

Abstract
Since its original discovery as a negative regulator of neuronal differentiation, the repressor element (RE)-1 silencing transcription factor (REST), also known as the neuron-restrictive silencer factor, has been implicated in novel processes such as maintenance of embryonic stem cell pluripotency and self-renewal and regulation of mitotic fidelity in non-neural cells. REST expression and activity is tightly controlled by transcriptional and post-transcriptional mechanisms in a cell and developmental stage-specific manner and perturbations in its levels or function are associated with various pathological states. REST differentially influences target-gene expression through interaction with a wide variety of cellular cofactors in a context-dependent manner. However, the influence of the microenvironment on REST-mediated regulation of gene expression is poorly understood. This review will present our current understanding of REST signaling with a greater focus on its emerging ties with noncoding RNAs and novel interacting partners, as well as its roles in embryonic stem cell self-renewal, cellular plasticity and oncogenesis/tumor suppression.
Keywords: Down’s syndrome, embryonic stem cells, medulloblastoma, neural stem cells, neuronal differentiation, non-neural stem cells, oncogenic activity, pluripotency, regulation of self-renewal, REST/NRSF, tumor-suppressor activity
The repressor element (RE)-1 silencing transcription factor (REST), also known as the neuron-restrictive silencer factor, was originally identified as a transcriptional repressor of neuronal differentiation genes [1,2]. In mice, REST is expressed ubiquitously in developing non-neural tissues between embryonic days E8.5 and 9.5 where it is thought to act as a molecular restraint for precocious neuronal differentiation [3]. Knockout studies demonstrated that mice homozygous null for REST (REST-/-) showed brain malformation at embryonic day E9.5, followed by extensive apoptosis and lethality by E11.5. Additionally, increased expression of type III β-tubulin, a REST target gene, was observed in non-neural tissues of these embryos. These results suggested an important role for REST in brain development and in the regulation of neuronal differentiation [3]. An alternative approach using retroviral transduction of a dominant-negative mutant of REST in chicken embryo showed derepression of only a subset of REST target genes, suggesting that release from REST-mediated repression must be followed by either transcriptional activation or relief from other repressive activities to turn on other REST target genes [3]. Consistent with its function as a repressor of neuronal differentiation, REST is expressed in embryonic stem cells (ESCs), neural stem cells (NSCs) and non-neural cells, and is absent in most, but not all, differentiated neurons. Several reviews have discussed the biology of REST in great detail [4-14]. Here, we focus on recent studies that have shed light on emerging links between REST and non-coding RNAs, novel protein-protein interactions as well as on its role in stem/progenitor cells and in cancers.
Identification of REST target genes
REST is a 116-kDa zinc finger protein that contains a central DNA-binding domain flanked by two independent repressor domains located at the amino and carboxy termini. REST binds to a 21-23 base pair sequence termed the RE1 element, found in the regulatory regions of target genes [2,15]. Several different approaches have been taken to define the RE1 element and to identify REST target genes, ranging from bioinformatics to genome-wide binding experiments and study of diseases. In probably the first bioinformatics-based genome-wide approach to the study of REST targets, Bruce et al. identified a number of genes whose regulatory regions housed an RE1 site and had the potential to be REST-regulable genes [16]. While several of these genes had functions in neuronal development, a number of these putative targets were involved in functions far removed from neurogenesis. Thus, this study not only increased the number of possible REST target genes, but it also raised the intriguing possibility that REST may be involved in the regulation of other cellular functions.
However, this approach did not confirm actual occupancy of these RE1 sites and more recent efforts have turned to genome-wide binding studies, such as serial analysis of chromatin occupancy (SACO) and large-scale chromatin immunoprecipitation (ChIP-Seq), to ask questions regarding the binding of REST to the RE1 elements across the genome [17-29]. Interestingly, these studies revealed the existence of noncanonical RE1 bipartite sequences in addition to the canonical RE-1 sequence, which had two half-sites separated by 11 base pairs. In these noncanonical sites, the two half-sites were separated by either 10 or 16-19 base pairs from their centers [18,19]. Significant REST binding to these noncanonical sequences was demonstrated in vivo, pointing to a vast underestimation of REST-regulable genes in the genome by previous studies. Significantly, these high-throughput genome-wide analyses also revealed variability in the occupancy of the RE1 elements in different cell types [23-27]. This may have stemmed from the sensitivity of the assays used, with the more sensitive assays, such as SACO and ChIP-Seq methodologies, being able to detect even weak or transient interactions. Alternatively, it could be a reflection of actual variation in REST binding to its target genes based on its affinity for the target RE1 elements or based on the presence or absence of co-activators/repressors in these cell types. Indeed, a recent report by Jothi and colleagues has identified core residues within the RE1 element that are important for stronger binding [24]. Differential REST binding to its target genes could also result from nucleosome structure or occupancy of adjacent sequences by other DNA-binding proteins, causing REST to be included or precluded at these sites.
When REST binding to various targets in ESCs, NSCs and hippocampal neurons was evaluated, it was observed that distinct REST complexes occupied RE1 elements at various target genes and a number of REST target genes were expressed at low levels in ESCs and NSCs but at higher levels in neurons, supporting the hypothesis that the interaction of REST with its target genes was distinct in these cell types [25,26]. To further address if significant differences in REST binding occurred in a cell type-specific manner, Johnson et al. took a ChIP-chip approach in conjunction with a ChIP paired-end tag (ChIP-PET) strategy to evaluate REST binding to various sites [27]. They further validated genes identified by this method as bonafide REST targets through comparison of their expression patterns in the presence of REST and upon knockdown of its activity in mouse ESCs, NSCs and fibroblasts (a differentiated non-neural cell type). Despite the fact that one of the most important functions of REST in all three cell types is to prevent precocious neurogenesis, its binding repertoire in NSCs and fibroblasts was more similar than in ESCs. These experiments also revealed that REST bound to a core group of genes in all three cell types; however, in addition it bound to more stem cell-specific targets in ESCs. These targets overlapped with that of other pluripotency regulators, such as Oct4, Sox2 and Nanog. An RE1 element was found in the regulatory regions of Nanog, REST itself, ZF206, ZF281, Lin28 and genes in the Wingless (Wnt) signaling cascade - all targets with known roles in the control of pluripotency [27].
Other studies using the ChIP-Seq approach in human T-lymphoblast cell lines demonstrated REST binding to genes critical for pancreatic islet β-cell development, such as HNF4a, HNF6 and Hes1, alluding to a potential role for REST in pancreatic development [19]. Further validation of whether these genes are targets of REST’s repressive activity under physiological conditions is necessary. Thus, these studies suggest regulatory roles for REST along distinct lineages; however, as stated previously, questions regarding the mechanism(s) by which decisions are made to occupy a specific RE1 in one cell type, but not in another cell type, remain largely unanswered.
