
Protein extractions were performed in RIPA buffer and samples were separated on 15% SDS polyacrylamide gels and electrophoretically transferred to PVDF membranes (Millipore, Burlington, MA). The primary antibodies (anti-JNK, anti-pJNK and anti-β Actin) and secondary antibodies were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA).
Results
Activation and expansion of Mart-127-35 reactive CTL
Mart-127-35 epitope-reactive CTL were expanded from PBMC of healthy donors using peptide-pulsed autologous mature dendritic cells (Figure 1A). Within 12 days Mart-127-35 T cells expanded two hundred fold (from 0.06% to 12%), as measured by staining with HLA-A2 tetramers loaded with Mart-127-35, and secreted significant amounts of IFN-γ when stimulated with their cognate antigen (Figure 1B). Functional Mart-127-35 reactive T cells prepared in this manner were used throughout our studies to understand mechanisms of AICD in primary human antigen-reactive CTL.
Figure 1. Expansion of Mart-127-35 reactive CTL and induction of AICD.

(A) Mart-127-35 CTL precursors (left) and CTL expanded with autologous DC pulsed with peptides were tracked by FACS using staining for CD8 and tetramer reagents. (B) IFN-γ secreted into co-culture supernatant collected after overnight incubation of Mart-127-35 CTL stimulated with cognate or control peptide-pulsed T2 cells. (C) Expanded CTL were restimulated with cognate or control epitope-pulsed T2 cells and were stained after 4 hours with tetramer, CD8, and Annexin V. Histogram depict fluorescence intensity for Annexin V in tetramer-gated CD8+ CTL when unstimulated (grey filled), stimulated with T2 cells pulsed with control peptide (thin black), and stimulated with T2 cells pulsed with cognate peptide (thick black). All data represents one of at least seven separate experiments with similar results.
Sensitivity of expanded Mart-127-35 reactive CTL to AICD
To evaluate the effect on viability of TCR ligation on the Mart-127-35- reactive CTL upon exposure to cognate antigen, cells were stained with the relevant tetramer and Annexin V. Figure 1C shows increased Annexin V binding, indicating decreased viability of tetramer-positive cells upon exposure of the TCR to its cognate antigen (thick black overlays), as compared to the control peptide (thin black lines) or unstimulated cells (gray filled overlay). Thus, the cell death observed in this model is antigen-specific and consistent with the idea that repeated TCR-mediated activation of CTL results in AICD (11, 12).
TCR dependent superoxide generation
Fluorescence intensity of the intracellular stain HE, that tracks superoxide levels (15, 22), showed a 35% to 50% increase upon TCR engagement with the cognate peptide as compared to a control peptide in Mart-127-35- CTL (Figure 2A). Preincubation of Mart-127-35- CTL with the antioxidant MnTBAP prevented this increase. These data suggested that superoxide levels increase upon TCR stimulation with the cognate epitope, leading us to further examine the role of ROS in AICD of CTL.
Figure 2. Increased endogenous ROS levels after TCR stimulation and rescue of Mart-127-35 reactive CTL from AICD by ROS inhibition.

Mart-127-35 epitope-reactive CTL were stimulated with control or cognate peptide-pulsed T2 cells (1 μg/ml) or first preincubated with 400 μM of MnTBAP or 25 μM SP600125 for 30 min and then stimulated with their cognate epitope. (A). Staining with hydro ethidium (HE) was performed 4 hours later. Histogram represents fluorescence intensity of HE on the antigen-reactive CTL. Numbers represent mean fluorescence intensity (MFI). (B). Induction of AICD in CTL was evaluated by staining for tetramer, CD8, and Annexin V 4 hours later and analysis using FACS. Histogram overlays depict Annexin V expression on tetramer-gated CTL stimulated with T2 cells pulsed with control peptide (thin black) and relevant peptide (thick black), respectively. MAGE-3 (control epitope); Mart-127-35 (cognate epitope). (C) Another representation of how Mart-127-35 reactive CTL were rescued from death (and thus remained Annexin V negative) to various degrees in response to pretreatment with SP600125 and MnTBAP before cognate antigen exposure. * p<0.05, as compared to untreated CTL stimulated with T2 cells pulsed with Mart-1 peptide. (D) Matched IFN-γ secretion from the setup in (C) measured by ELISA. All data represent one of three separate experiments with similar results.
