

Abstract
CTLs eliminate virus-infected and tumorigenic cells through exocytosis of cytotoxic agents from lytic granules. While insights into the intracellular mechanisms facilitating lytic granule release have been obtained through analysis of loss-of-function mutations in humans and mice, there is a paucity of information on negative regulators of secretory lysosome release at the molecular level. By generating and analyzing estrogen receptor–binding fragment-associated antigen 9–KO (Ebag9 KO) mice, we show here that loss of EBAG9 confers CTLs with enhanced cytolytic capacity in vitro and in vivo. Although loss of EBAG9 did not affect lymphocyte development, it led to an increase in CTL secretion of granzyme A, a marker of lytic granules. This resulted in increased cytotoxicity in vitro and an enhanced cytolytic primary and memory T cell response in vivo. We further found that EBAG9 interacts with the adaptor molecule γ2-adaptin, suggesting EBAG9 is involved in endosomal-lysosomal biogenesis and membrane fusion. Indeed, granzyme B was sorted to secretory lysosomes more efficiently in EBAG9-deficient CTLs than it was in WT CTLs, a finding consistent with the observed enhanced kinetics of cathepsin D proteolytic processing. While EBAG9 deficiency did not disrupt the formation of the immunological synapse, lytic granules in Ebag9–/– CTLs were smaller than in WT CTLs. These data suggest that EBAG9 is a tunable inhibitor of CTL-mediated adaptive immune response functions.
Introduction
CTLs and NK cells employ regulated exocytosis of perforin and granzymes, cytotoxic agents from specialized secretory lysosomes (also referred to as lytic granules) to eliminate virus-infected and tumorigenic transformed cells (1). While genetic experiments with gene-deleted mice or in T cells derived from patients with hereditary diseases have provided most of the current evidence for the consequences of a loss-of-function mutation in the adaptive immune response, the characterization of negative regulators in CTL function has thus far received little attention (2–4).
To maintain cytolytic effector functions in CTLs, a constant supply with newly synthesized or recycled effector molecules has to be ensured. The delivery of newly synthesized acid hydrolase precursors, among them granzymes and cathepsins, beyond the Golgi complex depends on the association of these enzymes with the mannose 6-phosphate receptor (M6PR) (5). This complex recruits clathrin adaptors, followed by the formation of clathrin-coated vesicles or carriers that transport the hydrolase-M6PR complex to endosomes. After dissociation of the complex in endosomes, the hydrolase is targeted to lysosomes, while the receptor recycles to the trans-Golgi network (TGN). Sorting of such complexes at the TGN is intimately linked with the function of multi-component adaptor protein complexes 1–4 (AP1–4) and members of the monomeric Golgi-localized, γ-ear–containing ADP-ribosylation factor–binding (ARF-binding) protein (GGA) family of adaptors (6). Selective transport of cargo proteins from the TGN to endosomes is under the control of the ubiquitously expressed AP1 complex and monomeric GGA adaptor proteins. Based on the homology to the large subunit of AP1, γ1-adaptin, the isoform γ2-adaptin has been previously described (7–9). γ2-Adaptin appears to function independently of AP1 and γ1-adaptin, and major differences include subcellular localization, lack of dependency on the GTPase ARF1 for recruitment onto TGN membranes, and the inability of γ2-adaptin to substitute for the loss of γ1-adaptin in gene-deleted mice (7–9). Thus, γ2-adaptin may not act as a typical vesicle-forming adaptor, and γ2-adaptin–containing AP complexes have not yet been identified. While information on the physiological role of γ2-adaptin is rare, support for a sorting function within the endosomal-lysosomal compartment came from an infection model in which γ2-adaptin controlled hepatitis B virus (HBV) production and virion secretion (10, 11).
We previously reported that the tumor-associated antigen estrogen receptor–binding fragment-associated antigen 9 (EBAG9), upon overexpression in PC12 cells, imposed an inhibitory effect on synaptic-like vesicle release (12). In these neuroendocrine cells EBAG9, also referred to as receptor binding cancer antigen expressed on SiSo cells (RCAS1), was shown to interact with Snapin, an enhancer of synaptic vesicle priming which acts through recruitment of synaptotagmin I to the synaptic soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex (13). According to the SNARE hypothesis, target and vesicle SNAREs form a core complex that brings vesicle and target membrane together, ultimately leading to membrane fusion (2, 14–16). In addition to this vesicle-priming step in neurons, a second function for Snapin in exocytosis has been suggested, as Snapin was described to be a member of the biogenesis of lysosome-related organelles complex-1 (BLOC-1) (17).
EBAG9 overexpression was previously recorded in several carcinoma entities, and accordingly this molecule has been suggested to be an independent prognostic marker for disease-specific survival (18–20). However, its broad tissue distribution, and occurrence in benign tissues at all developmental stages make it unlikely that the molecule has evolved for tumor immune escape only, as had been initially suggested (20, 21). Protein expression of EBAG9 is estrogen inducible and has been shown in cell lines and in estrogen-treated mice (22–24). In general, the physiological pleiotropic effects of estrogens are well known in many aspects of development, but their effector mechanisms at the molecular level and their potential pathogenic effects on immunity are not fully understood (25, 26).
To resolve the functional relevance of Ebag9 in vivo, we developed a gene-deleted mouse strain. In this model, we revealed a physiological immunoregulatory function of EBAG9. We focused on CTL equipped with secretory lysosomes that undergo polarized transport and exocytosis in a Ca2+-dependent manner (27). Loss of EBAG9 amplified release of lytic granule content and facilitated enhanced cytolytic capacity in vitro and in vivo. We identified what we believe is a novel interaction partner of EBAG9, γ2-adaptin, which suggests that EBAG9 is required for the control of the endosomal-lysosomal trafficking route in cytotoxic T cells. These data define a critical role for EBAG9 as an estrogen-responsive repressor of T cell cytolytic capacity during adaptive immune responses.
Results
Generation of EBAG9-deficient mice.
To study the physiological function of EBAG9, we generated Ebag9-deficient mice by homologous recombination in ES cells (Figure 1A). The deleted region contained the translation start site as well as exons 2–4, encoding aa 1–107. Southern blot hybridization verified the homologous recombination event, yielding the expected fragment of approximately 4.5 kb in mutant genomic DNA (Figure 1B). Western blot analysis of lymph node lysates confirmed the absence of EBAG9 protein in homozygous mutant mice (Figure 1C). Ebag9–/– mice were healthy and fertile without any apparent morphological abnormalities. In the C57BL/6 strain background, animals exhibited a black coat color. Analysis of genotypes at weaning revealed that the mutated allele segregated at a normal Mendelian frequency of Ebag9+/+ (25%), Ebag9+/– (50%), and Ebag9–/– (25%) mice (matings, n > 20). Additionally, matings of EBAG9-deficient mice produced normal litters, which strongly argues against a postulated role of EBAG9 in maintaining pregnancy at early stages of embryonic development by downregulation of the maternal immune response (28). Furthermore, EBAG9 was suggested to regulate erythroid development by modulating apoptosis of erythroid progenitor cells (29). However, EBAG9-deficient mice exhibited normal peripheral blood cell counts and erythroid progenitors (Ter119+CD71+) in the bone marrow were unaltered (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI37760DS1). Development of lymphocytes from secondary lymphoid organs was not affected by the EBAG9 mutation because gene-deleted mice displayed comparable numbers (data not shown) and subsets of lymphocytes (Supplemental Table 2).
Figure 1. Generation of Ebag9 mutant mice.

(A) Schematic representation of the targeting strategy employed to disrupt the Ebag9 gene. The WT Ebag9 locus (top), the targeting vector (middle), and the mutated locus (Ebag9Δ) after homologous recombination (bottom) are shown. The Ebag9 gene consists of 7 exons; exons removed after homologous recombination are shown in red. The predicted fragment sizes after XhoI (X) and BamHI (B) digestion of genomic DNA are indicated. Neo, neomycin resistance cassette; Tk, thymidine kinase cassette. (B) Southern blot analysis of XhoI/BamHI-digested genomic DNA from F1 animals using the probe indicated in A. (C) Western blot analysis of EBAG9 expression in lymph nodes from adult mice, using polyclonal anti-EBAG9 serum. (D) Differential expression of the EBAG9 protein in various tissues analyzed by immunoblot. (E) Immunoblot analysis of EBAG9 expression in WT CTLs and NK cells.
In immunoblot analysis a broad tissue distribution of EBAG9 (from WT mice) was obtained, among them lymphoid organs (Figure 1D). Ex vivo cultures from WT animals showed that EBAG9 protein was expressed in CTLs and NK cells (Figure 1E). In this study, we focused on CTLs and NK cells, since their regulated secretory pathway used for the release of cytotoxic mediators from lytic granules exhibits extensive mechanistic analogy to the neuroendocrine cell system (2).
Deletion of Ebag9 leads to an enhanced release of granzyme A from CTLs, resulting in an increased cytotoxicity in vitro.
To investigate the role of EBAG9 in the secretory pathway of CD8+ T cells, we generated CTLs from Ebag9+/+ and Ebag9–/– splenocytes (H-2b haplotype) in a mixed lymphocyte reaction (MLR). Numbers of CD8+ (88% ± 5%, mean ± SD) and CD4+ (3% ± 3%, mean ± SD) T cells obtained were comparable between Ebag9–/– and Ebag9+/+ animals (n = 7 experiments; data not shown). Induced secretion of the lytic granule marker granzyme A from EBAG9-deficient CTLs was significantly increased compared with WT (45%) (Figure 2A). Total intracellular enzymatic activity of granzyme A in Ebag9+/+ and Ebag9–/– CTLs was comparable (data not shown). Flow cytometry analysis revealed a stimulation-dependent increase in the cell surface expression of CTLA4, but a difference between Ebag9+/+ and Ebag9–/– CTLs was not seen (Figure 2B). Likewise, we found no significant differences in the release of IFN-γ (Figure 2C), which is rapidly secreted by the constitutive secretory pathway after TCR activation (30), or the chemokine RANTES (Supplemental Figure 1), which is released by a mechanism distinct from lytic granule exocytosis or IFN-γ secretion (31).
Figure 2. Secretory granule release in CTLs.