The high-throughput studies discussed above also indicated that REST might have links to some noncoding RNAs (ncRNAs). Although ncRNAs, such as microRNAs (miRNAs), were previously known to be involved in the regulation of neurogenesis, the underlying mechanism remained unclear until recent research added some of these molecules to the portfolio of REST targets [10]. One of the earliest miRNAs identified as a direct REST target based on binding studies and expression analyses was miR-124a [20,21]. In fact, miR-124a appears to be part of a regulatory network that controls REST activity in NSCs and non-neural cells. These studies demonstrated that in NSCs and non-neural cells, REST repressed the expression of the proneural miR-124a, thus preventing neurogenesis and also facilitating the continued expression of non-neural genes [21]. Upon onset of neuronal differentiation, declining REST levels in NSCs accompanied by miR-124a-mediated downregulation of the REST-associated small C-terminal domain phosphatase-1, allowed the transition from a progenitor state to a differentiated neuronal phenotype (Figure 1) [21]. Since these initial reports, the list of putative REST-target miRNAs has grown substantially. Computational analyses, genome-wide binding studies and ChIP analyses have collectively identified several neuron-specific miRNAs, including miR9-1, miR9-3, miR-29a/miR-29b, miR-95, miR-132, miR-133, miR-135b, miR-139, miR-153, miR-218-212, miR-346, miR-375 and miR-455, to have the potential for regulation by REST [18,20,22-24,27]. Since each miRNA controls the expression of multiple genes, link-up with these ncRNAs affords REST potential control over a vast regulatory circuit. The significance of REST-mediated control of miRNA expression is best illustrated in Huntington’s disease (HD), a neurodegenerative disease that is characterized by CAG expansion in the gene encoding the Huntingtin protein (Htt). The normal function of Htt is to sequester REST in the cytoplasm and allow expression of the prosurvival brain-derived neurotrophic factor (BDNF) in hippocampal neurons. In HD this neuroprotective function is lost as a result of the inability of the mutant Htt protein to sequester REST and prevent its nuclear localization. As a result, BDNF expression is repressed and that of other REST target genes is deregulated in striatal neurons [30,31]. More recently, aberrant REST localization has been correlated with significant misexpression of a number of brain-specific miRNAs, including miR-29a, miR-124a, miR-132 and miR-135b and targets of the well-studied miR-124a, in mouse HD models [32]. In human brain tissue from HD patients, a decrease in miR-132 levels was observed, whereas miR-29a and miR-330 levels were significantly upregulated. Levels of the miR-132 target, p250GAP, were also significantly elevated in patient samples, supporting the hypothesis that misregulated REST expression causes widespread deregulation of gene expression through disruption of miRNA circuitry [32].
Figure 1. Overview of REST function in self-renewal of embryonic stem cells and neurogenesis in neural and non-neural cells.
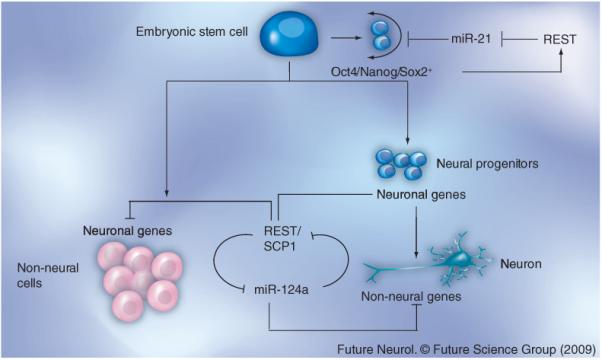
In embryonic stem cells, REST represses the expression of miR-21, a negative regulator of proteins involved in self-renewal including Sox2, Nanog and Oct4. In the absence of miR-21, the expression of Sox2, Nanog and Oct4 in embryonic stem cells is maintained, which in turn feeds back and activates REST expression and contributes to self-renewal in these cells [85]. In non-neural cells, the REST/SCP1 complex represses the expression of neuronal genes and the proneuronal miR-124. In neurons, REST is absent and, consequently, miR-124 is expressed [20,21]. This not only promotes neuronal gene expression, but also represses non-neuronal gene expression [20,21].
miRNA: Micro-RNA; REST: Repressor element silencing transcription factor; SCP1: Small CTD phosphatase-1.
Ties between REST and miRNA have been extended to proteins important for REST-associated repressive activity. For example, MeCP2 has the potential to be regulated by miR9*, miR-124a, miR-132 and miR-212 [22]. Similarly, Co-REST, yet another component of the REST complex, is a putative target of the miRNAs miR-29b, miR-124a, miR-135b, miR-153 and miR-218 [22,23]. Finally, REST itself may be subject to regulation by the brain-specific miR-153, indicative of a feedback loop [22,23].
REST has also been hypothesized to regulate another class of ncRNAs termed macroRNAs. In a recent study, 18 candidate macroRNAs were studied and of these, two appeared to be regulated by REST in mouse NSCs. Notably, DCGR5, a macroRNA associated with DiGeorge syndrome, was regulated by REST through an upstream RE1 element, suggesting that REST-dependent macroRNAs may be potentially involved in the etiology of such diseases [29].
REST has been implicated in pathological conditions of neural and non-neural cells and studies of these diseases have identified a number of target genes. A discussion of several REST targets has been presented elsewhere [4,6,9,33-38]. Here we have focused on the more recently identified target genes involved in pathological states. REST expression is known to be activated during ischemia-induced neuronal death, causing suppression of the AMPA-receptor subunit GluR2 in hippocampal CA1 pyramidal neurons [39]. Modulation of REST-dependent metabolic changes in BDNF gene expression is also known to affect the frequency of seizures [39,40]. The μ-opioid receptor (MOR)-1 has also been identified as a novel REST target gene. MOR-1 is normally expressed in basket cells and inhibitory interneurons of the CA1 area. However, under conditions of ischemic death, elevated REST expression promotes an increase in the repressive dimethyl histone H3-K9 mark at the MOR-1 promoter [41]. Likewise, REST has been linked to abnormal neuronal differentiation and depressive disorders in the presence of alcohol [42,43]. In rat models, alcohol is known to decrease neurogenesis in the dentate gyrus of the hippocampus. This decline in neuronal differentiation has been attributed to a decrease in the DNA-binding ability of REST, pointing to a possible role for REST in ethanol-associated neuronal dysfunction [43]. Tryptophan hydroxylase (TPH)-2 is an enzyme involved in serotonin biosynthesis, and polymorphisms in the cognate gene have been implicated in susceptibility to psychiatric disorders. Recent work by Patel and colleagues determined the expression of the TPH2 gene to be under the control of REST [44]. The cocaine- and amphetamine-regulated transcript (CART), which is widely expressed in the CNS and peripheral nervous system as well as in endocrine cells, is thought to be important in physiological processes such as feeding, stress and fear, anxiety and sensory processing. However, mechanism(s) regulating CART expression had not been elucidated. Studies by Li and colleagues indicate that REST binds the RE1 element in the promoter of the CART gene and controls transcription of this gene in endocrine cells, raising the possibility of a regulatory role for REST in the above processes [45].
Myotilinopathy, a myofibrillar myopathy characterized by aggregation of cytoskeletal proteins and ubiquitin, has more recently been shown to express neuron-related proteins such as the ubiquitin carboxy-terminal hydrolase LI (UCHL1), synaptophysin, α-internexin and synaptosomal-associated protein-25. Detection of UCHL1 in diseased muscle was surprising since its expression is mostly restricted to the brain and testes. This observation was explained in a recent report by Barrachina et al., who showed that REST was expressed at lower levels in muscle fibers of patients with myotinilopathy [46]. The inverse correlation between REST and UCHL1 gene and protein expression observed in these samples taken together with their finding that REST bound the RE1 elements in the UCHL1 promoter suggested that misregulated REST expression may contribute to the disease phenotype.