Inhibition of ROS and AICD
The effect of pretreating the CTL with a ROS scavenger, MnTBAP, was next evaluated. As shown in Figure 2B, Mart-127-35- reactive CTL preincubated with MnTBAP exhibited less increase in Annexin V staining than untreated CTL upon cognate antigen stimulation, as compared to control peptide. Protection from AICD by MnTBAP indicates that there is a role of ROS in AICD. Addition of 2-mercaptoethanol alone to the co-culture media had no rescuing effect on T cells undergoing AICD (data not shown). The data obtained with MnTBAP were similar to our previous observation with the JNK inhibitor SP600125 (Figure 2B, C) (11, 12). One drawback to using SP600125 is that JNK inhibition also inhibits cytokine secretion (11, 12). Therefore, we examined the impact of MnTBAP on IFN-γ production. As shown in Figure 3D, in contrast to SP600125 antioxidant MnTBAP had no impact on the ability of Mart-127-35- reactive T cells to secrete IFN-γ. Moreover, CTL pretreated with MnTBAP, and thus rescued from AICD, accumulated to higher numbers when left in culture for five days (data not shown). As a further demonstration that CTL could be rescued from AICD by superoxide quenching, several other commercially available cell-permeable ROS inhibitors (L-NAC, MnTPyP, Tiron, and D1417) were tested (24). All these compounds rescued CTL from AICD (data not shown). These results provide proof-of-principle that antioxidants can be used to rescue CTL from TCR-mediated AICD. Furthermore, it indicates that T-cell effector function and AICD signaling pathways are separate. Thus, rescuing T cells from AICD by antioxidants may increase the overall persistence of CTL while preserving effector function.
Figure 3. Effect of TCR stimulation and ROS inhibition on the membrane potential (Δψ) and DNA degradation.

Mart-127-35 reactive CTL preincubated with MnTBAP (400 μM) and SP600125 (25 μM) were stimulated with cognate Mart-127-35 and control Mage-3 peptide-pulsed T2 cells for induction of AICD. (A) Histogram represents DiOC6 staining on tetramer-gated CTL. Numbers in the upper left corner represent MFI for DiOC6 staining and in the lower left corner represent the percentage of cells that have low membrane potential. (B) ss-DNA levels in tetramer-positive cells were determined with ss-DNA-specific antibody. Data presented show one representative experiments of two.
Mechanism of ROS inhibiton in rescue from AICD
During apoptosis, mitochondrial membrane permeability changes are accompanied by release of various proteins from the inter-membrane space that initiate and maintain a caspase cascade, chromatin condensation, and DNA fragmentation. Release of these proteins results in permeabilization of the inner mitochondrial membrane and dissipation of Δψ (25), which can be assessed through FACS analysis with a fluorescent dye like DiOC6 (26). The mitochondrial membrane integrity of CTL undergoing AICD and the effect of MnTBAP pretreatment on this event was thus evaluated. Figure 3A shows that stimulation of Mart-127-35- CTL with cognate epitope resulted in about 50% decrease in DiOC6 staining compared to controls, indicating an antigen driven loss of Δψ. Pretreatment with the ROS inhibitor MnTBAP or JNK inhibitor SP600125 resulted in less decrease in Δψ (28% and 22% respectively). Another means of measuring apoptosis is DNA fragmentation caused by frequent single-strand cuts (27). An antibody that recognizes deoxycytidine of single-stranded (ss)-DNA of at least 25-30 bases in length, in the absence of any reactivity to double-stranded DNA, and specifically detects apoptotic, but not necrotic, cells was used (28). As shown in Figure 3B, restimulation of Mart-127-35- reactive CTL with their cognate peptide to induce AICD resulted in around 70% increase of cells with nicked ss-DNA as compared to controls. In contrast, CTL pretreated with SP600125 or MnTBAP before the exposure to cognate antigen had only marginally higher fractions of cells with ss-DNA than CTL restimulated with the control peptide. Thus, pretreatment of epitope-specific CTL with superoxide inhibitor MnTBAP rescued them from AICD by reducing damage to mitochondria and DNA.
