

Abstract
Conventional chemotherapeutics may induce immunogenic cancer cell death or stimulate immune effectors via so-called off-target effects. The study by Besch et al. in this issue of the JCI now demonstrates that agents designed to stimulate the innate immune system by activating intracellular pattern recognition receptors can kill cancer cells in a direct, cell-autonomous fashion (see the related article beginning on page 2399). The authors show that ligation of viral RNA sensors, such as RIG-I or MDA-5, by viral RNA mimetics triggers mitochondrial apoptosis in human melanoma cells in an IFN-independent fashion. The data suggest that tumor cell killing and immunostimulation may synergize for optimal anticancer immunochemotherapy.
Adjuvants are natural or synthetic compounds that stimulate the immune response, mostly by interacting with receptors of the innate immune system (i.e., pattern recognition receptors [PRRs]) that have evolved to recognize viral or bacterial structures known as pathogen-associated molecular patterns (PAMPs) (1). One of the hallmarks of cancer is the avoidance or suppression of antitumor immune responses (2), and the introduction of adjuvants into tumors, alone or in combination with cytotoxic agents, has been attempted in an effort to stimulate anticancer immune responses. Local instillation of bacterial extracts (e.g., Bacillus Calmette-Guérin) is now the standard therapy for noninvasive bladder cancer (3), and superficial basal cell carcinoma is usually eradicated by topical application of a synthetic PRR activator, imiquimod (which activates the PRR known as TLR7) (4). Innate effectors of the immune system (such as DCs) are activated by PAMPs to stimulate NK and CTL responses. In addition, tumor cells that are exposed to PAMPs can produce chemokines and cytokines (such as type I IFNs), which attract immune effectors into the tumor bed (1). Most immunoadjuvants have been generated to stimulate surface-exposed or lysosomal TLRs, which constitute the first class of PRRs to be discovered. However, recent studies have been evaluating the possibility of targeting cytosolic PRRs that detect the presence of viral genomes in infected cells (1, 5). Endogenous RNA present in the cytoplasm is largely single stranded and lacks 5′-triphosphate RNA (pppRNA) moieties because the RNA transcribed from nuclear host DNA is processed to remove them (for instance by 5′ capping and splicing) before the RNA is exported from the nucleus. In contrast, viral RNA stimulates specific cytosolic receptors by virtue of the presence of 5′-triphosphate residues (which interact with the cytoplasmic PRR retinoic acid–inducible gene I [RIG-I]) or that of double-stranded structures (which are mostly recognized by a RIG-I homolog, melanoma differentiation–associated antigen 5 [MDA-5]) (1, 5). Upon recognition of viral RNA mimetics such as synthetic pppRNA or polyinosinic-polycytidylic acid [poly(I:C), an artificial double-stranded RNA], RIG-I or MDA-5, respectively, bind to the adapter protein IFN-β promoter stimulator 1 (IPS-1, also known as CARDIF, MAVS, or VISA), which is tethered to the outer mitochondrial membrane. This interaction then triggers a complex signal transduction cascade culminating in the activation of transcription factors (in particular, NF-κB and IFN regulatory factor 3 [IRF-3]) and the production of cytokines including class I IFNs (1, 5) (Figure 1).
Figure 1. Lethal signaling induced by intracellular delivery of viral RNA mimetics.

pppRNA activates RIG-I, while poly(I:C) activates MDA-5 and may also interact with PKR and TLR7. RIG-I and MDA-5 then interact with IFN-β promoter stimulator 1 (IPS-1) on the surface of mitochondria. As shown by Besch et al. in their study in this issue of the JCI (6), IPS-1 is required for both the induction of IFN-α production (via the activation of the transcription factor IRF-3) and the transactivation of the genes coding for several BH-only proteins (Noxa, Puma). Among these, Noxa is activated in a p53-independent fashion and required for the subsequent mitochondrial outer membrane permeabilization (MOMP); the release of cytochrome c (cyt c) from the mitochondrial intermembrane space; and the activation of the apoptosome (a molecular complex composed of cytochrome c, apoptotic peptidase–activating factor 1 [Apaf-1], and caspase-9), resulting in apoptosis. Note that IRF-3 is not required for cell death induction by RIG-I and MDA-5 ligands, indicating that the signal transduction cascades resulting in IFN-α production and apoptosis induction bifurcate downstream of IPS-1. Dotted lines indicate hypothetical pathways.