REST: repression by networking
REST-associated repressor domains located at the amino (N)- and the carboxy (C)-terminus of the protein modulate the expression of target genes through interactions with other co-repressor and chromatin remodeling enzymes. Previous reviews have extensively discussed the interactions of the N- and C-terminal repression domains with the mSin3A/histone deacetylase (HDAC) and Co-REST/LSD1 histone H3-K4 demethylase/Brg1 co-repressor complexes, respectively, to remodel chromatin and negatively regulate gene expression [7-9,11,47-49]. Likewise, the association of REST with G9a histone H3-lysine 9 (H3-K9) methyltransferase, CtBP, DNA methyltransferase 1, DNA-methyl-CpG-binding protein-2, the SWI/SNF chromatin remodeling complex, the TATA box binding protein and small RNA polymerase II carboxyl-terminal domain phosphatases have also been extensively discussed in other excellent reviews [7-9,11,47-49]. Some of the newly identified interactions have been outlined below and their implications for different cellular processes have also been described.
The association of REST with the proteins SMCX and Mediator, has drawn attention to X-linked mental retardation disorders such as Opitz-Kaveggia (FG) syndrome and Lujan syndrome [50-52]. JARID1C/SMCX was first identified as a jumonji domain-containing protein that demethylated histone H3-K-4 to di- and mono-methylated products, an event known to be important for chromatin remodeling and changes in gene expression. Its expression in the postsomitogenesis zebrafish was observed to be restricted to the brain and its loss during embryogenesis and in mammalian neurons decreased neuronal survival and dendritic development, respectively. SMCX is part of a transcriptional repressor complex containing HDACs, the histone methyl transferase G9a and REST [51]. Using chromatin immunoprecipitation assays, SMCX and REST were shown to co-occupy promoters of REST target genes in HeLa cells. Importantly, RNA interference-mediated knockdown of SCMX caused an increase in histone H3-K4 trimethylation of these target genes that correlated with their derepression. Six of these genes have been implicated in other neurological disorders, such as epilepsy, autism and schizophrenia. These findings suggest that REST, through its interaction with SMCX, may play a role in impaired neuronal gene expression in X-linked mental retardation and perhaps other neurologic disorders [51].
Mediator, a conserved multiprotein complex and a major player in RNA polymerase II-dependent transcription, was recently found to modulate the transcriptional repressive properties of G9a histone methyl transferase and REST at neuronal genes [52]. MED12, a component of the Mediator complex, is important in neural crest and brain development in zebrafish and MED12 polymorphisms have been linked to schizophrenia and psychosis, while missense mutations in the gene are thought to be causative of X-linked mental retardation and Lujan’s syndrome [52]. Interestingly, these mutations disrupted the recruitment of REST-associated G9a to the Mediator complex in vitro, compromising the dimethylation of histone H3-K9, a repressive mark at several RE1 elements. This correlated well with the derepression of these REST target genes in a wide range of human cell types.
The Chromodomain on Y-Like (CDYL) protein was also recently identified as a REST-interacting protein in HeLa cells using co-immunoprecipitation and mass-spectrometric approaches [53]. CDYL has been proposed to act as a bridge between REST and G9a based on the observation that knockdown of CDYL blocked the co-immunoprecipitation of REST and G9a in vitro and in vivo. The CDYL, REST and G9a-bound target genes were enriched for dimethyl histone H3-K9, consistent with a previous report that the N-terminal chromodomain of CDYL-bound methylated H3-K9. Since G9a is responsible for mono- and di-methyl marks of H3-K9 found on euchromatin and heterochromatin, respectively, the enrichment for dimethyl histone H3-K9 marks at REST target genes suggest a preferential association of REST with heterochromatin-associated G9a.
The telomere repeat factor (TRF)2 is critical for telomere integrity and regulation of genomic stability. TRF2 was previously demonstrated to modulate neurogenesis; however, the mechanism(s) that link TRF2 to neurogenesis was not understood until a recent study by Zhang et al. demonstrated that countering TRF2 activity or knockdown of TRF2 expression in neuroblastoma and human embryonal carcinoma stem cell lines induced neuronal differentiation [54]. This study showed that in undifferentiated cells, REST existed in a complex with TRF2, the majority of which displayed strong focal nuclear staining and localization to promyelocytic leukemia (PML)-nuclear bodies. By contrast, a diffuse nuclear staining for both proteins with little PML association was observed in differentiating cells. Using co-immunoprecipitation assays, expression analyses and rescue experiments, it was demonstrated that the association of REST with TRF2 in PML bodies prevented the ubiquitin proteasome-mediated degradation of REST in undifferentiated stem/progenitor cells. These results indicate a nontelomeric role for TRF2. An extension of this argument would be that in cancer cells, TRF2 could maintain cells in an undifferentiated, progenitor-like state by maintaining REST levels.
Homozygous mutations in PRICKLE1 have been implicated in the etiology of autosomal-recessive progressive myoclonus epilepsy-ataxia (PME) syndrome [55]. Prickle 1, also known as the REST-interacting LIM-domain protein (RILP), is a component of the noncanonical Wnt signaling pathway. It was independently identified as a REST-interacting protein and, as the name suggests, has functions in nuclear localization of the latter protein [56]. Interestingly, the PRICKLE1 mutation observed in patients with PME abrogates its interaction with REST and alters its subcellular localization in vitro [57]. The mutation in PRICKLE1 also caused abnormal gastrulation and somitogenesis in zebrafish embryos in overexpression studies [57]. It would be interesting to examine the consequence of this mutation in the chromosomal context and under stochiometric conditions to evaluate if REST is involved in the gastrulation phenotype.
From the preceding discussion, it is increasingly evident that REST associates with a wide array of interacting partners in a cell type-specific manner. This may in part allow for a cell- and context-dependent regulation of gene expression. These effects could be potentially fine-tuned through the binding of other transcriptional activators and repressors within promoters containing RE1 sequences [11].
Regulation of REST activity/function
Work from several groups has demonstrated that REST levels are significantly different in ESCs, neural progenitors and non-neural cells [7-9,11-14]. REST levels and activity appear to be regulated by transcriptional and post-transcriptional mechanisms depending upon the cell type, as outlined in Figure 2. Previous studies have shown Wnt signaling to be important for the transcriptional activation of REST expression in human embryonic carcinoma and in the chick spinal cord [58,59] ESCs, REST is expressed at high levels and these cells are induced to differentiate into neural progenitors, REST levels are regulated through its proteasomal degradation [60]. Westbrook colleagues showed the involvement of the β-TRCP E3 ubiquitin ligase in the degradation of REST during neuronal differentiation of neural progenitors to neurons. Whether this is relevant to degradation of REST in ESCs is not known [61] Ballas et al. also demonstrated that the binding of the unliganded retinoic acid receptor complex to the REST promoter negatively regulated its transcription as neural progenitors differentiated into neurons. Although REST is largely thought of as a repressive activity, its interaction with a double-stranded RNA molecule with homology to the RE1 sequence converts it into a transcriptional activator in adult NSCs [62]. REST splice variants, such as REST4, have been suggested to play an inductive role in controling glucocorticoid-mediated glutamine synthetase expression in neural tissues, whereas full-length REST represses its expression in non-neural tissues [63]. These splice variants also act as dominant-negative regulators of full-length REST function by competing with it for binding to the LIM-domain protein and nuclear transporter, RILP (Prickle1) [56]. RILP has also been shown to interact with Htt and Dynactin p150Glued proteins, suggesting a potential role for these proteins in the subcellular localization of REST [57]. In non-neural cells, REST levels are regulated in a cell cycle, β-TRCP-dependent manner and, as discussed later, disruption of this control contributes to genetic instability [64]. Consistent with a post-transcriptional mode of regulation of REST and perhaps its activity, a highly variable pattern of migration of the protein is seen upon polyacrylamide gel electrophoresis and western blotting. Some of this variability in migration could be attributed to REST phosphorylation and perhaps ubiquitination. However, information on the kinase or the trigger that a ctivates this kinase is lacking at this time [61,64].