(A) CTLs were generated in a 3- to 6-day MLR. Release of granzyme A from CTLs was induced by cross-linking of the TCR with anti-CD3ε mAb for 4 hours. Enzymatic activity in supernatants was determined. Secretion is expressed as percentage of maximal activity, measured for Triton X-100 cell lysates. Data are mean ± SD of 7 independent experiments performed in triplicate. *P < 0.02, Student’s t test. (B) Cell surface translocation of CTLA4. CTLs were stimulated for 4 hours with plate-bound mAbs (anti-CD3ε, 5 μg/ml; anti-CD28, 3 μg/ml) (+CD3) or left untreated (-CD3). Cells were stained for CTLA4 and CD8 expression without permeabilization, and viable cells were gated for analysis of CTLA4 fluorescence by flow cytometry. One representative example of 6 independent experiments is shown. Gray histogram shows the isotype control. (C) Release of IFN-γ upon TCR activation. Supernatants from A were analyzed by ELISA. Data are mean ± SD of 6 independent experiments. (D) Cytolytic activity of CTLs (H-2b haplotype) at different ratios of effector cells to a fixed number of allogeneic P815 target cells (H-2d haplotype). Data represent mean ± SD of quadruplicate experiments. One representative experiment of 8 is shown.
In an allogeneic cytotoxicity assay against Fas-negative P815 target cells (H-2d haplotype) (Figure 2D), Ebag9–/– CTLs exhibited a profound advantage in target cell lysis, visible as a 3-fold increase in lytic potency at lower effector to target ratios. To confirm that the enhanced killing capability of the EBAG9-deficient CTLs relies on the improved release of granzyme-containing lytic granules, the allogeneic cytotoxicity assay was repeated in the presence of lysosomal acidification inhibitor concanamycin A and the granzyme B inhibitor Z-AAD-CMK. In the presence of these drugs, Ebag9+/+ and Ebag9–/– CTLs showed comparable abilities for target cell lysis (Supplemental Figure 2). In summary, deletion of EBAG9 provides CTLs with an enhanced capacity for in vitro cytotoxicity.
Enhanced primary and secondary CD8+ T cell responses in EBAG9-deficient mice in vivo.
To examine the functional significance of enhanced lytic granule release for primary CD8+ T cell responses in vivo, we made use of a syngeneic antigen-specific SV40 T antigen (Tag) mouse model (32). First, we investigated the influence of EBAG9 deletion on the antigen-specific elimination of cells loaded with the dominant Tag epitope–derived peptide IV in primary T cell responses. For this in vivo kill assay, Ebag9+/+ and Ebag9–/– mice were immunized with Tag-bearing Co16.113 cells. After 7 days, non-loaded and Tag peptide IV–loaded spleen cells were injected into Tag-immunized or control naive WT mice (Figure 3A). Tag peptide IV–loaded spleen cells were eliminated in Tag-immunized mice, but not in naive WT animals. In Ebag9–/– mice the decrease of Tag peptide–loaded cells was significantly enhanced, corresponding to an increased killing potency of Ebag9–/– CTLs (Figure 3B) (WT, 21.5% ± 10.5%; KO, 53.5% ± 13.6%; mean ± SD). Another systemic model to study primary CTL responses in vivo is the clearance of the bacterium Listeria monocytogenes. After injection, these bacteria migrate to various target organs, including liver, where they enter hepatocytes. Ebag9–/– and Ebag9+/+ mice were challenged with L. monocytogenes, and the CTL response was analyzed by determining the bacterial titer in liver 6 days later (Figure 3C). Significantly lower bacterial titers were obtained from Ebag9–/– livers, suggesting that gene-deleted animals exhibited an improved CTL-mediated clearance function.
Figure 3. Enhanced antigen-specific CD8+ T cell responses in vivo.

(A) Immunized (I) WT and KO mice were challenged at day 7 with non-loaded and peptide-loaded spleen cells labeled with different amounts of CFSE. The ratio between the 2 populations was determined by flow cytometry 4 hours later. One representative example of 7 is shown. N, naive non-immunized C57BL/6 mice. (B) Primary CD8+ T cell response. The percentage of specific killing of peptide-loaded cells in WT (filled squares) and KO (open triangles) mice is indicated. Shown are combined data from 2 independent experiments (n = 7). **P < 0.001, Student’s t test. (C) Ebag9+/+ and Ebag9–/– were infected with L. monocytogenes. Bacterial titers (day 6) are indicated. *P < 0.05, Mann-Whitney test. (D) Ebag9+/+ and Ebag9–/– (H-2b haplotype) mice were inoculated i.v. with CFSE-labeled H-2d and H-2b splenocytes. After 4 hours, the ratio between the 2 populations in the spleen was determined. Specific killing of allogeneic H-2d splenocytes is indicated. Shown are combined data of 2 independent experiments (n = 5). (E) Cytolytic activity of NK cells against a fixed number of YAC-1 cells. Data are mean ± SD of quadruplicate experiments. One representative example of 2 experiments is shown. (F) Ebag9+/+ and Ebag9–/– mice were primed as described in A, followed by a boost after 30 days. On day 4 after rechallenge, a cytotoxicity assay was performed as described in A. Shown are combined data of 2 independent experiments (n = 7). **P < 0.001, Student’s t test. Horizontal bars in B, C, D, and F indicate mean values.
The occurrence of EBAG9 in the NK cell lineage prompted us to compare their cytolytic capacity in Ebag9+/+ and Ebag9–/– animals. In an in vivo kill assay against allogeneic target splenocytes, no significant difference between WT and mutant mice was observed (Figure 3D). Likewise, in a standard NK cell in vitro cytotoxicity assay against YAC-1 target cells, the cytolytic capacity between both groups was comparable (Figure 3E). To further rule out a confounding effect of NK cells in our antigen-specific Tag peptide model, we treated recipient EBAG9 mutant mice with anti–IL-2Rβ mAb (TM-β1). Depletion of NK cells did not attenuate the magnitude of Tag peptide–loaded target cell elimination, thus supporting our conclusion that EBAG9 plays a significant role in this CTL-dependent model in vivo (Supplemental Figure 3).
To investigate CTL activity in secondary T cell responses, Ebag9+/+ and Ebag9–/– mice were challenged again after more than 30 days. Four days after secondary immunization, the elimination of Tag peptide–loaded spleen cells was measured as described above. Ebag9–/– mice exhibited a substantially stronger cytolytic memory T cell response compared with Ebag9+/+ or naive mice (Figure 3F) (WT, 17.1% ± 9.4%; KO, 47% ± 15.57%; mean ± SD). Frequencies of antigen-specific CD8+ T cells were assessed using H-2Kb dimers loaded with the Tag peptide at day 6 after the second challenge (boost). Under these restimulation conditions, no significant differences in the frequency of antigen-specific CTLs between WT and KO mice were seen (WT, 1.39% ± 0.76%; KO, 1.8% ± 0.93%; mean ± SD; P = 0.22, Mann-Whitney test) (Supplemental Figure 4).
Priming and expansion of naive antigen-specific T cells.
The enhanced cytolytic capacity of EBAG9-deficient T cells could be related to the frequency of antigen-specific T cells. As noted before (Supplemental Table 2), total numbers and composition of lymphocyte subsets in spleen and lymph nodes from WT and KO animals were indistinguishable.
The frequency of effector T cells is intimately linked with the priming phase, a process in which DCs play a leading role through the presentation of antigen and provision of costimulation (33, 34). When we analyzed the frequencies of antigen-specific CD8+ T cells using H-2Kb dimers loaded with the Tag peptide at day 8 after immunization, no significant differences between KO and WT animals were observed (Figure 4A). Frequencies of Tag dimer–positive T cells in non-immunized Ebag9+/+ and Ebag9–/– were indistinguishable. Those results were confirmed by the same frequencies of IFN-γ+CD8+ T cells after immunization (Figure 4B). To exclude the possibility that KO CTLs had an inherent proliferative advantage, we assessed the proliferation rate of Tag dimer–positive T cells during a primary immune response (Figure 4, C and D). We observed an identical proliferation rate of Tag dimer– and IFN-γ–positive CD8+ T cells, as determined by BrdU uptake.
Figure 4. Priming and expansion of antigen-specific CD8+ T cells.