Sensitivity of TIL to AICD and rescue by MnTBAP treatment
A high proportion of melanoma patients were recently shown to exhibit objective clinical responses when given non-myeloablative chemotherapy prior to the transfer of autologous TIL (29). If ROS inhibition could also rescue TIL from AICD it would have direct clinical application, by improving ex vivo expansion of TIL for immunotherapy. Importantly, ROS inhibition can reduce telomere shortening and restrict replicative senescence (30), and thereby potentially also increase in vivo persistence of TIL. Like PBMC-derived CTL, TIL1235 also underwent AICD upon antigen stimulation as measured by Annexin V staining (Figure 4A). Importantly, MnTBAP treatment efficiently rescued TIL from AICD without impairing effector functions. Cytokine secretion (Figure 4B), cytolytic activity as measured using degranulation assay (Figure 4C) (23) and the proliferative potential of T cells as measured using Ki-67 staining (31) (data not shown) were all intact in MnTBAP pretreated cells. As expected, the JNK inhibitor SP600125 impaired the ability of TIL to secrete cytokines (Figure 4B) without affecting their degranulation (12) (Figure 4C). Thus, TIL are also prone to ROS-mediated AICD and benefit from antioxidant treatment.
Figure 4. Pretreatment with MnTBAP protects TIL from AICD without impairing function.

Mart-1- reactive TIL1235 preincubated with media alone, MnTBAP (250-500 μM) and SP600125 (25 μM) were co-cultured with target cells. (A) After 4 hours co-culture with T2 cells pulsed with cognate or control peptide (1 μg/ml) cells were stained with Annexin V, 7AAD and CD8 and analyzed by FACS. Histogram represent the log fluorescence of Annexin V on CD8+ gated TIL. Numbers in the upper right corner represent MFI and in the lower right corner represent the percentage of cells that stained positive for Annexin V. (B) IFN-γ release by and (C) CD107a expression on TIL1235 upon co-culture with HLA-A2-positive (MEL624) or matched HLA-A2-negative (MEL624-28) melanoma cells. All data shown are from one representative experiment of at least two.
Effect of superoxide inhibition on AICD of ‘non-self’ influenza matrix epitope reactive CTL
Since tumor associated human melanoma “self” epitope Mart-127-35 reactive T cells may behave differently than “non-self” epitope reactive T cells, we evaluated the impact of superoxide inhibition on influenza matrix epitope MP58-66 reactive CTL. These “non-self” reactive CTL were also expanded from PBMC of healthy donors using peptide-pulsed autologous DC (Figure 5Ai), and secreted significant amounts of IFN-γ when stimulated with their cognate antigen relative to controls (Figure 5Aii). As with Mart-127-35 reactive CTL, restimulation of MP58-66- reactive CTL resulted in an increase in Annexin V staining of tetramer-positive cells upon exposure to the cognate antigen (thick black overlays), as compared to the control peptide (thin black overlays) or un-stimulated cells (grey filled overlay) (Figure 5B). In addition, fluorescence intensity of HE, tracking intracellular superoxide levels (22), showed almost 30% increase in MP58-66- reactive CTL after TCR engagement with relevant peptide compared to the control peptide (Figure 5C). An analysis of ss-DNA revealed that pretreatment with MnTBAP reduced the extent of DNA fragmentation upon cognate antigen encounter (Figure 5D), as observed with Mart-127-35 reactive CTL. Thus, these observations suggest that in vitro expanded tumor epitope Mart-127-35 and viral epitope MP58-66 reactive CTL undergo AICD by similar mechanisms and could be rescued by inhibiting endogenous ROS.
Figure 5. Effect of MnTBAP on AICD of influenza epitope MP58-66 reactive T cells.

(Ai). Tetramer staining to track CTL precursors (left) that were expanded using MP58-66 peptide-pulsed DC and stained on day 12 (right). (Aii) IFN-γ secretion after overnight incubation of the MP58-66 CTL stimulated with cognate or control peptide gp100209 pulsed T2 cells. Data shown in (A) represent one of more than five separate experiments with similar results. (B) Histogram overlays depict fluorescence intensity for Annexin V in MP58-66 tetramer-gated CTL when unstimulated (grey filled), stimulated using T2 cells pulsed with control Mage-3 peptide (thin black), and stimulated using T2 cells pulsed with cognate MP58-66 peptide (thick black). (C) MP58-66 -reactive CTL untreated or pretreated with MnTBAP were stimulated with control Mage-3 or cognate MP58-66 peptide-pulsed (1 μg/ml) T2 cells. Staining was performed 4 hours later. Histogram represents fluorescence intensity of HE on the antigen-reactive CTL. Numbers in upper left corner represent MFI. Data in (B) and (C) represent one of three separate experiments with similar results. (D). Untreated, MnTBAP and SP600125 pretreated MP58-66 epitope-reactive CTL were exposed to the cognate (MP58-66) and control (Mart-127-35) peptide-pulsed T2 cells for induction of AICD. Histogram represents ss-DNA levels in tetramer-positive cells determined after staining with ss-DNA specific antibody. Data from one representative experiment of two is shown.