Figure 2. Regulation of REST by transcriptional and post-transcriptional mechanisms.
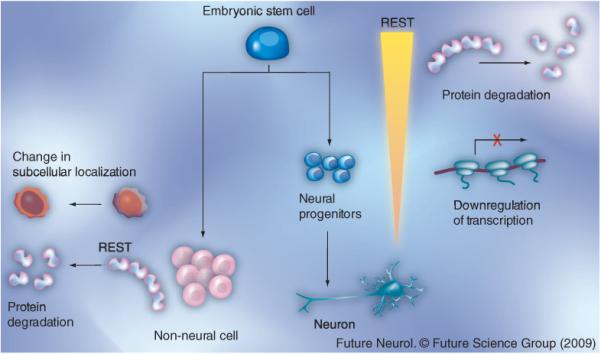
REST is expressed at high levels in embryonic stem cells. A decline in REST levels is observed during the specification of embryonic stem cells into neural progenitors. REST levels decline through ubiquitin-mediated proteasomal degradation. In neural progenitors, REST is transcriptionally downregulated by the unliganded retinoic acid receptor [60]. The failure to appropriately downregulate REST during induction of neurogenesis may contribute to brain tumors such as medulloblastoma. In non-neural cells, REST function is regulated by changes in its subcellular localization between the nucleus and the cytoplasm mediated by the Huntingtin/RILP/Dynactin p150Glued proteins [56,57]. REST expression is also negatively regulated by phosphorylation and proteasomal degradation through a SCF (β-TRCP)-dependent mechanism [62,64].
REST: Repressor element silencing transcription factor.
REST as a tumor suppressor & an oncogene
REST can function both as a tumor suppressor and an oncogene depending upon the cellular context [6,9,12]. It is normally expressed in non-neural tissues, such as the lung, breast and colon epithelium. A lack of normal REST function in these cells has been postulated to promote tumorigenesis, indicating that, in addition to perhaps repressing neurogenesis in these cells, REST may also have a tumor-suppressor function [6,9]. Indeed, this possibility has been addressed by Westbrook and colleagues, who elegantly demonstrated that lack of REST function in telomerase and SV40 T-antigen immortalized human mammary epithelial cells (TLM-HMEC) resulted in their transformation [65]. However, this study did not provide a mechanistic understanding of how REST functions as a tumor suppressor. The more recent identification of an association between REST, the chromodomain protein CDYL and G9a histone methyl transferase has provided a glimpse into the molecular machinery that may regulate REST’s role as a tumor suppressor [53]. The study demonstrated that the absence of CDYL not only prevented the recruitment of G9a to REST target genes, but that it also promoted transformation of TLM-HMEC through derepression of the proto-oncogene TrkC. A similar phenotype was obtained upon knockdown of G9a, suggesting that these two proteins are important for REST-associated tumor-suppressive activity. Interestingly, loss of heterozygosity of 6p25, which houses the CDYL locus, is a frequent event in cervical cancers. Inhibition of CDYL-enhanced human papillomavirus E6 and E7 oncoproteins mediated transformation of normal primary human squamous epithelial cells. These results suggest that REST acting through CDYL may play a role in preventing cellular transformation [53].
An oncogenic role for REST was initially suggested in the pediatric brain tumor, medulloblastoma [66]. REST expression is aberrantly maintained in human medulloblastoma samples and in established tumor cell lines, and maintenance of REST expression in these tumors correlated well with a lack of neuronal differentiation [66,67]. However, REST alone was insufficient for tumorigenesis and cooperates with the Myc oncogene in this process [68]. This finding was validated by the observation that several REST-overexpressing medulloblastoma samples examined also exhibited elevated Myc expression. This was also consistent with previous findings that mice constitutively expressing REST develop normally apart from some axonal guidance problems [69]. Furthermore, while Myc overexpression alone was insufficient for maintenance of proliferation of NSCs cultivated under neurogenic conditions, constitutive REST expression forced NSCs to proliferate and prevented their entry into the differentiation program. In vivo, REST and Myc overexpressing NSCs forced cerebellar tumors in immunodeficient mice, but similar injections into the forebrain did not support tumor formation, implying a superimposition of the microenvironment in REST-associated tumorigenic effects [68]. Finally, these findings also raise questions regarding the mechanism(s) by which REST expression is deregulated in these tumors and the effectors through which REST promotes its tumorigenic effects. One possibility is that aberrant activation of developmental pathways (e.g., Shh, Wnt and Notch) that are important in cerebellar development may contribute to abnormal REST expression [70]. A recent study demonstrated misregulated expression of a panel of miRNAs with links to REST in medulloblastoma samples [71]. Whether this has any role in the misexpression of REST in tumor cells has yet to be determined.
Thus, these studies with medulloblastoma highlight the importance of REST in neural tumors and suggest that aberrant persistence of REST expression in these cells probably contributes to oncogenesis, at least in part through restriction of cellular specification. It is intriguing that Guadavaccaro and colleagues suggest an oncogenic role for REST in non-neural cells [64]. Their studies demonstrated that failure to degrade REST in the G2/M phase of proliferating non-neural cells promoted genomic instability through persistent repression of Mad2, a REST target and spindle checkpoint gene. These results not only illustrate variability in REST requirement and function in different cell types, but also within the same cell based on its cell cycle phase.
REST facilitates cell-fate specification
Although REST is critical for repression for multiple neuronal differentiation genes, as discussed earlier, mere dismissal of REST from the RE1 sites is not adequate for derepression of several REST target genes [3]. This indicates a subsequent need for binding by other transcriptional activators or relief from other repressive activities at these loci to turn on gene expression [3,72]. These possibilities were assessed in experiments where ESCs were differentiated into neurons under controlled conditions. A careful evaluation of occupancy of target gene promoters by the REST complex revealed the expression of class I neuronal genes (e.g., type III β tubulin) to involve a simple dismissal of the REST-corepressor complex from the RE1 sites of promoter regions [60]. Class II genes (synapsin and glutamate receptor), on the other hand, were regulated through a multistep process wherein the REST-corepressor complex was first discharged from the RE1 sites of target genes. However, since the adjacent MeCP2-associated chromatin DNA was occupied by a corepressor complex that lacked REST, it resulted in continued target gene repression. Other cellular events, such as membrane depolarization, triggered chromatin remodeling and activated transcription. These findings were recapitulated in cortical progenitors and cortical neurons isolated from mouse embryos [60]. A requirement for relief from additional repressive activities was not demonstrated in these studies, but, nevertheless, it remains a formal possibility.