(A) Frequency of antigen-specific CTLs is unaltered in Ebag9–/– mice. Mice were immunized as described in Figure 3A, and splenocytes (on day 4) were cultured in the presence of Tag peptide (1 μg/ml) for another 4 days. Cells were stained with anti-CD8a mAb and H-2Kb:Ig dimers loaded with Tag peptide, followed by flow cytometry analysis. Shown is the percentage of Tag dimer–positive CD8+ T cells. n = 7 Ebag9+/+ (filled squares) and n = 8 Ebag9–/– mice (open triangles). Data are from 2 independent experiments. (B) For intracellular IFN-γ staining, cells were stimulated as described in A and stained as described in Methods. The percentage of IFN-γ–positive CD8+ cells at day 8 is shown (n = 4 animals per group; data are from 2 independent experiments). (C) The proliferative capacity of Ebag9–/– Tag-specific CD8+ T cells was unaltered. Mice were immunized as described above, followed by in vitro culture of splenocytes (on day 4) in the presence of Tag peptide for another 4 days. For the last 24 hours, 10 μM BrdU was included in the culture medium. Splenocytes were surface stained with H-2Kb dimers and CD8a, followed by intracellular anti-BrdU staining. The percentage of proliferating cells was assessed by flow cytometry. (D) As described in C, the proliferative rate (percentage) of CD8+IFN-γ+ effector T cells was assessed by intracellular BrdU staining. Horizontal bars indicate the mean value.
DCs are considered the most potent APC subpopulation, as they are highly potent in antigen uptake and concentrated in peripheral lymphoid and nonlymphoid organs (35). In a coculture system, we first compared the priming capacity of bone marrow–derived DCs using OVA-specific CD4+ T cells from OT-II (H-2b haplotype) and DO11.10 (H-2d haplotype) mice as responders. DC stimulators from KO and WT animals, backcrossed onto either the C57BL/6 or BALB/c background, exhibited comparable stimulatory capacity in this system, as measured by CFSE dilution in T cells (Figure 5, B and C). Likewise, priming of OT-I CD8+ T cells (H-2b haplotype), which depends on cross-presentation of the OVA protein–derived peptide SIINFEKL (36), was similar in the presence of KO and WT DCs (Figure 5A).
Figure 5. Similar efficiency of EBAG9-deficient and WT DCs for priming of OVA-specific T cells in vitro.

(A) DCs derived from Ebag9+/+ and Ebag9–/– mice (H-2b haplotype) were pulsed overnight with 100 μg/ml OVA protein. CFSE-labeled naive OT-I T cells (1 × 105) were stimulated with pulsed or unpulsed DCs (1 × 104) for 3 days. Cells were analyzed by flow cytometry, and T cells were gated for the expression of the clonotypic TCR or CD8a and CFSE. Shown is a representative FACS plot of gated CD8+ cells. Proliferation of T cells was evaluated according to CFSE dilution. Numbers indicate the percentage of proliferated CD8+ T cells within the gate. Representative data from 2 independent experiments with DCs from at least 6 animals per group. No significant differences were seen. (B) DCs (H-2d haplotype) were generated and pulsed with OVA protein (75 μg/ml) as described in A, followed by a 3-day coculture with CFSE-labeled OT-II CD4+ T cells. T cells were gated as described in A. Numbers indicate the percentage of proliferated CD3+ T cells within the gate. Shown are representative data from 3 independent experiments. DCs were taken from at least 6 animals per group. (C) DCs (H-2d haplotype) were pulsed for 2 hours with OVA-derived peptide (5 μg/ml), followed by a coculture with DO11.10 CD4+ T cells. T cells were gated as described in A. Shown are representative data from 4 independent experiments. DCs were taken from at least 9 animals per group. Percentages indicate the percentage of proliferated CD3+ T cells within the gate.
To explore the priming phase of OVA-specific T cells in vivo, we employed adoptive transfer of CFSE-labeled DO11.10 and OT-II T cells into Ebag9–/– and Ebag9+/+ recipients (data shown for DO11.10). Three days after immunization with OVA, the proliferation rate was assessed by CFSE dilution of transgenic CD4+ T cells (Figure 6A). In agreement with our in vitro data, WT recipients exhibited the same priming efficacy as EBAG9-deficient animals. Likewise, total numbers of proliferating T cells were indistinguishable (data not shown). Finally, to test whether antigen-specific Ebag9–/– CTLs had a proliferative advantage in vivo, we backcrossed OT-I TCR-transgenic mice onto the Ebag9–/– background and adoptively transferred OT-I WT and OT-I KO T cells (CD45.2 congenic mouse strain) into congenic hosts (CD45.1 congenic mouse strain). Again, following immunization with OVA, a proliferative advantage of antigen-specific Ebag9–/– CTLs was not seen (Figure 6B). Taken together, it seems unlikely that the magnitude of the cytolytic response in Ebag9–/– animals in vivo is a consequence of altered T cell effector frequencies.
Figure 6. Loss of EBAG9 does not affect the proliferation of naive, OVA-specific transgenic T cells in vivo.

(A) CFSE-labeled naive CD4+ DO11.10 T cells (1 × 106) were adoptively transferred into Ebag9–/– and Ebag9+/+ recipients (Balb/c, H-2d haplotype). Eighteen hours later, mice were immunized by s.c. injection of 100 μg OVA protein and 25 μg CpGs. Control WT and KO animals received T cells but no OVA protein (naive). Proliferation of transferred T cells in draining lymph nodes was analyzed by flow cytometry at day 3. Representative FACS plots of gated CD4+ cells from pooled draining lymph nodes (WT, n = 4; KO, n = 5 mice). Numbers indicate the percentage of proliferated CFSE-labeled KJ1.26+ responder T cells in WT or KO recipients within the gate. No differential stimulation of responder T cells was observed in WT or KO recipients. (B) Naive OT-I/Ebag9–/– (n = 4 mice) and OT-I/Ebag9+/+ CD8+ (n = 4 mice) T cells (CD45.2 congenic mouse strain) were CFSE labeled and transferred i.v. into congenic (CD45.1 congenic mouse strain) recipients (5 × 105 cells). Twenty hours later, animals were immunized i.v. with OVA protein (1 μg/g body weight) and CpGs (25 μg). Control groups included a non-immunized animal. Two days later, proliferation of transferred T cells in spleen was analyzed by flow cytometry. Transferred T cells were detected by anti-CD45.2 (LY-5.2) staining. Numbers indicate the percentage of proliferated CFSE-labeled CD45.2+ responder T cells in WT or KO recipients within the gate. Shown are representative data for 5 congenic recipients of OT-I/Ebag9–/– T cells and 4 congenic recipients of OT-I/Ebag9+/+ T cells.
EBAG9 redistributes to the plasma membrane upon TCR activation.
To address the subcellular localization of EBAG9 in primary CD8+ T cells, OT-I TCR transgenic T cells were transduced with a retrovirus encoding an EBAG9-GFP fusion protein (Figure 7A). The major proportion of EBAG9-GFP localized together with the Golgi marker GM130 (>86% overlap; Figure 7B) in a perinuclear localization. In contrast, quantification of colocalization of EBAG9-GFP with the endosomal or lysosomal compartment, as determined by staining of the late endosome/lysosome membrane marker Lamp1, the lysosomal protease cathepsin D, or the lytic granule marker perforin, showed that only a minor proportion of vesicular structures overlapped with EBAG9-GFP localization (Lamp1, 28%; cathepsin D, 20%; perforin, 13%). We noted that a significant pool of EBAG9 molecules redistributed toward the plasma membrane after TCR cross-linking induced by incubation of T cells on anti-CD3/CD28–coated coverslips (Figure 8A). Under these nonpolarized stimulatory conditions, comparison of EBAG9-GFP with the plasma membrane marker CD8a revealed some overlap of both fluorescence signals (Pearson’s correlation coefficient, r = 0.55 ± 0.11). In contrast, in untreated T cells EBAG9-GFP exhibited a predominantly perinuclear staining pattern (r = 0 ± 0).
Figure 7. Subcellular localization of EBAG9 in primary T cells.