Adjuvants with direct cytotoxic effects
In this issue of the JCI, Besch et al. (6) report that introduction of viral RNA mimetics into human melanoma cells can stimulate a novel signal transduction pathway that triggers activation of the mitochondrial pathway of apoptosis, culminating in melanoma cell death. Exposure of human melanoma cell lines to liposome-encapsulated pppRNA or poly(I:C), ensuring intracellular delivery of the RIG-I or MDA-5 ligands, caused both IFN-α production and cell death in an IPS-1–dependent fashion (Figure 1). However, the mere recruitment of RIG-I or MDA-5 to mitochondria is not sufficient to induce apoptosis. For this, RIG-I or MDA-5 must activate the expression of the BH3-only protein Noxa, which then causes mitochondrial cytochrome c release and consequent activation of the apoptotic peptidase–activating factor 1– (Apaf-1–) and caspase-9–dependent apoptotic pathway (Figure 1). The exact mechanisms that lead to the activation of a proapoptotic transcriptional program remains elusive, apart from the fact that it is independent of the tumor protein p53 (6). Of note, although a panel of nontransformed cells overexpress Noxa in response to RIG-I or MDA-5 ligation, they do not succumb to apoptosis (6, 7), presumably because the antiapoptotic protein Bcl-xL, a close homolog of Bcl-2, prevents mitochondrial permeabilization (6). Thus, RIG-I and MDA-5 ligands exhibit a vague tumor specificity that requires further mechanistic and toxicological exploration.
Importantly, Besch et al. (6) show that poly(I:C) encapsulated into liposomes can reduce the growth of pulmonary melanoma metastases in vivo, in xenografted mice. Since this effect is observed in immunodeficient NOD/SCID mice (which lack both NK and T cells), it is likewise mediated by direct effects on the tumor cells. Although indirect effects mediated by IFN cannot be ruled out as participating in the anticancer effects of poly(I:C) in vivo, IFN does not contribute to RIG-I/MDA-5–induced killing in vitro. Thus, knockdown of the type I IFN receptor or blockade of IFN production (by depletion of IRF-3) failed to reduce apoptosis induction by RNA mimetics (6).
Ligation of one particular TLR, TLR3, has been shown to induce apoptosis of mammary carcinoma cells (8) and melanoma cells (9), establishing a precedent with respect to how the activation of PRRs can induce apoptosis in tumor cells. However, poly(I:C), which can interact with another TLR, TLR7 (which is present on lysosomes), as well as with RNA-activated protein kinase (PKR), was shown by Besch et al. (6) to kill melanoma cells solely by its action on MDA-5, not TLR7 or PKR. This contrasts with the fact that intracellular delivery of poly(I:C) requires PKR for optimal killing of HeLa cells (10), pointing to possible heterogeneities in the response of tumors to viral RNA mimetics. Of note, in specific circumstances, the ligation of PRRs (such as TLR4 on human head and neck squamous cell carcinoma cells) may have tumor-promoting effects (11), meaning that each PAMP should be subjected to a careful case-by-case analysis. From the virologist’s point of view, it is not surprising that the ligation of PRRs by PAMPs causes apoptosis. Indeed, one of the phylogenetically ancient “immune responses” against viral infection is programmed cell death of individual virus-infected cells, which thus limits viral replication and spread (12). However, from an oncologist’s point of view, it is surprising that activation of a PRR can trigger a tumor-specific, cell-intrinsic lethal signal transduction pathway. Thus, the discovery reported by Besch et al. (6) should stimulate active research on other synthetic PAMPs for their potential to activate tumor cell death programs. Small molecules such as the synthetic retinoid CD437 may stimulate RIG-I–dependent apoptosis in melanoma cells as well, yet do not require liposome encapsulation for intracellular delivery (13), opening an avenue toward the development of viral RNA mimetics with improved pharmacokinetics.