The use of two distinct mechanisms to regulate REST target gene expression was also confirmed through a slightly different approach. Here, the recombinant protein REST-VP16, which contains the REST DNA-binding domain fused to the transactivator domain of the herpes simplex virus VP16, was used to compete with endogeneous REST for DNA binding and drive activation of target genes in a variety of cell types [72,73]. In neural stem cell lines derived from cerebellar progenitor cells, conditional REST-VP16 expression activated REST target genes giving rise to differentiated cells with properties of mature neurons, such as synaptic vesicle recycling and glutamate-induced calcium influx [73]. These results indicate that the simple process of activating the expression of REST target genes was sufficient to convert neural stem cells into a neuronal phenotype.
Cell fate decisions can also be impacted in non-neural progenitors, such as myoblasts, through alteration of REST activity [74]. The conditional expression of REST-VP16 activated the expression of a number of REST target genes and supported their conversion into cells with neuron-like characteristics, including depolarization-dependent calcium influx and synaptic vesicle recycling [74]. Injection of these engineered myoblasts into the cerebellum also allowed assimilation into the cerebellar environment in a nontumorigenic manner and with expression of neuronal differentiation markers. Whether countering REST function in differentiated cells of non-neural origin will allow a similar switch in lineage is not clear. It would also be interesting to find out if the microenvironment can fine-tune such specification decisions. For example, can REST-VP16-expressing myoblasts be converted into glutamatergic neurons in the cerebellum and be directed to form motor neurons in the neuromuscular junction?
This potential for reprogramming gene expression and altering cell fate has been demonstrated by a number of recent studies where introduction of specific transcription factors promoted somatic cells, such as human and murine fibroblasts, to dedifferentiate into pluripotent cells and simulating an embryonic cell-like state [75-77]. These induced pluripotent cells appear to not only have similar gene-expression profiles but are also functionally comparable to ESCs [75-77].
REST in ESC self-renewal & pluripotency
Embryonic stem cells are pluripotent, self-renewing cells with the ability to differentiate into all cells and tissues in the body. A core network of transcription factors, including Oct4, Sox2, Nanog, Brg1 and TCF3 [79-87], not only control self-renewal in these cells, but also regulate specification in ESCs by repressing the expression of lineage-specific genes. REST is expressed at very high levels in ESCs and a number of recent reports in the literature have attempted to dissect the role of REST in ESCs. The data emerging from these studies paint a conflicting picture of REST biology in ESCs. The following section will attempt to present these studies and their differing interpretation of REST function in normal ESCs and in diseases arising from defects in ESCs [14,28,85-91].
In a recent study by Singh et al., it was demonstrated that mouse ESCs haplosufficient for REST had downregulated expression of Oct4, Nanog, Sox2, Tbx3 and c-myc [85,86]. These findings were corroborated by small interfering RNA-mediated knockdown of REST expression in ESCs that not only lead to a loss of self-renewal, but also promoted expression of markers of different lineages. Conversely, exogenous REST restored self-renewal in these cells (Figure 1). These findings were supported by other studies that showed a direct correlation between loss of self-renewal and pluripotency and a decline in REST levels [84,87]. Knockdown of Brg1 in mouse blastocyst also resulted in changes characteristic of loss of pluripotency and self-renewal, including downregulated expression of Oct4, Sox2, Nanog, Sall4 and REST and subsequent premature differentiation [87]. Collectively, these reports favor a role for REST in ESC self-renewal and pluripotency. However, experiments by Sun and colleagues provide a slightly different twist to this story [28]. These authors suggest that while REST haploinsufficiency did not cause any phenotypic changes characteristic of loss of pluripotency or precocious differentiation, a complete loss of REST in ESCs interfered with the generation of late (Sox+/Nestin-) NSCs, neural progenitors and neurons and also increased apoptosis in neurons derived from such ESCs. These observations are consistent with the findings that REST haploinsufficient mice do not display any major phenotypic differences from REST wild-type mice. Additionally, in this study, the loss of REST in ECSs appeared to recapitulate the massive apoptosis observed in the brain of E9.5 REST-null embryos preceding embryonic lethality [3]. The most striking feature of neurons derived through differentiation of REST-null ESCs was defects in adhesion and cell migration that could be rescued by expression of laminins (and laminin-directed signaling). In vivo, REST-null embryos appear to have significantly impaired levels of a number of laminin genes, including Lama1, b1, c1 and a2, suggesting that the decrease in neurons may be the result of improper adhesion and consequent cell death [28].
In an effort to understand the mechanism by which REST maintains self-renewal and pluripotency, Singh and colleagues demonstrated that in REST heterozygous ESCs and in embryoid bodies that have lower REST expression compared with ESCs, a subset of miRs with the potential to affect expression of Nanog, Sox2, Tbx3 and c-myc, such as miR-21, miR-27a and miR-93, were upregulated. An inverse correlation between REST levels and that of miR-21 in differentiated mouse ESCs and undifferentiated control cells was also observed in this study [85]. A subsequent analysis of the Genomatix database followed by chromatin immunoprecipitation assays identified miR-21, as a direct repressive target of REST. Thus, this report suggests that REST maintains self-renewal and pluripotency in ESCs through mechanisms involving repression of the microRNA, miR-21 (Figure 1) [85]. The notion that miR-21 is a direct target of REST has not been validated by other high-throughput analyses carried out with other cell types, although miR-21 is expressed upon retinoic acid-mediated differentiation of ESCs [18,19,22,24,27]. This is not entirely surprising since REST binding to RE1 elements is cell type- and differentiation stage-dependent and it is possible that the association of REST with the miR-21-associated RE1 element is restricted to ESCs. Another possible explanation may be that ES cultures in vitro may not comprise a unique cell population and instead have a more hetero genous composition, as indicated by a recent study [91]. The binding of REST to the miR-21-associated RE1 element may be a characteristic feature of a minor subset of cells and may account for the small but statistically significant changes in REST binding to the miR-21-regulatory region observed by Singh and colleagues. Alternatively, as Sun and colleagues suggest, REST may control this self-renewal/pluripotency through regulation of Wnt signaling and or other regulators of pluripotency [28]. Clearly, the reason(s) for these variable findings are not fully clear and further studies are necessary.
Finally, although REST is co-expressed with other self-renewal markers, such as Oct4, Sox2 and Nanog, in the inner cell mass of mouse blastocysts from which ESCs cells are derived, its role in the self-renewal of these cells is more ambiguous. REST+/- haploinsufficient mice were normal, much like animals that were heterozygous for Oct4, Nanog and Sox2 and the LIF-STAT3-c-Myc pathway [78-84,88]. Moreover, since REST-/- exhibits a lethal phenotype between embryonic days 9.5 and 11, one interpretation is that REST may not play a role at the blastocyct stage and in the self-renewal and pluripotency of the ICM cells [3].