(A) Splenocytes from OT-1–transgenic C57BL/6 mice were stimulated and transduced with retrovirus encoding EBAG9-GFP as described in Methods. Cells were fixed, permeabilized, and stained with antibodies directed against GM130, cathepsin D, Lamp1, or perforin and were analyzed by confocal microscopy. Arrows indicate regions of interest. Insets show zoomed magnifications of 2- to 3-fold. Scale bars: 10 μm. (B) Quantitative analysis of EBAG9-GFP structures overlapping with the indicated organelle-specific marker. Colocalization was defined as r > 0.5. The percentage overlapping markers is the percentage of organelles that had r > 0.5. n > 28 cells for each marker analyzed in at least 2 independent experiments (for EBAG9-GFP and anti-GFP, rmax = 0.81; for anti-GM130 and -CD8a, rmin = 0).
Figure 8. Activation-dependent intracellular redistribution of EBAG9 in CTLs.

(A) EBAG9 redistributes toward the plasma membrane upon T cell activation. For non-polarized activation via the TCR, EBAG9-GFP–transduced T cells were cultured on plate-bound anti-CD3ε and anti-CD28 mAbs (+CD3) or left untreated (–CD3). Cells were fixed and stained with an anti-CD8a antibody without permeabilization. Colocalization between EBAG9 and CD8a was quantified by calculating the Pearson’s correlation coefficient. At least 10 cells were analyzed in 2 independent experiments. Scale bar: 10 μm. (B) EBAG9 moves to the immunological synapse upon polarized stimulation of CTLs. CTLs were generated and transfected with EBAG9-GFP as described in Methods. CTLs were mixed with CD3/CD28-precoated Dynabeads, incubated for 5 minutes at 37°C, plated on coverslips, and incubated for another 30 minutes before fixation and permeabilization. Lytic granules were stained with anti–cathepsin D antibody (red). Representative images of non-conjugated and non-polarized T cells are shown in the top row. Polarized T cells with conjugated beads are shown in bottom 3 rows. Data are from 1 experiment with CTLs from 3 animals. At least 20 conjugates were analyzed (r = –0.10 ± 0.18). Scale bars: 5 μm. Insets have additional magnifications of 2- to 3-fold.
To investigate the redistribution of EBAG9-GFP upon formation of the immunological synapse, we made use of anti-CD3/CD28 mAb–coated microbeads, which acted as surrogate APCs (Figure 8B). Granule polarization to the contact site between T cell and CD3/CD28-coated microbeads was assessed by anti–cathepsin D staining. Whereas in nonpolarized CTLs cathepsin D–positive organelles were distributed clearly separate from the perinuclear staining pattern of EBAG9-GFP, upon polarization EBAG9-GFP was relocated toward the synapse. The EBAG9-GFP–positive organelles surrounded the cathepsin D–containing granules, but a fusion between both compartments was rarely seen (r = –0.10 ± 0.18). Taken together, since many organelles including early endosomes cluster in the perinuclear region adjacent to the Golgi complex, the calculated colocalization with GM130 might indicate an association with several organelles in close proximity. However, the data exclude the localization of EBAG9-GFP to preformed secretory lysosomes in resting T cells. This is in agreement with our previous experiments in epithelial and neuronal cell lines, in which we used confocal microscopy and electron microscopy (12, 37).
EBAG9 interacts with the adaptor protein γ2-adaptin.
To gain mechanistic insight into the cellular function of EBAG9, we scored for novel interaction partners employing the yeast 2-hybrid system. Screening of a human lymph node library yielded 3 interacting clones, BLOC-1, subunit 2 (BLOS2), Snapin, and γ2-adaptin (Table 1). BLOS2 and Snapin were shown to be subunits of the BLOC-1 complex, a multisubunit protein complex essential for endosomal-lysosomal sorting and lysosome-related organelle formation (17). In coimmunoprecipitation experiments from cell lines (data not shown), we failed to verify a physical interaction among EBAG9, BLOS2, and Snapin (data not shown).
Table 1 .
Yeast 2-hybrid screening

To assess whether EBAG9 and the novel interaction partner γ2-adaptin also associate in eukaryotic cells, we performed pulldown experiments with ear domain of γ2-adaptin (γ2-ear) GST fusion protein studies (Figure 9, A and B). A selective interaction between EBAG9-GFP and the ear domain of γ2-adaptin was observed in binding assays using recombinant fusion proteins. In contrast, the ear domain of γ1-adaptin failed to bind EBAG9-GFP (Figure 9A). The association was further confirmed by coimmunoprecipitation of endogenous γ2-adaptin (but not γ1-adaptin) and EBAG9 in stably transfected MDA-MB435-EBAG9-GFP cells (Figure 10B). Next, using truncated GFP-fusion proteins of EBAG9, the relevant domain for binding to the γ2-adaptin ear domain was mapped to aa 30–175, a region of the EBAG9 protein linking the membrane proximal domain (aa 8–27) and the predicted coiled-coil domain (aa 176–206) (Figure 9B and Figure 10A). In confocal microscopy analysis, we examined the subcellular localization of EBAG9-GFP and γ2-adaptin in Jurkat T cells. Consistent with the pulldown and coimmunoprecipitation experiments, we found significant overlap between EBAG9-GFP and γ2-adaptin in perinuclear structures (mean r = 0.469; Figure 9C). Collectively, these data indicate that EBAG9 interacted physically with γ2-adaptin.
Figure 9. EBAG9 interacts with γ2-adaptin in vitro.

(A) Jurkat cells (2 × 106) were transiently transfected with EBAG9-GFP and solubilized in Triton X-100–containing lysis buffer. Cell lysate was incubated with the indicated immobilized GST-fusion protein, and bound EBAG9-GFP was analyzed by SDS-PAGE, followed by immunoblotting with biotinylated anti-GFP antibody. An aliquot (1/6 volume of the reaction mixture) of the cell lysates was immunoblotted for expression of EBAG9-GFP (input). (B) The membrane proximal attachment domain (MA) alone (aa 8–27) and the coiled-coil domain (CC; aa 176–206) were dispensable for the interaction of EBAG9-GFP with the ear domain of γ2-adaptin. MDA-MB435 cells (2 × 106) transfected with EBAG9-GFP (WT) or deletion mutants (MA, ΔCC, ΔMA, ΔMAΔCC) were lysed in Triton X-100–containing lysis buffer, and GST pulldowns were performed as described in A. Input was 1/6 volume of the reaction mixture. To control for an equal loading of GST proteins, membranes were stained with Ponceau (input GST). At right is a schematic representation of EBAG9-GFP and its deletion mutants.
Figure 10. Coimmunoprecipitation of EBAG9 and γ2-adaptin in vivo.

(A) GFP or EBAG9-GFP stably expressing MDA cells were transiently transfected with γ2-adaptin–HA. Cells were solubilized in Triton X-100 lysis buffer and subjected to immunoprecipitation with anti-GFP mAb. Coimmunoprecipitated γ2-adaptin–HA was detected by immunoblotting with an anti-HA mAb. EBAG9-GFP and GFP were detected with biotinylated anti-GFP antibody (input GFP). Input was 1/100 volume of the reaction mixture. Lane 1, MDA-MB435-EBAG9-GFP; lane 2, MDA-MB435-GFP plus γ2-adaptin–HA; lane 3, MDA-MB435-EBAG9-GFP plus γ2-adaptin–HA. (B) Stably transfected MDA-MB435-EBAG9-GFP and GFP-transfected control cells (1 × 107) were solubilized in Triton X-100–containing lysis buffer and subjected to immunoprecipitation with anti-GFP mAb. Coimmunoprecipitated endogenous γ2-adaptin was detected in immunoblotting with a γ2-adaptin–specific antibody. EBAG9-GFP and GFP were detected with biotinylated anti-GFP antibody (input GFP). (C) Colocalization of EBAG9-GFP and γ2-adaptin in a perinuclear localization. Jurkat cells were cotransfected with EBAG9-GFP and γ2-adaptin–HA. Cells were fixed, permeabilized, and then stained with anti-HA mAb. Images were analyzed by confocal microscopy, and colocalization was assessed by calculating the Pearson’s correlation coefficient r of at least 30 cells analyzed in 2 independent experiments (mean r = 0.469). For EBAG9-GFP and anti-GM130, mean r = 0.63 (for EBAG9-GFP and anti-GFP, rmax = 0.95). Scale bar: 5 μm.
EBAG9 deficiency affects the endosomal-lysosomal trafficking pathway.
The association of EBAG9 with the adaptor molecule γ2-adaptin might indicate a modulatory function of EBAG9 on the trafficking route toward the endosomal or lysosomal compartment. To assess the consequences of EBAG9 deletion on this section of the secretory pathway, immunoblot analysis of Ebag9+/+ and Ebag9–/– CD8+ total cell lysates was performed. Comparable expression levels were observed of several SNARE and SNARE-associated molecules (SNAP23; VAMP7; Snapin; syntaxin 4, 7, 8, and 13; synaptotagmin VII), which are involved in lytic granule release at the plasma membrane or localized to the endosomal-lysosomal pathway (Figure 11A). Thus, EBAG9 deletion did not result in any apparent compensatory changes in the expression of those proteins.
Figure 11. Redistribution of the endosomal vesicle marker syntaxin 7.