Effect of superoxide inhibition on JNK phosphorylation
Our previous data demonstrated that JNK is activated in CTL undergoing apoptosis (11, 12). Other studies have also suggested that ROS play a role in JNK activation (32, 33). To determine the role of JNK in the MnTBAP mediated inhibition of AICD of CTL we examined the level of JNK phosphorylation in untreated or inhibitor pretreated MP58-66 reactive T cells stimulated using cognate and control pentamer reagents. Cell lysates were prepared and analyzed by Western blot using antibodies specific for JNK and phosphorylated JNK. The total level of JNK was relatively constant regardless of the pretreatment and the antigenic stimulation (Figure 6A). In contrast, the level of phosphorylated JNK increased upon exposure to the cognate antigen as compared to the control. This increase was completely blocked by MnTBAP and SP600125 pretreatment (Figure 6B), but not by ERK inhibitor PD098059, which served as specificity control. Since AICD induced in MP58-66 reactive T cells using cognate pentamer negates the role of costimulatory signal, the observed phosphorylation of JNK is TCR-complex specific. These data suggest that ROS could control CTL viability by regulating the activity of pro- and anti-apoptotic proteins through JNK phosphorylation.
Figure 6. Effect of ROS inhibition on JNK phosphorylation.
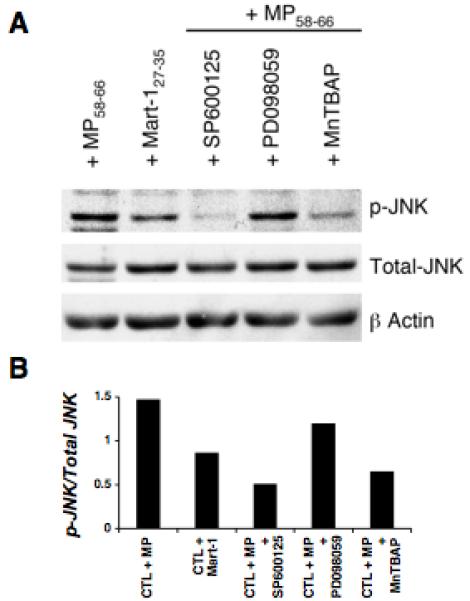
MP58-66 reactive SP600125, MnTBAP or PD098059 (ERK inhibitor) pretreated CTL were exposed to the cognate epitope MP58-66 and untreated CTL were exposed to both cognate epitope MP58-66 and control epitope Mart-127-35 for induction of AICD. (A) Western blot depicts total and phosphorylated (p-)JNK levels using lysates made from the MP58-66 reactive CTL. (B) Densitometric analysis of the blot in (A). Bands were scanned into the computer and band intensity was quantitated using the National Institutes of Health (NIH) Image 1.61 software (free software available from the NIH). Values represent the ratio of p-JNK over total JNK.
Discussion
Upon antigen stimulus, naive resting T cells go through a process of activation, proliferation, and differentiation. Interestingly, activated T cells may die through AICD after re-encountering their cognate antigen (9). It is not clear if AICD contributes to homeostasis, but it plays an important role in preventing untoward side effects of an uncontrolled or persistent effector response (34). However, for the host’s protection against cancer and infection AICD may be detrimental as disease states might be prolonged if potentially therapeutic CTL are deleted.