The heated discussion surrounding REST in self-renewal, pluripotency and lineage decisions is reflected in studies on Down’s syndrome, a genetic disease associated with trisomy 21, reduced neuronal numbers, decreased brain size, mental retardation, diminished neuronal plasticity and elevated neurodegeneration. Studies by Bahn et al. suggested that the neurological deficits observed in patients with Down’s syndrome stemmed from diminished REST expression in ESCs of these individuals and the consequent precocious neuronal differentiation, apoptosis and loss of neurons [92]. This observation is supported by the increase in cell death seen in neurons derived from REST-null mice described by Sun and colleagues [28]. A different study proposed a REST-independent mechanism and attributed the abnormalities in Down’s syndrome to diminished expression of the transcription factor AP4 in the fetal brain of patients with Down’s syndrome [90]. A complex containing AP4, geminin, SMRT and HDAC3 is thought to repress the expression of the dual specificity tyrosine-phosphorylated and regulated kinase-1 (DYRK1A), a key player in Down’s syndrome. The authors suggest that decreased expression of AP4 may contribute to the premature activation of DYRK1A kinase and the neuronal abnormalities observed in Down’s syndrome [90]. Consistent with this, Sohn et al. demonstrated that the brain of normal individuals and those with Down’s syndrome did not reveal a substantial difference in REST levels [93]. However, REST has been brought back into the Down’s syndrome picture with the recent suggestion that the decrease in REST levels seen in ESCs and brain-derived neurospheres as well as in the whole adult brain of Down’s syndrome mice promoted downregulated expression of Oct4, Nanog and Sox2, a phenotype similar to that observed in REST+/- ESCs described by Singh and colleagues [85,89]. The upregulated expression of transcription factors, such as GATA4, GATA6, FOXA2, PITX2 and SNAI1, that drive specific embryonic lineages suggested premature cell-fate specification differentiation in ESCs from Down’s syndrome mice. Precocious neuronal differentiation and REST target gene expression in embryoid bodies derived from mice with Down’s syndrome also suggested that a decrease in REST levels in these cells perturbs the balance between self-renewal and differentiation, again consistent with the experimental outcome observed by Singh and colleagues [85,89]. Thus, further work is needed to reconcile these differing views on the role of REST in ESC self-renewal and pluripotency.
Executive summary.
■ Although repressor element silencing transcription factor (REST) was originally identified as a regulator of neurogenesis, a number of recently identified REST target genes appear to have non-neurogenic functions.
■ Noncoding RNAs have emerged not only as novel REST targets, but also as potential regulators of REST. This provides REST with the opportunity to regulate a larger set of genes than would be expected based on the presence of the RE1 element alone.
■ REST interacts with a number of different proteins in different cell types to regulate target gene expression. Differences in the composition of the REST complex at different target genes are observed even within the same cell type.
■ REST can function as an oncogene and a tumor suppressor depending upon the cell type.
■ REST has been implicated in the regulation of self-renewal and pluripotency in embryonic stem cells. How REST achieves this has been the subject of much debate.
Conclusion
REST has been studied by a number of laboratories in recent years and a growing body of literature has provided insights into several novel aspects of REST biology. Although REST was originally identified as being important in brain development and in the negative regulation of neurogenesis in neural progenitors and non-neural cells, clearly it has relevance in other cellular functions. This has been validated in different cell types by demonstration of functions such as maintenance of ESC self-renewal, neural crest cell migration, control of cardiac gene expression, regulation of mitosis, genomic stability and oncogenesis in neural progenitors and tumor suppression in non-ESCs. Another important theme is that the levels of REST in ESCs, neural progenitors and non-neural cells are significantly different and perturbations in these levels lead to various disorders ranging from Down’s syndrome to cancers. This underscores the need for a tight control of REST expression and function in these different cell types. In conclusion, while rapid advances have been made in our understanding of REST biology over the past decade, it is clear that this is just the tip of the iceberg. Further research will reveal the depth of REST’s influence on cellular processes.
Future perspective
Genome-wide studies have suggested a significant underestimation of REST target genes. Although at the present time, a number of REST target genes are those involved in neuronal differentiation, a role for REST in the regulation of genes involved in other cellular functions, such as self-renewal, cell motility and migration, cellular metabolism, angiogenesis, the cell division cycle, DNA repair and genomic stability, is becoming increasingly apparent. How REST coordinates these myriad functions with its known role in regulation of neuronal differentiation is intriguing. ncRNAs with their well-established ability to control several cellular processes may provide an avenue for REST to cast a wider net on genes that lack REST-binding sequences. Differential affinity of REST for RE1 elements in different genes, variation in its levels in different cell types, association with different binding partners and finally the influence of the microenvironment could all contribute to the ability of REST to control different cellular processes. The same argument could potentially explain the conflicting roles for REST in oncogenesis and tumor suppression.
Finally, REST appears to be a potential target for therapeutic intervention in neurological diseases and other pathological conditions discussed in this review [94]. However, the modulation of REST expression or activity must be achieved in a tissue-dependent manner, given its dual role as an oncogene and a tumor suppressor.
Acknowledgement
The author is grateful to Sadhan Majumder for his editorial comments.
Footnotes
Financial & competing interests disclosure
The author would like to thank the Children’s Brain Tumor Foundation, the National Brain Tumor Foundation and the American Cancer Society Institutional Research Grant for grant support. Our work is also supported in part through the SPORE grant- P50CA127001 from the National Cancer Institute. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Chong JA, Tapia-RamiRez J, Kim S, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 2.Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 3.Chen ZF, Paquette AJ, Anderson DJ. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998;20:136–142. doi: 10.1038/2431. [DOI] [PubMed] [Google Scholar]
- 4.Roopra A, Huang Y, Dingledine R. Neurological disease: listening to gene silencers. Mol. Interv. 2001;1:219–228. [PubMed] [Google Scholar]
- 5.Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Coulson JM. Transcriptional regulation: cancer, neurons and the REST. Curr. Biol. 2005;15:R665–R668. doi: 10.1016/j.cub.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh J, Gage FH. Chromatin remodeling in neural development and plasticity. Curr. Opin. Cell. Biol. 2005;17:1–8. doi: 10.1016/j.ceb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Lunyak VV, Rosenfeld MG. No rest for REST: REST/NRSF regulation of neurogenesis. Cell. 2005;121:499–501. doi: 10.1016/j.cell.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Majumder S. REST in good times and bad: roles in tumor suppressor and oncogenic activities. Cell Cycle. 2006;5:1929–1935. doi: 10.4161/cc.5.17.2982. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Yeo G, Muotri AR, et al. Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 2006;29:77–103. doi: 10.1146/annurev.neuro.29.051605.112839. [DOI] [PubMed] [Google Scholar]
- 11.Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat. Rev. Genet. 2007;8:544–554. doi: 10.1038/nrg2100.■■ Excellent review on repressor element silencing transcription factor (REST) interactions and mechanism of REST-mediated regulation of gene expression.
- 12.Weissman A. How much REST is enough? Cancer Cell. 2008;13:381–383. doi: 10.1016/j.ccr.2008.04.011.■ Good review on REST regulation and its implications for cancer.
- 13.Ooi L, Wood IC. Regulation of gene expression in the nervous system. Biochem. J. 2008;414:327–341. doi: 10.1042/BJ20080963. [DOI] [PubMed] [Google Scholar]
- 14.Hermanson O. Stem Cells have different needs for REST. PLoS Biol. 2008;6:2094–2097. doi: 10.1371/journal.pbio.0060271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraner SD, Chong JA, Tsay HJ, Mandel G. Silencing the type II sodium channel gene: a model for neural-specific gene regulation. Neuron. 1992;9:37–44. doi: 10.1016/0896-6273(92)90218-3. [DOI] [PubMed] [Google Scholar]
- 16.Bruce AW, Donaldson IJ, Wood IC, et al. Genome-wide ana lysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl Acad. Sci. USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101.■■ One of the first bioinformatics approaches to identifying REST target genes, excellent website to search for repressor element (RE)-1 elements throughout the genome.
- 17.Johnson R, Gamblin RJ, Ooi L, et al. Identification of REST regulon reveals extensive transposable element-mediated binding site duplication. Nucleic Acids Res. 2006;34:3862–3877. doi: 10.1093/nar/gkl525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto SJ, McCorkle SR, Hover J, et al. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J. Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007.■ Demonstration of a noncanonical RE1 element at REST target genes.