(A) Identical expression levels of SNARE and SNARE-associated proteins in WT and EBAG9-deficient CTLs. Equal amounts of CD8+ T cell extracts were analyzed by SDS-PAGE, followed by immunoblotting with the indicated antibodies. Synaptotagmin VII occurred in several splice variants. One representative experiment of 9 animals analyzed (per group) is shown. (B) A postnuclear supernatant from 8 × 107 to 1 × 108 Ebag9+/+ or Ebag9–/– CTLs (day 6–7) was centrifuged at 100,000 g for 45 minutes to obtain a microsomal fraction. Microsomes were further resolved on a continuous OptiPrep gradient. Fractions (0.75 ml) were collected from the top of each gradient, and proteins were precipitated with TCA. Samples were resolved by SDS-PAGE and analyzed by immunoblot using polyclonal antibodies as indicated. Data are representative of 3 experiments performed, with pooled CTLs from 9 mice per group. (C) Protein marker distribution was quantified by densitometric scanning and expressed as percentage of the total amount present in the microsomal pellet.
To examine whether endogenous SNARE molecules were associated with distinct membrane compartments in Ebag9+/+ and Ebag9–/– CD8+ T cells, a microsomal membrane fraction was further separated by Optiprep gradient centrifugation and analyzed in conjunction with immunoblotting. Whereas we found no significant difference in the distribution of the plasma membrane SNARE molecules syntaxin 4 or SNAP23 (data not shown) or in the distribution of the recycling endosome marker syntaxin 13, the late endosome marker syntaxin 7 was present in lighter membrane fractions in KO CD8+ T lymphocytes compared with WT T cells (Figure 11, B and C). To validate the resolution capacity of the Optiprep gradient, enzymatic activity of the conventional lysosome marker β-hexosaminidase and the Golgi complex marker GM130 was determined. Peak activity for the lysosomal marker was seen in fractions 6–8, and GM130 exhibited major peaks in fractions 3–5 in both genotypes (Supplemental Figure 5).
To study the potential role of EBAG9 in the process of granule biogenesis and maturation, we examined the morphology of the lytic granule compartment by electron microscopy. CTLs were labeled overnight (for 18 hours) with the endocytic tracer horseradish peroxidase to load lytic granules. Mature lytic granules, characterized by their typical electron-dense cores, were analyzed and measured by randomly selecting 90 cells from independent sections and mice (n = 4 per group) (Figure 12A). Relative size distribution revealed a statistically significant shift toward smaller granule diameters in EBAG9-deficient CTLs (Figure 11B; P < 0.005, ANOVA). The diameters of lytic granules decreased on average from 1,637 nm (± 35.7 SEM) in WT CTLs to 1,461 nm (± 31.2 SEM) in Ebag9–/– CTLs, corresponding to a 30% volume decrease (P < 0.001, Student’s t test). These results indicate that EBAG9 modulated the ultimate generation of secretory granules in CTLs.
Figure 12. Altered size distribution of secretory lysosomes in EBAG9-deficient CTLs.

(A) CTLs (day 6–7) obtained from MLR were labeled overnight with horseradish peroxidase to load the lytic granules. Cells were fixed and then DAB stained. Ultrathin sections were analyzed by electron microscopy. Two representative images of Ebag9+/+ and Ebag9–/– CTLs are shown. Original magnification, ×2,000. (B) The diameter of each DAB-stained lytic granule was measured and is expressed as relative frequency distribution. For WT CTLs, 281 lytic granules were examined, and for EBAG9-deficient CTLs, 356 lytic granules were examined. Four different animals per group were analyzed (shown are mean values).
Formation of the immunological synapse remains unaffected in Ebag9–/– CTLs.
To investigate the formation of the immunological synapse, we made use of anti-CD3 mAb–coated microbeads. We analyzed the polarization of the lytic granule marker perforin to the site of contact between T cell and microbeads by confocal microscopy analysis (Figure 13, A and B). In this assay, granule polarization to the contact site between T cell and CD3-coated microbeads was first assessed in differential interference contrast images, which allowed the identification of T cell–bead conjugates showing clear cell-bead contact. Whereas at the 0-minute time point we observed almost no focused localization of perforin with beads at the contact site, there was clear localization after 10 minutes, and localization was even more focused at the 30-minute time point. In EBAG9-deficient CTLs, the lytic granules polarized toward the immunological synapse at the same rate as the granules from WT CTLs.
Figure 13. EBAG9 deletion does not affect formation of the immunological synapse.

(A) CTLs obtained from MLR (day 6–7) were mixed with the anti-CD3–coated latex beads and incubated for 0, 10, or 30 minutes at 37°C. Conjugates were plated on coverslips, fixed, and permeabilized. Perforin was stained with a biotinylated anti-perforin antibody. Polarization of perforin toward the contact site of CTLs and microbeads was assessed by confocal microscopy and quantified by random selection of conjugates. Those conjugates showing a distinct perforin immunofluorescence at the T cell–bead contact site were considered polarized. Beads are visible in Brightfield images as opaque round structures. One representative image of WT CTLs is shown. Scale bars: 5 μm. (B) Bars show statistical analysis from 3 independent experiments with at least 100 cells evaluated. Data represent mean ± SD.
To obtain further proof for the mobilization of cytolytic granules to the immunological synapse, anti–cathepsin D staining was used as an alternative marker (38). Furthermore, to address potential kinetic differences in conjugate formation, association between T cells and CD3/CD28-coated microbeads was allowed to form for only 15 minutes, followed by the analysis of conjugate formation and granule reorientation. Whereas unconjugated CD8+ cells displayed discrete, punctate staining of intracellular vesicles containing cathepsin D in a nonpolarized pattern, effector T cells conjugated to beads polarized cathepsin D toward the T cell–bead interface in a clearly distinguishable pattern (Supplemental Figure 6). Among the conjugates analyzed (n = 30 for each genotype), comparable numbers of conjugates showed a tight polarization of lytic granules toward the immunological synapse. In addition, simultaneous detection of lytic granule reorientation and localization of the signaling zone molecule Lck was performed in a separate experiment. CTLs were allowed to form conjugates for 15 minutes, followed by anti–granzyme B and anti-Lck staining. Synapse formation for bead-conjugated T cells was assessed by the reorientation of both markers. Clusters of coalesced granzyme B–stained lytic granules could be identified exclusively in conjugated T cells, surrounded by Lck staining (Supplemental Figure 7B). This pattern differed markedly from unconjugated, nonpolarized T cells in which granules were scattered at the cell periphery (Supplemental Figure 7A). Consistent with the shorter length of conjugate formation (15 minutes), conjugated T cells in rare cases contained single, nonpolarized lytic granules. Among the conjugates analyzed (n = 10 for each group), similar numbers of granules showed a tight polarization of the majority of granules at the synapse. We conclude that EBAG9 is not involved in the secretion process at the plasma membrane itself, but rather acts upstream through regulation of trafficking steps and maturation of secretory lysosomes.
Altered distribution of a lysosomal membrane marker and lysosomal hydrolases in Ebag9+/+ and Ebag9–/– CTLs.
Next, we wanted to determine whether loss of EBAG9 influenced the supply of secretory lysosomes with cytolytic effector molecules. We examined the distribution of the late endosome/secretory lysosome membrane marker Lamp1 and the intraluminal hydrolase granzyme B in CD8+ T cells of Ebag9+/+ and Ebag9–/– mice by quantitative confocal microscopy analysis. Colocalization of granzyme B and Lamp1 with the Golgi complex marker GM130 or lysosomal marker cathepsin D was quantified by randomly selecting dots of granzyme B or Lamp1 immunofluorescence. Subsequently, the overlap with cathepsin D or GM130 was quantified by calculating the Pearson’s correlation coefficient for each dot. As shown in the histogram of Figure 14A, the Pearson’s correlation coefficient distribution between granzyme B and cathepsin D shifted to larger r values in Ebag9–/– CTLs, reflecting a more efficient sorting of granzyme B to the lysosomal compartment in Ebag9–/– CTLs (mean r = 0.182) compared with Ebag9+/+ CTLs (mean r = 0.059; P < 0.0001). Similar results were obtained for the grade of colocalization between perforin and cathepsin D (data not shown), suggesting that perforin as well as granzyme B undergo a more efficient targeting to secretory lysosomes in Ebag9–/– CTLs.
Figure 14. EBAG9 controls targeting of lysosomal marker molecules toward the secretory lysosome compartment.