Given that the effectiveness of T cells may be significantly improved by protection from AICD, understanding its mechanism in human antigen-reactive CTL is important. We have earlier demonstrated that a fraction of human CTL reactive to Mart-127-35 and MP58-66 underwent AICD upon re-encountering their respective cognate peptides and that JNK inhibition prevented AICD but also impaired the cells ability to secrete cytokines (11, 12). The current study shows that inhibition of ROS rescues CTL from AICD without impairing their effector functions. In support of this idea, both tumor and viral epitope-reactive primary human CTL activated by their cognate epitope contained more superoxide than their counterparts exposed to a control epitope. Since superoxide is a byproduct of the mitochondrial electron transport chain, elevated levels may reflect increased respiration within activated cells (35). Furthermore, antigen-reactive primary CTL and TIL escaped AICD when treated with MnTBAP. MnTBAP has been shown to protect cells from superoxide-mediated toxicity in neural cells and endothelial cells, through its mimicry of superoxide dismutase and/or catalase (36, 37). Loss of Δψ has been shown to be a key event in apoptosis of mouse thymocytes exposed to various apoptotic stimuli (38) and in peripheral T cells undergoing SEA induced apoptosis (15). Additionally, death of thymocytes has been accompanied by the production of ROS as Δψ dissipates (39). Our data indicate that similar ROS-dependent mechanisms cause opening of the permeability transition pore and regulate loss of the Δψm and AICD in human CTL. In addition to decreasing superoxide levels, MnTBAP pretreatment rescued the CTL from loss of Δψ, DNA fragmentation, and death upon recognition of cognate antigens. Moreover, enhanced accumulation of MnTBAP pretreated, as compared to untreated, CTL after restimulation with cognate epitope indicate that they indeed remained viable for a longer time.
TCR activation has been shown to increase ROS within T cells (40), although how excess ROS accumulates is unclear. One explanation is that increased demand for ATP production is imposed on T cells by their conversion from resting precursors to rapidly dividing effectors. In addition to the rate of production, ROS levels depend on the detoxifying activities of antioxidants. For example, superoxide dismutase (SOD) converts superoxide into oxygen and H2O2 (41). In turn, H2O2 is detoxified by glutathione peroxidase and/or catalase (42). Thus, the severity of ROS damage is ultimately dependent on both the levels and types of ROS and the levels and activities of antioxidants. Because MnTBAP mimics both SOD and catalase activity, it ensures the complete detoxification of superoxide and its downstream metabolites (15). Hence, MnTBAP may be particularly useful in preventing cellular damage in which superoxide is the initiating ROS.
As such, a role for MnTBAP or other antioxidants in rescuing CTL from death is understandable. However, the exact pathways these agents modulate to protect cells from AICD is not clearly understood. The intracellular concentration of ROS can lead to selective activation of AP-1 transcription factors determining cell fate, i.e. survival versus death (43). Activation of MAP kinases (ERK, p38, and JNK) has also been observed in response to changes in the cellular redox state (44) and the balance between ERK and JNK activation appears to be a key determinant of cell survival. A decrease in ERK and an increase in JNK activity are required for apoptosis (45). Hence, SP600125, MnTBAP, and other antioxidants might in fact rescue CTL from AICD by a common mechanism, i.e. blocking of JNK-driven apoptotic signaling. Our data support the interpretation that AICD observed in ex vivo expanded CTL reactive for tumor ‘self’ antigen or viral ‘non-self’ antigen is mediated through ROS and JNK activation, and involves mitochondrial membrane collapse that can be prevented by antioxidants such as MnTBAP.
The importance of CTL persistence for successful adoptive T-cell therapy has been highlighted in both experimental models (5) and in clinical trials involving cancer patients (7). Preventing AICD of TIL may benefit their ex vivo expansion as well as increasing their persistence and efficacy in vivo. Thus, we investigated the level of sensitivity of TIL to AICD when re-encountering cognate antigen and its modulation by ROS inhibition. Pretreatment of TIL1235 with MnTBAP rescued them from AICD upon cognate-antigen encounter. While SP600125 concomitantly impaired their ability to secrete cytokines, MnTBAP treatment did not interfere with CTL functionality. A better understanding of how ROS mediate their effects (46) is likely to unveil novel targets that can be exploited to increase persistence of CTL and thereby improve immunotherapies.