- 19.Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 20.Conaco C, Otto S, Han JJ, et al. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl Acad. Sci. USA. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103.■ Demonstration of a link between REST and microRNAs.
- 21.Visvanathan J, Le S, Le B, et al. The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 2006;21:744–749. doi: 10.1101/gad.1519107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Xie X. Comparative sequence ana lysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2007;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortazavi A, Leeper Thompson EC, et al. Comparative genomics modeling of the NRSF/REST network: from single conserved sites to genome-wide repertoire. Genome Res. 2006;16:1208–1221. doi: 10.1101/gr.4997306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jothi R, Cuddapah S, Barski A, et al. Genome-wide identification of in vivo protein DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun YM, Greenway DJ, Johnson R, et al. Distinct profiles of REST interaction with its target genes at different stages of neuronal development. Mol. Cell. Biol. 2005;16:5630–5638. doi: 10.1091/mbc.E05-07-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenway DJ, Street M, Jeffries A, Buckley NJ. RE1 silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells. 2007;25:354–363. doi: 10.1634/stemcells.2006-0207. [DOI] [PubMed] [Google Scholar]
- 27.Johnson R, Teh CH-L, Kunarso G, et al. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008;6:2205–2219. doi: 10.1371/journal.pbio.0060256.■ Elegant demonstration of distinct REST target genes in embryonic stem cells and neural stem cells (NSCs).
- 28.Sun YM, Cooper M, Finch S, et al. REST-mediated regulation of extracellular matrix is crucial for neural development. PLoS ONE. 2008;3:E3656. doi: 10.1371/journal.pone.0003656.■ Suggests that REST does not regulate pluripotency, but instead controls the production of nestin-positive NSCs.
- 29.Johnson R, Teh CH, Jia H, et al. Regulation of neural macroRNAs by the transcriptional repressor REST. RNA. 2009;15:85–96. doi: 10.1261/rna.1127009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuccato C, Tartari M, Crotti A, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 31.Zuccato C, Belyaev N, Conforti P, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson R, Zuccato C, Belyaev ND, et al. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008;29:438–445. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Qiang M, Rani CS, Ticku MK. Neuron-restrictive silencer factor regulates the N-methyl-d-aspartate receptor 2B subunit gene in basal and ethanol-induced gene expression in fetal cortical neurons. Mol. Pharmacol. 2005;67:2115–2125. doi: 10.1124/mol.104.010751. [DOI] [PubMed] [Google Scholar]
- 34.Cheong A, Bingham AJ, Li J, et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell. 2005;20:45–52. doi: 10.1016/j.molcel.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 35.Ohba T, Watanabe H, Takahashi Y, et al. Regulatory role of neuron-restrictive silencing factor in the expression of TRPC1. Biochem. Biophys. Res. Commun. 2006;351:764–770. doi: 10.1016/j.bbrc.2006.10.107. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa Y, Kuwahara K, Harada M, et al. Class II HDACs mediate CaMK-dependent signaling to NRSF in ventricular myocytes. J. Mol. Cell. Cardiol. 2006;41:1010–1022. doi: 10.1016/j.yjmcc.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Kuratomi S, Kuratomi A, Kuwahara K, et al. NRSF regulates the developmental and hypertrophic changes of HCN4 transcription in rat cardiac myocytes. Biochem. Biophys. Res. Commun. 2007;353:67–73. doi: 10.1016/j.bbrc.2006.11.119. [DOI] [PubMed] [Google Scholar]
- 38.Ding N, Tommori-Sato C, Sato S, et al. MED19 and MED26 are synergistic functional targets of the RE1 silencing transcription factor in epigenetic silencing of neuronal gene expression. J. Biol. Chem. 2009;284:2648–2656. doi: 10.1074/jbc.M806514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calderone A, Jover T, Noh KM, et al. Ischemic insults derepress the gene silencer REST in neurons destined to die. J. Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garriga-Canut M, Scoenike B, Qazi R, et al. 2-deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 41.Formisano L, Noh KM, Miyawaki T, et al. Ischemic insults promote epigenetic reprogramming of μ opioid receptor expression in hippocampal neurons. Proc. Natl Acad. Sci. USA. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tateno M, Ukai W, Hashimoto E, et al. Implication of increased NRSF/REST binding activity in the mechanism of ethanol inhibition of neuronal differentiation. J. Neural Transm. 2006;113:283–293. doi: 10.1007/s00702-005-0320-6. [DOI] [PubMed] [Google Scholar]
- 43.Ishii T, Hashimoto E, Ukai W, et al. Lithium induced suppression of transcription factor NRSF/REST: effects on the dysfunction of neuronal differentiation by ethanol. Eur. J. Pharmacol. 2008;593:36–43. doi: 10.1016/j.ejphar.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 44.Patel PD, Bochar DA, Turner DL, et al. Regulation of tryptophan hydroxylase-2 gene expression by a bi-partite RE-1 silencer of transcription/neuron restrictive silencing factor (REST/NRSF) binding motif. J. Biol. Chem. 2007;282:26717–26724. doi: 10.1074/jbc.M705120200. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Liu Q, Yang Y, et al. Regulatory role of neuron-restrictive silencing factor in the specific expression of cocaine- and amphetamine-regulated transcript gene. J. Neurochem. 2008;106:1314–1324. doi: 10.1111/j.1471-4159.2008.05487.x. [DOI] [PubMed] [Google Scholar]
- 46.Barrachina M, Moreno J, Juves S, Moreno D, Olive M, Ferrer I. Targets of neuron-restrictive silencing factor are abnormally up-regulated in human myotilinopathy. Am. J. Pathol. 2007;171:1312–1323. doi: 10.2353/ajpath.2007.070520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee MG, Wynder C, Cooch N, et al. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 48.Yeo M, Lee SK, Lee B, et al. Small CTD phosphatases function in silencing neuronal gene expression. Science. 2005;307:596–600. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe H, Mizutani T, Haraguchi T, et al. SWI/SNF complex is essential for NRSF-mediated suppression of neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene. 2006;25:470–479. doi: 10.1038/sj.onc.1209068. [DOI] [PubMed] [Google Scholar]
- 50.Iwase S, Lan F, Bayliss P, et al. The X-linked mental retardation gene SMCX/JARID1 defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 51.Tahiliani M, Mei P, Fang R, et al. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 52.Ding N, Zhou H, Esteve PO, et al. Mediator links epigenetic silencing of neuronal gene expression with X-linked mental retardation. Mol. Cell. 2008;3:347–359. doi: 10.1016/j.molcel.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulligan P, Westbrook TF, Ottinger M, et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol. Cell. 2008;32:718–726. doi: 10.1016/j.molcel.2008.10.025.■■ Identified CDYL as being critical for REST-mediated tumor suppression.