Representative immunofluorescence images of Ebag9+/+ and Ebag9–/– CTLs, activated by incubation on anti-CD3e/anti-CD28–coated culture plates (day 4–5). CD8+ cells were purified by negative selection with magnetic beads. Colocalization between the indicated proteins was quantified by calculating r. Image sets to be compared between WT and KO samples were acquired during the same session using the same acquisition settings. Dots of granzyme B or Lamp1 immunofluorescence were randomly selected and set as regions of interest. In this figure legend, n indicates the number of vesicles analyzed in at least 3 independent experiments. (A) Colocalization of granzyme B with the lysosomal marker cathepsin D. Ebag9+/+, mean r = 0.059 (n = 179); Ebag9–/–, mean r = 0.182 (n = 196). The difference between Ebag9+/+ and Ebag9–/– mice was statistically significant (P < 0.0001, Student’s t test). Scale bar: 10 μm. (B) Colocalization of Lamp1 with cathepsin D. Ebag9+/+, mean r = 0.352 (n = 475); Ebag9–/–, mean r = 0.176 (n = 367). The difference between Ebag9+/+ and Ebag9–/– mice was statistically significant (P < 0.0001, Student’s t test). Colocalization is expressed as relative frequency distribution, whereby r was plotted against the frequency of vesicles counted for 1 class of r values.
In contrast, the grade of colocalization between Lamp1 and cathepsin D decreased in Ebag9–/– T cells (Figure 14B; Ebag9+/+, mean r = 0.352; Ebag9–/–, mean r = 0.176; P < 0.0001), indicating a perturbed targeting of Lamp1 to lysosomes in Ebag9–/– CD8+ T cells. Of course, with each lytic granule marker there were also single cells with a high level of colocalization. We measured overlap between granzyme B and GM130 as a control and found almost no overlap, which was comparable in Ebag9+/+ (mean r = –0.026) and Ebag9–/– (mean r = –0.017; P > 0.7) CTLs (Supplemental Figure 8A). Similar results were obtained for the grade of colocalization between Lamp1 and GM130 (Ebag9+/+, mean r = 0.074; Ebag9–/–, mean r = 0.082; P > 0.5; Supplemental Figure 8B).
Deletion of EBAG9 enhances sorting of the lysosomal hydrolase cathepsin D.
To examine the involvement of EBAG9 in the dynamics of protein transport, we determined the intracellular transport rate of newly synthesized proteins. CTLs from WT and EBAG9-deficient mice were pulse-labeled for 5 minutes with [35S] methionine/cysteine, washed, and transferred to chase medium (39). Cells were further incubated on uncoated or CD3/CD28-precoated plates to distinguish constitutive and inducible release. At the time points indicated in Supplemental Figure 9, the amount of labeled protein was determined in the medium. Constitutive discharge of radiolabeled protein was indistinguishable between WT and KO CTLs (data not shown). However, starting 90 minutes after transfer onto CD3/CD28-coated plates, the increase of regulated release was significantly higher in EBAG9-deficient CTLs (Supplemental Figure 9; P ≤ 0.05, Mann-Whitney test). These data suggested that the intracellular transit time of secretory proteins, as modified specifically through TCR activation, is changed in EBAG9-deficient T cells.
To obtain a more specific kinetic description of endosomal-lysosomal protein sorting, we used pulse-chase labeling in combination with subcellular fractionation and cathepsin D–specific immunoprecipitation. Cathepsin D, which is commonly appreciated as a secretory lysosome marker, is synthesized as a 53-kDa precursor form and is proteolytically converted within the endosomal-lysosomal compartment via a 47-kDa intermediate into the mature lysosomal form (31 kDa) (40, 41). A postnuclear supernatant from CTLs was separated in a 2-step procedure to enrich the lysosomal compartment. Distribution of radioactivity changed with time, but corresponding Ebag9+/+ and Ebag9–/– samples (taken after 0 and 100 minutes of chase) exhibited comparable amounts of counts over the gradient (Figure 15C). At the 100-minute chase point, we recovered significantly higher amounts of the lysosomal form of cathepsin D (31 kDa) in fractions 6 and 7 from EBAG9-deficient mice (Figure 15B). Based on densitometric scanning, we calculated the ratios of precursor/intermediate/lysosomal form in fraction 7 at 100 minutes of chase to be 0.9:1:1.1 for Ebag9+/+ and 0.9:1:2.2 for Ebag9–/–. At 0 minutes of chase, we obtained only minor amounts of cathepsin D from this region, but no lysosomal form (Figure 15A). By immunoblot analysis, total amounts and the distribution of granzyme B were not discernible, and peak fractions 6 and 7 overlapped with the recovery of lysosomal cathepsin D from these fractions. Thus we infer that loss of EBAG9 causes enhanced sorting of the precursor form of cathepsin D to secretory lysosomes.
Figure 15. EBAG9 controls intracellular sorting and processing of cathepsin D.