An earlier study demonstrated that the natural free radical scavenger vitamin E suppresses the activity of the transcription factors NF-κB and AP-1, thus blocking expression of CD95L and preventing T cell AICD (47). Since AICD is a major cause of T cell depletion in AIDS, a disease associated with high levels of oxidative stress, this study examined 35 HIV-1 positive individuals and found that their T cells were more susceptible to AICD as compared to T cells isolated from healthy controls. Administration of vitamin E suppressed CD95L mRNA expression and protected T cells of HIV-1-infected individuals from CD95-mediated apoptosis (47). In addition a role for inducible nitric oxide synthase in regulating T cell death and immune memory has been shown (48). These studies taken together with our data suggest that inhibition of free radical generation/oxidative stress can affect T cell survival and merits further clinical investigation. Since MnTBAP itself has been used in vivo in pre-clinical models (49) and other antioxidants already are approved for clinical use, these findings have direct implications for current immunotherapeutic treatments. Further, engineering T cells to over-express antioxidant enzymes has recently been shown to protect T cells from exogenous ROS (50). Similar approaches may also protect CTL from AICD and are underway in our laboratory to improve T cell persistence and the results of immunotherapy.
Acknowledgments
The work was supported by CA 83130 & Dowling Foundation grant (BM), CA 102280 & CA 104947 (MN), Charlotte-Geyer Foundation grant & laboratory start-up funds (SM) and Abney Foundation post-doctoral fellowship (HN).
References
- 1.Jaattela M. Programmed cell death: many ways for cells to die decently. Ann Med. 2002;34:480–8. doi: 10.1080/078538902321012423. [DOI] [PubMed] [Google Scholar]
- 2.Hildeman D, Jorgensen T, Kappler J, Marrack P. Apoptosis and the homeostatic control of immune responses. Curr Opin Immunol. 2007;19:516–21. doi: 10.1016/j.coi.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu B, Finn OJ. T-cell death and cancer immune tolerance. Cell Death Differ. 2008;15:70–9. doi: 10.1038/sj.cdd.4402274. [DOI] [PubMed] [Google Scholar]
- 4.Whiteside TL. Apoptosis of immune cells in the tumor microenvironment and peripheral circulation of patients with cancer: implications for immunotherapy. Vaccine. 2002;20(Suppl 4):A46–51. doi: 10.1016/s0264-410x(02)00387-0. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Khong HT, Dudley ME, et al. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28:258–67. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Res. 2005;65:2001–8. doi: 10.1158/0008-5472.CAN-04-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robbins PF, Dudley ME, Wunderlich J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaattela M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003;4:416–23. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 9.Budd RC. Activation-induced cell death. Curr Opin Immunol. 2001;13:356–62. doi: 10.1016/s0952-7915(00)00227-2. [DOI] [PubMed] [Google Scholar]
- 10.Mehrotra S, Stevens R, Zengou R, et al. Regulation of melanoma epitope-specific cytolytic T lymphocyte response by immature and activated dendritic cells, in vitro. Cancer Res. 2003;63:5607–14. [PubMed] [Google Scholar]
- 11.Mehrotra S, Chhabra A, Chattopadhyay S, Dorsky DI, Chakraborty NG, Mukherji B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: implications in cancer immunotherapy. J Immunol. 2004;173:6017–24. doi: 10.4049/jimmunol.173.10.6017. [DOI] [PubMed] [Google Scholar]
- 12.Mehrotra S, Chhabra A, Hegde U, Chakraborty NG, Mukherji B. Inhibition of c-Jun N-terminal kinase rescues influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death. J Leukoc Biol. 2007;81:539–47. doi: 10.1189/jlb.0706479. [DOI] [PubMed] [Google Scholar]
- 13.Murahashi H, Azuma H, Zamzami N, et al. Possible contribution of apoptosis-inducing factor (AIF) and reactive oxygen species (ROS) to UVB-induced caspase-independent cell death in the T cell line Jurkat. J Leukoc Biol. 2003;73:399–406. doi: 10.1189/jlb.0702335. [DOI] [PubMed] [Google Scholar]
- 14.Papa L, Gomes E, Rockwell P. Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis. 2007;12:1389–405. doi: 10.1007/s10495-007-0069-5. [DOI] [PubMed] [Google Scholar]
- 15.Hildeman DA, Mitchell T, Teague TK, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–44. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 16.Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15035–40. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannoni E, Buricchi F, Grimaldi G, et al. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008;15:867–78. doi: 10.1038/cdd.2008.3. [DOI] [PubMed] [Google Scholar]
- 18.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–74. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg SA, Dudley ME, Restifo NP. Cancer immunotherapy. N Engl J Med. 2008;359:1072. doi: 10.1056/NEJMc081511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JC, Childs R. Immunotherapy for renal cell cancer. J Clin Oncol. 2006;24:5576–83. doi: 10.1200/JCO.2006.08.3774. [DOI] [PubMed] [Google Scholar]