- 54.Zhang P, Pazin M, Schwartz CM, et al. Non-telomeric TRF2-REST interaction modulates neuronal gene silencing and fate of tumor and stem cells. Curr. Biol. 2008;18:1489–1494. doi: 10.1016/j.cub.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bassuk AG, Wallace RH, Buhr A, et al. A homozygous mutation in PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 2008;83:572–581. doi: 10.1016/j.ajhg.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimojo M, Hersh LB. REST/NRSF-interacting LIM domain protein, a putative nuclear translocation receptor. Mol. Cell. Biol. 2003;23:9025–9031. doi: 10.1128/MCB.23.24.9025-9031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimojo M. Huntingtin regulates repressor element-1 silencing transcription factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain interacting protein (RILP) and dynactin p150Glued. J. Biol. Chem. 2008;283:34880–34886. doi: 10.1074/jbc.M804183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Willert J, Epping M, Pollack R, et al. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2002;2:8–15. doi: 10.1186/1471-213x-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishihara S, Tsuda L, Ogura T. The canonical Wnt pathway directly regulates NRSF/REST expression in chick spinal cord. Biochem. Biophys. Res. Commun. 2003;311:55–63. doi: 10.1016/j.bbrc.2003.09.158. [DOI] [PubMed] [Google Scholar]
- 60.Ballas N, Grunseich C, Lu D, et al. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 61.Westbrook TF, Hu G, Ang XL, et al. SCFβ-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–374. doi: 10.1038/nature06780.■ Shows phosphorylation to be important for the proteasomal degradation of REST.
- 62.Kuwabara T, Hsieh J, Nakashima K, et al. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- 63.Abramovitz L, Shapira T, Ben-Dror I, et al. Dual role of NRSF/REST in activation and repression of the glucocorticoid response. J. Biol. Chem. 2008;283:110–119. doi: 10.1074/jbc.M707366200. [DOI] [PubMed] [Google Scholar]
- 64.Guardavaccaro D, Frescas D, Dorrello NV, et al. Control of chromosome stability by the β-TrCP-REST-Mad2 axis. Nature. 2008;452:365–369. doi: 10.1038/nature06641.■ Shows control of mitotic spindle checkpoint by REST, which is significant for genomic instability in cancer.
- 65.Westbrook TF, Martin ES, Schlabach MR, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033.■■ First demonstration of REST as a tumor suppressor.
- 66.Lawinger P, Venugopal R, Guo ZS, et al. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat. Med. 2000;6:826–831. doi: 10.1038/77565.■■ One of the earliest reports demonstrating a role for REST in cancers.
- 67.Fuller GN, Su X, Price RE, et al. Many human medulloblastoma tumors overexpress repressor element-1 silencing transcription (REST)/neuron-restrictive silencer factor (NRSF), which can be functionally countered by REST-VP16. Mol. Cancer Ther. 2005;4:343–349. doi: 10.1158/1535-7163.MCT-04-0228. [DOI] [PubMed] [Google Scholar]
- 68.Su X, Gopalakrishnan V, Stearns D, et al. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol. Cell. Biol. 2006;26:1666–1678. doi: 10.1128/MCB.26.5.1666-1678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paquette AJ, Perez SE, Anderson DJ. Constitutive expression of the neuron-restrictive silencer factor (NRSF)/REST in differentiating neurons disrupts neuronal gene expression and causes axon pathfinding errors in vivo. Proc. Natl Acad. Sci. USA. 2000;97:12318–12323. doi: 10.1073/pnas.97.22.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fogarty MP, Kessler JD, Wechsler-Reya RJ. Morphing into cancer: the role of developmental signaling pathways in brain tumor formation. J. Neurobiol. 2005;64:458–475. doi: 10.1002/neu.20166. [DOI] [PubMed] [Google Scholar]
- 71.Ferretti E, De Smaele E, Po A, et al. MicroRNA profiling in human medulloblastoma. Int. J. Cancer. 2009;124:568–577. doi: 10.1002/ijc.23948. [DOI] [PubMed] [Google Scholar]
- 72.Immaneni A, Lawinger P, Zhao Z, et al. REST-VP16 activates multiple neuronal differentiation genes in human NT2 cells. Nucleic Acids Res. 2000;28:3403–3410. doi: 10.1093/nar/28.17.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su X, Kameoka S, Lentz S, et al. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol. Cell. Biol. 2004;24:8018–8025. doi: 10.1128/MCB.24.18.8018-8025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watanabe Y, Kameoka S, Gopalakrishnan V, et al. Conversion of myoblasts to physiologically active neuronal phenotype. Genes Dev. 2004;18:889–900. doi: 10.1101/gad.1179004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi Y, Desponts C, Do JT, et al. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 76.Nishikawa S, Goldstein RA, Nierras CR, et al. The promise of human induced pluripotent stem cells for research and therapy. Nat. Rev. Mol. Cell. Biol. 2008;9:725–729. doi: 10.1038/nrm2466. [DOI] [PubMed] [Google Scholar]
- 77.Huangfu D, Osafune K, Maehr R, et al. Induction of pluripotent stem cells from primary human fibroblasts. Nat. Biotech. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Rao S, Chu J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 79.Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 80.Kim J, Chu J, Shen X, et al. An extended network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aoi T, Yae K, Nakagawa M, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 83.Yu J, Thomson JA. Pluripotent stem cell lines. Genes Dev. 2008;22:1987–1997. doi: 10.1101/gad.1689808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lunyak VV, Rosenfeld MG. Epigenetic regulation of stem cell fate. Hum. Mol. Genet. 2008;17:R28–R36. doi: 10.1093/hmg/ddn149. [DOI] [PubMed] [Google Scholar]
- 85.Singh SK, Kagalwala MN, Perker-Thornburg J, et al. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863.■ Shows REST to be important in the regulation of self-renewal and pluripotency of embryonic stem cells.
- 86.Kagalwala MN, Singh SK, Majumder S. Stemness is only a state of the cell. Cold Spring Harb. Symp. Quant. Biol. 2009 doi: 10.1101/sqb.2008.73.042. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 87.Kidder BL, Palmer S, Knott JG. Swi/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells. 2008 doi: 10.1634/stemcells.2008-0710. DOI: 10.1634/stemcells.2008-0710. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 88.Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell specific microRNAs. Dev. Cell. 2003;5:351–358. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 89.Canzonetta C, Mulligan C, Deutsch S, et al. DYRK1-A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic cell fate in Down syndrome. Am. J. Hum. Genet. 2008;83:388–400. doi: 10.1016/j.ajhg.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim MY, Jeong BC, Lee JH, et al. A repressor complex AP4, transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc. Natl Acad. Sci. USA. 2006;103:13074–13079. doi: 10.1073/pnas.0601915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carter MG, Stagg CA, Falco G, et al. An in situ hybridization-based screen for heterogeneously expressed genes in mouse ES cells. Gene Expr. Patterns. 2008;8:181–198. doi: 10.1016/j.gep.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bahn S, Mimmack M, Ryan M, et al. Neuronal target genes of the neuron restrictive silencer factor in neurospheres derived from fetuses with Down syndrome: a gene expression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- 93.Sohn SY, Weitzdoerfer R, Mori N, Lubeg G. Transcription factor REST-dependent proteins are comparable between Down syndrome and normal brains: challenging a hypothesis. J. Neural Transm. Suppl. 2003;67:59–66. doi: 10.1007/978-3-7091-6721-2_5. [DOI] [PubMed] [Google Scholar]
- 94.Leone S, Mutti C, Kazantsev A, et al. SAR and QSAR study on 2-aminothiazole derivatives, modulators of transcriptional repression in Huntington’s disease. Bioorg. Med. Chem. 2008;16:5695–5703. doi: 10.1016/j.bmc.2008.03.067. [DOI] [PubMed] [Google Scholar]