The concept of immunochemotherapy
Besch et al. (6) found that an artificial PAMP can directly kill cancer cells and hence act like a classical chemotherapeutic agent. Working with mouse melanomas or colon carcinomas implanted in immunocompetent mice, the same group recently reported that ligation of RIG-I (with an oligonucleotide that contains a 5′-triphosphate moiety and that has been encapsulated into polyethyleneimine liposomes) can stimulate the DC-dependent production of IFN-α and induce an IFN-α–dependent and NK-mediated anticancer effect (14). In light of the data presented in this issue of the JCI (6), it is tempting to speculate that ligation of MDA-5 would also stimulate a therapeutically useful antitumor immune response. When the RIG-I–stimulating 5′-triphosphate moiety was introduced into an oligonucleotide that mediates RNA interference of Bcl-2 (a key protein in the cell death pathway due to its ability to maintain the integrity of mitochondrial membranes), the therapeutic effect was greatly improved, and the combination of RIG-I ligation and Bcl-2 targeting turned out to be more efficient than either of the two strategies alone (14). In other words, simultaneous targeting of tumor cells for apoptotic destruction (chemotherapy) and activation of the immune system (immunotherapy) may be advantageously combined into immunochemotherapy (15) (Figure 2A). Supporting this contention in clinical terms, retrospective analyses of patients treated with a monotherapy of polyadenylic-polyuridylic acid [poly(A:U)], a TLR3 ligand, highlighted that only TLR3-expressing (as opposed to TLR3-negative) breast cancers responded to this treatment (16).
Figure 2. Three mechanisms of immunochemotherapy mediated by single agents.
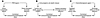
(A) An agent may simultaneously induce tumor cell apoptosis and act as a PAMP, thus activating immunostimulatory PRRs. This is the case for RIG-I and MDA-5 ligands, which, in addition to activating the innate immune system, can kill cancer cells, resulting in a combined immunochemotherapeutic effect. (B) An agent may induce tumor cell death in such a way that dying cells release danger-associated molecular patterns (DAMPs), thereby indirectly activating PRRs in innate immune effectors. For example, anthracyclines induce the exposure and release of several DAMPs on dying tumor cells (17, 18). (C) Alternatively, an anticancer agent may directly kill tumor cells and mediate an immunostimulatory off-target effect on immune cells. For example, imatinib mesylate kills tumor cells that are addicted to constitutively activated tyrosine kinases (such as Bcr/Abl in chronic myeloid leukemia and mutated c-Kit or PDGF-Ra in gastrointestinal sarcoma) at the same time that it activates innate immune effectors through an effect on c-Kit in DCs (19).
Other types of immunochemotherapy
Conventional chemotherapeutic agents may have indirect effects on the immune system that improve their therapeutic efficacy. For example, anthracyclines can stimulate tumor cells to expose and secrete danger-associated molecular patterns (DAMPs), which like PAMPs act as endogenous immunoadjuvants to elicit anticancer immune responses (17). Prominent DAMPs are the preapoptotic exposure of calreticulin on cancer cells (which acts as an engulfment signal for DCs) (17) and the apoptotic release of HMGB1 (which acts on TLR4 present on DCs to stimulate optimal presentation of tumor antigens) (18). In breast cancer patients treated with anthracyclines, a loss-of-function allele of TLR4 indeed has a negative impact and accelerates disease progression (18). Thus, some cytotoxic agents may de facto elicit an immunochemotherapeutic effect by virtue of their capacity to stimulate immunogenic cell death (12, 15) (Figure 2B).
Yet another example is provided by imatinib mesylate (Glivec, Gleevec), which targets the oncogenic tyrosine kinases Bcr/Abl, c-Kit, and PDGF-Ra and which is used for the standard therapy of Philadelphia chromosome–positive chronic lymphoid leukemia (which depends on Bcr/Abl) and gastrointestinal sarcoma (which depends on either c-Kit or PDGF-Ra). Imatinib mesylate also activates NK cells, presumably through an indirect action on DCs (which express c-Kit), and both DCs and NK cells are indispensable for imatinib mesylate–mediated anticancer effects in suitable mouse models (19). In patients with gastrointestinal sarcoma, the efficacy of therapeutic responses clearly correlates with the induction of IFN-γ production by NK cells (20). Thus, even targeted anticancer agents may stimulate the antitumor immune response by off-target effects on innate immune effectors (Figure 2C), thereby initiating an immunochemotherapeutic effect.