- 21.Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6458–62. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banki K, Hutter E, Gonchoroff NJ, Perl A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J Immunol. 1999;162:1466–79. [PMC free article] [PubMed] [Google Scholar]
- 23.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. Journal of immunological methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 24.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–70. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267–71. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 26.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 27.Peitsch MC, Muller C, Tschopp J. DNA fragmentation during apoptosis is caused by frequent single-strand cuts. Nucleic acids research. 1993;21:4206–9. doi: 10.1093/nar/21.18.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frankfurt OS, Robb JA, Sugarbaker EV, Villa L. Monoclonal antibody to single-stranded DNA is a specific and sensitive cellular marker of apoptosis. Exp Cell Res. 1996;226:387–97. doi: 10.1006/excr.1996.0240. [DOI] [PubMed] [Google Scholar]
- 29.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. Journal of cell science. 2004;117:2417–26. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- 31.Schmitt F, Tani E, Tribukait B, Skoog L. Assessment of cell proliferation by Ki-67 staining and flow cytometry in fine needle aspirates (FNAs) of reactive lymphadenitis and non-Hodgkin’s lymphomas. Cytopathology. 1999;10:87–96. doi: 10.1046/j.1365-2303.1999.00065.x. [DOI] [PubMed] [Google Scholar]
- 32.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–39. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ. 2007;14:1001–10. doi: 10.1038/sj.cdd.4402088. [DOI] [PubMed] [Google Scholar]
- 34.Lenardo MJ. Molecular regulation of T lymphocyte homeostasis in the healthy and diseased immune system. Immunol Res. 2003;27:387–98. doi: 10.1385/IR:27:2-3:387. [DOI] [PubMed] [Google Scholar]
- 35.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–90. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 36.Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–55. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 37.Day BJ, Shawen S, Liochev SI, Crapo JD. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced endothelial cell injury, in vitro. J Pharmacol Exp Ther. 1995;275:1227–32. [PubMed] [Google Scholar]
- 38.Zamzami N, Marchetti P, Castedo M, et al. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–72. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–77. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med. 2004;37:1144–51. doi: 10.1016/j.freeradbiomed.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 41.Halliwell B. Biochemical mechanisms accounting for the toxic action of oxygen on living organisms: the key role of superoxide dismutase. Cell Biol Int Rep. 1978;2:113–28. doi: 10.1016/0309-1651(78)90032-2. [DOI] [PubMed] [Google Scholar]
- 42.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–11. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 43.Kriehuber E, Bauer W, Charbonnier AS, et al. Balance between NF-kappaB and JNK/AP-1 activity controls dendritic cell life and death. Blood. 2005;106:175–83. doi: 10.1182/blood-2004-08-3072. [DOI] [PubMed] [Google Scholar]
- 44.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–96. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 45.Rincon M, Flavell RA, Davis RA. The JNK and P38 MAP kinase signaling pathways in T cell-mediated immune responses. Free Radic Biol Med. 2000;28:1328–37. doi: 10.1016/s0891-5849(00)00219-7. [DOI] [PubMed] [Google Scholar]
- 46.Kovacic P, Somanathan R. Integrated approach to immunotoxicity: electron transfer, reactive oxygen species, antioxidants, cell signaling, and receptors. J Recept Signal Transduct Res. 2008;28:323–46. doi: 10.1080/10799890802305217. [DOI] [PubMed] [Google Scholar]
- 47.Li-Weber M, Weigand MA, Giaisi M, et al. Vitamin E inhibits CD95 ligand expression and protects T cells from activation-induced cell death. J Clin Invest. 2002;110:681–90. doi: 10.1172/JCI15073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vig M, Srivastava S, Kandpal U, et al. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. The Journal of clinical investigation. 2004;113:1734–42. doi: 10.1172/JCI20225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laniewski NG, Grayson JM. Antioxidant treatment reduces expansion and contraction of antigen-specific CD8+ T cells during primary but not secondary viral infection. Journal of virology. 2004;78:11246–57. doi: 10.1128/JVI.78.20.11246-11257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ando T, Mimura K, Johansson CC, et al. Transduction with the antioxidant enzyme catalase protects human T cells against oxidative stress. J Immunol. 2008;181:8382–90. doi: 10.4049/jimmunol.181.12.8382. [DOI] [PubMed] [Google Scholar]