(A and B) CTLs were pulse-labeled with 500 μCi [35S]methionine/cysteine for 15 minutes and chased for 0 minutes (A) and 100 minutes (B). A postnuclear supernatant was enriched for a light microsomal fraction, including lysosomes by centrifugation at 20,000 g for 20 minutes. Pellets were further resolved on a continuous OptiPrep gradient. Fractions (0.9 ml) were collected from the top of each gradient and lysed in an equal volume of 2× NP-40 lysis buffer, and aliquots were immunoprecipitated with a mixture of anti–cathepsin D antibodies. Samples were analyzed by SDS-PAGE. Aliquots of each fraction were precipitated with TCA and analyzed by immunoblot using a polyclonal anti–granzyme B serum. Data are representative of 2 experiments performed, with pooled CTLs from 6 mice per group. (C) Distribution of radioactivity along the gradients is shown.
Discussion
The results presented here identify EBAG9 as an important player in the control of secretory lysosome release in CTLs. We not only present what we believe is a novel molecular component of the regulated secretory pathway within the immune system, but also provide evidence for a systemic immunosurveillance function of EBAG9 through genetic deletion in mice.
Apart from the genetic analysis of rare human disease-causing mutations and their corresponding murine mutant strains (3, 4), little is known about the proteins that mediate protein sorting into and fusion of lytic granules with the plasma membrane in CTL. In Ebag9–/– mice, we obtained an enhanced release of granzyme A when CTLs were triggered with anti-CD3 antibodies. This observation is consistent with the effect of EBAG9 upon overexpression in PC12 cells, since loss of EBAG9 might lower an inhibitory step in Ca2+-dependent regulated exocytosis (12). In contrast, the capacity of EBAG9 to regulate specifically the granzyme A pathway, but not CTLA4 targeting, is in agreement with results from Ashen mice, which have a loss of function in the Rab27a gene (42). The enhanced in vitro release of granzyme A translates into a substantially increased capacity of Ebag9–/– CTLs to kill allogeneic target cells. Since the frequency of allogeneic CTLs might confound the interpretation of this result, we additionally conducted an antigen-specific in vivo kill assay (32). CTL cytolytic capacity against syngeneic splenocytes that carry the dominant Tag peptide IV was assessed in primary and memory responses. In agreement with our in vitro results, Ebag9–/– mice killed their targets more efficiently than did Ebag9+/+ animals.
Since effector cell frequencies could affect the interpretation of the cytotoxicity assays (43, 44), we determined numbers of antigen-specific CTLs during the priming and effector phases using Kb:Tag peptide dimer staining. Furthermore, we addressed the priming capacity of KO and WT DCs in vitro and in vivo, but a difference between the genotypes was not seen. In conjunction with the comparable proliferation rate of EBAG9-deficient OT-I cells, we suggest that the magnitude of the cytolytic activity in EBAG9-deficient animals is due to an increased secretion of lytic granule content.
A contribution of NK cells to the elimination of peptide-pulsed splenocytes could be excluded, thus we infer that EBAG9 activity may define a discrete secretory mechanism in CTL, which is not found in NK cells. Furthermore, this finding indicates that the precise composition of the exocytosis machinery might vary between hematologic cell types (2). While NK cells can provide secretory granules upon activation or inhibition of cell surface receptors almost immediately, CTLs require a longer priming and maturation process prior to gaining cytolytic competence (45, 46). Striking differences between CTLs and NK cells include cell surface and lysosomal proteins involved in antigen presentation, capturing, and internalization, which enable NK cells to act as APCs to CD4+ T cells (47). Furthermore, differences between primary NK cells and NK cell lines with regard to protein profiles in secretory lysosomes were observed (48). Thus, it could be envisaged that CTLs and NK cells employ individual vesicular transport pathways to supply secretory lysosomes with cytolytic effector molecules. To our knowledge, a comparative proteomic approach addressing the composition of secretory lysosomes from primary NK cells and defined maturational stages of CTLs has not been demonstrated yet.
The immunological phenotype of EBAG9-deleted mice could be explained mechanistically through the identification of γ2-adaptin as an interaction partner of EBAG9. Despite the structural similarities to the γ1 subunit of the AP1 complex, which itself functions as an adaptor complex for transport carriers between the TGN and endosome, a unique function of γ2-adaptin was identified. To our knowledge, γ2-adaptin–containing AP complexes have not been identified so far. In addition, localization to the cytoplasm and perinuclear structures of the cis-Golgi apparatus as well as localization to CD63+ granules, late endosomes, and multivesicular bodies (MVBs) differed markedly from the localization described for AP1 (7, 8). Evidence for a sorting function within the endosomal-lysosomal sorting pathway came from virus infection models. For HBV maturation it was shown that γ2-adaptin interacted via its ear domain with the viral L-protein, whereas the head domain recruited the core particle (11, 49). Thus, γ2-adaptin may organize HBV assembly platforms on endosomal membranes. Functionally, depletion of γ2-adaptin correlated with diminished HBV virion release without compromising constitutive protein secretion. Unexpectedly, it was also shown that γ2 overexpression had the same effect on HBV and retroviral Gag protein release, which was partially explained by imposing a dominant inhibitory effect on endogenous γ2-adaptin (50). The unique function of γ2-adaptin may rely on its ubiquitin-binding activity, a common feature of monomeric adaptors (e.g., epsin and hepatocyte growth factor–regulated tyrosine kinase substrate), which control ubiquitin-dependent endocytotic sorting processes to the MVB/lysosome. Although differences between cellular physiology and virus infections could be a concern, from the HBV model it was inferred that γ2-adaptin may not act as a typical vesicle-forming adaptor, but functions in connecting HBV production and in the subsequent selection of cargo protein destined for inclusion into coated vesicles (11).
In support of a role in the endosomal/MVB sorting pathway, it was shown that γ2-adaptin depletion impaired the degradation of internalized epidermal growth factor and resulted in defective MVB morphology in liver and HeLa cells (51). More specifically, γ2-adaptin depletion induced an endosomal enlargement. Conversely, in EBAG9-deleted CTLs we obtained reduced secretory lysosome volumes, indicating that EBAG9 could act as a negative modulator on the endosomal transport route. In this scenario, EBAG9 inhibits γ2-adaptin cargo recruitment and sorting functions. As a potential consequence of this association, EBAG9 could restrain the upregulated sorting to the secretory lysosome compartment in CTLs. The absence of EBAG9 would allow enhanced activity of γ2-adaptin in these processes instead.
An interaction between EBAG9 and γ2-adaptin, as revealed by yeast 2-hybrid screening, was confirmed by GST–γ2-adaptin pulldown and coimmunoprecipitation studies. In the pulldown assay, the ear domain was sufficient to recruit EBAG9-GFP, which is consistent with the role of this domain to bind other accessory molecules (rabaptin-5, γ-synergin, enthoprotin, and NECAP1) (52). Using truncated versions of EBAG9, we identified a sequence between aa 30 and aa 175 as an interaction domain sufficient for γ2-ear domain binding. This sequence was predicted to be largely unstructured and therefore accessible for interactions with γ2-ear domains. In support of a physical interaction, strong colocalization between EBAG9-GFP and γ2-adaptin–HA was seen in a perinuclear region of T cells. This result is in agreement with the colocalization of EBAG9 and the cis-Golgi marker GM130 under resting conditions (12, 37) and with the reported colocalization of γ2-adaptin in epithelial cell lines with the cis-Golgi/intermediate compartment marker Helix pomatia lectin (8). Since it was also shown that γ2-adaptin resides together with the HBV L-protein at the Golgi complex, collectively these data support the notion that EBAG9 and γ2-adaptin interact physically and functionally.
An alternative interpretation of the functional role of EBAG9 in endosomal-lysosomal sorting and lysosome-related organelle formation involves an interaction with the BLOC-1 subunits BLOS2 and Snapin, both of which were retrieved in the yeast 2-hybrid assay. BLOC-1 regulates specific cargo-trafficking steps through interactions with other known trafficking regulators, including AP3 and BLOC-2 (53, 54). Within the secretory pathway, BLOC-1 is required for cargo-specific sorting from early endosomes toward lysosome-related organelles (55). However, a functional association between EBAG9 and the BLOC-1 complex is difficult to reconcile with our data. BLOC-1 defects, as shown in pallidin, muted, and sandy mouse strains, had no functional consequences on CTL cytolytic activity (55). Thus, it seems unlikely that CTL granules mature in the absence of those subunits while still dependent on Snapin and BLOS2. Furthermore, we failed to validate the interaction with Snapin and BLOS2 in coimmunoprecipitation experiments; in particular, we did not obtain evidence for the retrieval of those molecules within the context of an intact BLOC-1 complex.
EBAG9 localization within cytotoxic T cells is dynamic, as shown by the TCR-induced relocalization toward the immunological synapse. A similar activation-dependent redistribution toward the plasma membrane and the immunological synapse has been shown recently for the Wiskott-Aldrich syndrome protein-interacting protein (56), which turned out to be essential for lytic granule release in NK cells. We have previously shown that EBAG9, upon nerve growth factor–induced differentiation in neuroendocrine cells, redistributed toward secretory vesicles in the vicinity of the plasma membrane. Since EBAG9 exhibits a palmitoylation anchor and is subject to phosphorylation, we suggest that these modifications contribute to EBAG9 relocalization in T cells as well (12). The relocalization would also be consistent with the colocalization between EBAG9 and GM130 and the known polarization of the microtubule-organizing center and associated organelles toward the signaling platform at the T cell–target cell contact site. Under activated conditions, such trafficking would position EBAG9-containing structures in proximity to the endosomal-lysosomal compartment. More recently, it could be shown that lytic granule maturation is a stepwise process that involves the hMunc13-4–dependent fusion of Rab11-containing recycling endosomes with Rab27a+ late endosomal vesicles (57). This step is followed by a separate redistribution of these endosomal exocytic vesicles and lysosomal cytotoxic granules toward the immunological synapse. Colocalization between cytotoxic granules, containing perforin, and the assembled Rab11+/Rab27a+ endosomal structures occurred only when the granules were polarized, suggesting that a late step of granule maturation may proceed immediately prior to secretion. Notably, the efficiency of the latter process was strongly dependent on the expression levels of hMunc13-4, with a moderate efficiency in the presence of endogenous hMunc13-4 and a strong correlation when hMunc13-4 was overexpressed. Since EBAG9 in WT CTLs equipped with endogenous hMunc13-4 was found to reside mainly in a synapse compartment separate from polarized cytotoxic granules, these data suggest that the molecule could restrain sorting to the lytic granules from an upstream position.