The complex pathogenesis of malignancy, which juxtaposes cancer cell–intrinsic aberrations with profound effects on the host stroma, including innate and acquired immune effectors, demands a multi-pronged therapeutic strategy that targets tumor cells and improves or reestablishes antitumor immune responses. The aforementioned examples demonstrate that single agents (such as ligands of RIG-I and MDA-5, as well as anthracyclines and imatinib mesylate) can possess both direct and indirect immune-mediated anticancer effects. Thus, single agents with multi-pronged modes of action are establishing the utility and feasibility of immunochemotherapy. We anticipate that future immunochemotherapies will involve rational combinations of agents that target cancer cell–intrinsic pathways, on one hand, and elements of the immunologic pharmacopeia, including adjuvants, on the other hand.
Acknowledgments
The authors are supported by grants from the Ligue Nationale contre le Cancer, Fondation pour la Recherche Médicale, the European Union, Institut National du Cancer, and Agence Nationale pour la Recherche.
Footnotes
Conflict of interest: Laurence Zitvogel has received a research grant from Innate Pharma. Guido Kroemer has received a research grant and consultancy fees from Bayer Schering Pharma.
Nonstandard abbreviations used: IRF-3, IFN regulatory factor 3; MDA-5, melanoma differentiation–associated antigen 5; PAMP, pathogen-associated molecular pattern; PKR, RNA-activated protein kinase; poly(I:C), polyinosinic-polycytidylic acid; pppRNA, 5′-triphosphate RNA; PRR, pattern recognition receptor; RIG-I, retinoic acid–inducible gene I.
Citation for this article: J. Clin. Invest. 119:2127–2130 (2009). doi:10.1172/JCI39991
See the related article beginning on page 2399.
References
- 1.Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo J., Solimini N.L., Elledge S.J. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexandroff A.B., Jackson A.M., O’Donnell M.A., James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet. 1999;353:1689–1694. doi: 10.1016/S0140-6736(98)07422-4. [DOI] [PubMed] [Google Scholar]
- 4.Papadavid E., Stratigos A.J., Falagas M.E. Imiquimod: an immune response modifier in the treatment of precancerous skin lesions and skin cancer. Expert Opin. Pharmacother. 2007;8:1743–1755. doi: 10.1517/14656566.8.11.1743. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi O., Akira S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Besch R., et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon–independent apoptosis in human melanoma cells. J. Clin. Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirabayashi K., et al. Inhibition of cancer cell growth by polyinosinic-polycytidylic acid/cationic liposome complex: a new biological activity. Cancer Res. 1999;59:4325–4333. [PubMed] [Google Scholar]
- 8.Salaun B., Coste I., Rissoan M.C., Lebecque S.J., Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 9.Salaun B., Lebecque S.J., Matikainen S., Rimoldi D., Romero P. Toll-like receptor 3 expressed by melanoma cells as a target for therapy? Clin. Cancer Res. . 2007;13:4565–4574. doi: 10.1158/1078-0432.CCR-07-0274. [DOI] [PubMed] [Google Scholar]
- 10.McAllister C.S., Samuel C.E. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szczepanski M.J., et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69:3105–3113. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green D.R., Ferguson T., Zitvogel L., Kroemer G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan M., et al. Apoptosis induced by synthetic retinoic acid CD437 on human melanoma A375 cells involves RIG-I pathway. Arch. Dermatol. Res. 2009;301:15–20. doi: 10.1007/s00403-008-0902-x. [DOI] [PubMed] [Google Scholar]
- 14.Poeck H., et al. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat. Med. 2008;14:1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 15.Zitvogel L., et al. The anticancer immune response: indispensable for therapeutic success? . J. Clin. Invest. 2008;118:1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andre, F., et al. 2006. Toll like receptor 3 expression and efficacy of adjuvant treatment with polyadenylic-polyuridylic acid in patients with axillary node positive breast cancer: results from two randomized trials. Presented at the 2006 ASCO Annual Meeting. June 2–5. Atlanta, Georgia, USA. [Google Scholar]
- 17.Panaretakis T., et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Apetoh L., Tesniere A., Ghiringhelli F., Kroemer G., Zitvogel L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 2008;68:4026–4030. doi: 10.1158/0008-5472.CAN-08-0427. [DOI] [PubMed] [Google Scholar]
- 19.Borg C., et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J. Clin. Invest. 2004;114:379–388. doi: 10.1172/JCI21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menard C., et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009;69:3563–3569. doi: 10.1158/0008-5472.CAN-08-3807. [DOI] [PubMed] [Google Scholar]