Polarization of lytic granules toward the immunological synapse was unaltered in Ebag9–/– mice, and hence vesicle docking and tethering were apparently not influenced. Instead, in support of a pivotal function of EBAG9 in endosomal-lysosomal protein sorting, we found that loss of EBAG9 imposed a change in the steady-state distribution of the target SNARE syntaxin 7, which is required for both homotypic late endosome fusion and heterotypic fusion with lysosomes (58, 59). In contrast to syntaxin 7, in our subcellular fractionation experiments we observed no relocalization of the plasma membrane SNARE syntaxin 4 or the recycling endosome marker syntaxin 13.
Confocal microscopy analysis revealed a distinct colocalization pattern of lysosomal marker molecules Lamp1, granzyme B, and cathepsin D in EBAG9-deleted and WT CTLs. This suggested that granzyme B is prone to a more efficient trafficking to lysosomes in EBAG9-deficient CTLs. An important distinction between these markers includes their sorting pathways (6, 60). As an integral lysosomal membrane protein, Lamp1 does not require sorting receptors, since its cytosolic tail interacts directly with multimeric AP proteins, foremost AP3 for lysosomal targeting. In contrast, the acid hydrolases cathepsin D and granzyme B associate with the M6PR for proper delivery from the TGN to endosomes. Of course, the finding of a decreased targeting of Lamp1 to cathepsin D–containing lysosomes in EBAG9-deficient CTLs could be equally interpreted as a decreased targeting of cathepsin D to Lamp1-containing lysosomes. We consider this view to be unlikely, since loss of EBAG9 causes enhanced sorting of the soluble precursor form of cathepsin D to the lysosomal compartment, as shown by pulse-chase and subcellular fractionation analysis. A role for EBAG9 in the proper delivery of secretory lysosome content and lytic granule maturation is further substantiated by an altered vesicle size distribution with smaller granule volumes in EBAG9-deficient CTLs. In agreement with the involvement of EBAG9 in the regulated secretory pathway in CTLs, transit dynamics of secretory proteins upon TCR engagement was found to be enhanced. Taken together, we conclude that EBAG9 tightly cooperates with γ2-adaptin in the sorting of cytolytic effector molecules to secretory lysosomes. Immunologically, this modulation of endosomal-lysosomal trafficking is intimately linked with the cytolytic capacity of activated CTLs.
Finally, estrogen induces gene activation of EBAG9 (22–24). Since estrogen receptor expression on CD4+ and CD8+ T cells has been recorded, EBAG9 would be in a position to modulate the cytolytic T cell responsiveness toward antigenic target cells, depending on the prevalent estrogen levels (61). In view of the suggested immunosurveillance function of CTLs in cancer, the potential therapeutic benefit of estrogen activity inhibition warrants further study.
Methods
Generation of EBAG9-deficient mice.
Genomic Ebag9 DNA isolated from a 129/SV BAC genomic library was introduced into the pTV0 vector (62), and exons 2 to 4 were replaced by a neomycin cassette. Linearized vector was electroporated into E14-1 ES cells. Homologous recombination in ES cells was selected for using G418 and gancyclovir treatment and was verified by Southern blot hybridization. Two independent ES cell clones were expanded and used for microinjection into C57BL/6 blastocysts. Germline-mutated mice were bred to C57BL/6 and BALB/c backgrounds and used in generation of the F6 and F8 mouse strains, with identical results. In addition, Ebag9–/– mice were backcrossed with OT-I TCR transgenic mice and used in generation of the F7 mouse strain. Mice were further genotyped using PCR with following primer pairs: 5′-CGCTACCACTGAGGATGAGGA-3′ and 5′-GAGAAACCCCATCTAGTTCACATGC-3′ for the WT allele; 5′-GGCGCGGTCCCAGGTCCAC-3′ and 5′-GTTTGCCCAGTCATAGCCGAATAGCC-3′ for the mutant allele.
Mice.
C57BL/6 and BALB/c mice were obtained from Charles River Laboratories. TCR transgenic (CD4+) DO11.10 (H-2d haplotype) and OT-II (H-2b haplotype) were bred in our facility and recognized the OVA-derived peptide aa 323–339. OT-I mice (CD8+ TCR transgenic mice) recognized the OVA-derived cognate peptide SIINFEKL (63). All mice were maintained and bred under standard specific pathogen–free conditions. Experimental animal groups were sex and age matched. Animal experiments were reviewed and approved by the Berlin State review board at the Landesamt für Gesundheit und Soziales (registered Berlin LaGeTsi 0331/05).
Cell culture, transfection, and lymphocyte activation.
Jurkat human T cells and P815 cells were cultured in RPMI 1640 medium (PAA Laboratories) with 10% FCS, penicillin-streptomycin, glutamine, and Na-pyruvate (complete RPMI medium). MDA-MB435 cells were transfected using Lipofectamine 2000 (Invitrogen). Generation of stably transfected MDA-MB435 cell lines was done in selection medium containing 700 μg/ml geneticin-sulphate (G-418), and stable clones were maintained in the presence of 400 μg/ml G-418. Jurkat cells were transfected using an Amaxa electroporation system, according to the manufacturer's instructions.
CTLs were derived by activation of 4 × 106 splenocytes/ml from effector strain C57BL/6 (H-2b haplotype) (Ebag9+/+ and Ebag9–/–) with 4 × 106 irradiated (3,000 rads) splenocytes/ml as stimulators from BALB/c (H-2d haplotype) mice in complete RPMI medium supplemented with β-mercaptoethanol. After 3–6 days in culture, CTLs were purified over Biocoll (Biochrom), washed, and used for cytotoxicity and secretion assays as described below. For confocal microscopy analysis, naive CD8+ T cells were stimulated by culturing cells for 3–4 days with plate-bound anti-CD3ε mAb (3 μg/ml) and anti-CD28 mAb (2 μg/ml) in the presence of 50 U/ml IL-2. CD8+ cells were purified by negative selection with magnetic beads (Miltenyi Biotec or Dynal), followed by 1–2 days of culture in the presence of IL-2 and IL-7 (Peprotech; 50 U/ml). Purity of CD8+ cells was usually greater than 90%. For all functional and biochemical assays, comparable activation status of CTLs derived from KO and WT animals were confirmed by CD25 or CD69 expression.
NK cells were separated from spleen cells using anti-DX5 mAb–conjugated MACS microbeads (Miltenyi Biotec) according to the manufacturer’s instructions. DX5+ cells were cultured for 9 days in complete RPMI medium supplemented with IL-2 (1,000 U/ml). Purity was greater than 98%, as determined by flow cytometry (NK-1.1+ or CD49b+).
Antibodies.
A polyclonal rabbit anti-EBAG9 serum was described previously (12). Anti-B220–FITC mAb (clone RA3-6B2), anti-CD3ε mAb (clone 145-2C11), anti-CD3ε–biotin mAb (clone 145-2C11), anti-CD4–FITC mAb (clone H129.19), anti-CD8a–APC mAb (clone 53-6.7), anti-CD8a–FITC mAb (clone 53-6.7), anti-CD16/32 mAb (clone 2.4G2), anti-CD25–biotin mAb (clone 7D4), anti-CD28 mAb (clone 37.51), anti-CD49b (clone DX5), anti–NK-1.1 (clone PK136), anti-CTLA4–PE (clone UC10-4F10-11), anti-GM130 mAb (35), anti–IFN-γ–APC mAb (clone XMG1.2), and anti-Lamp1 mAb (clone 1D4B) were obtained from BD Biosciences. Polyclonal rabbit anti-Snapin, anti-SNAP23, anti–syntaxin 4, anti–syntaxin 7, anti–syntaxin 8, and anti–synaptotagmin VII sera were purchased from Synaptic Systems. Anti-VAMP7 (clone 1C7) was from Novus Biologicals. Anti–granzyme B, anti-granzyme B–Alexa Fluor 647 mAb (clone 16G6), and biotinylated anti-perforin antibody (clone JAW247) were purchased from eBiosciences. Anti-CD3ε–Alexa Fluor 488 mAb (clone 500A2), anti-CD8a–Alexa Fluor 647 mAb (clone 5H10), anti-CD8a–APC (clone CT-CD8a), and KJ1.26-APC were from Caltag Laboratories. Polyclonal goat anti–cathepsin D was obtained from Santa Cruz Biotechnology Inc. and R&D Systems. Polyclonal rabbit anti-Lck was from Abcam, biotinylated anti-GFP antibody was purchased from Vector Laboratories. Anti-Flag mAb (clone M5) was from Sigma-Aldrich. Anti-GFP mAb and anti-HA were from Roche. Cy5-labeled donkey anti-rat antibody, Cy5-conjugated donkey anti-chicken antibody, and streptavidin-Cy5 conjugate were obtained from Jackson ImmunoResearch Laboratories. Alexa Fluor 568–conjugated goat anti-rabbit antibody, goat anti-mouse, or donkey anti-goat antibodies were from Invitrogen. Appropriate isotype controls for flow cytometry were purchased from BD Biosciences or Caltag Laboratories. A TM-β1 antibody was provided by T. Schüler (Charité).
In vitro cytotoxicity assay.
Cytolytic activity of MLR-derived CTLs against allogeneic P815 cells was determined using Cytotox 96 nonradioactive kit (Promega) according to the manufacturer’s instructions. Percent lysis was calculated as 100 × (experimental – effectorspontaneous – targetspontaneous / effectormaximum – targetspontaneous). Cytotoxicity assays were performed in quadruplicate and were repeated at least 8 times. Cytolytic activity of NK cells against YAC-1 cells was determined as described above. Values for target cell lysis are the mean ± SD of quadruplicates.
In vivo cytotoxicity assay.
To investigate CD8+ T cell responses, splenocytes from C57BL/6 mice (1 × 107 cells/ml PBS) were loaded with 1 μM Tag peptide IV (VVYDFLKL; Biosyntan) or left without peptide for 15 minutes at 37°C. Cells were washed twice and labeled with different amounts of CFSE for 15 minutes at 20°C: 1 μM for the peptide-loaded population (CFSEhigh) and 0.1 μM for non-loaded population (CFSElow). Subsequently, cells were washed in complete RPMI medium and twice in PBS. In primary CTL responses, target cells (2 × 107) at a 1:1 ratio were injected i.v. on day 7 into mice that had been previously immunized (s.c. or i.p., on day 0) with the Tag-bearing tumor cell line Co16.113. After 4 hours, spleens of recipient mice were analyzed by flow cytometry. The specific cytolytic activity was calculated as 100 × (% of CFSEhigh cells / % of CFSElow cells) (32). NK cell activity was analyzed by monitoring lysis of H-2d–allogeneic splenocytes according to the procedure described above. Briefly, splenocytes from BALB/c mice (H-2d haplotype) and C57BL/6 (H-2b haplotype) were labeled with different amounts of CFSE and injected at a 1:1 ratio (i.v.) into Ebag9+/+ and Ebag9–/– mice. After 4 hours, recipient splenocytes were analyzed by flow cytometry for the relative survival of the donor allogeneic population (64).
Antigen-specific T cell proliferation assay.
Bone marrow–derived DCs were generated essentially as previously described (65, 66). After 8–9 days of culture, DCs were harvested and incubated either overnight with OVA protein (Sigma-Aldrich) including 0.1–1 μg/ml LPS or for 2 hours with OVA-derived peptide (aa 323–339). In some cases, DCs were stored frozen in liquid nitrogen prior to their usage. Equal quality of the DC preparation obtained from Ebag9+/+ and Ebag9–/– mice was always assessed by flow cytometry using anti-CD11c, CD86, and MHC class II antibodies. For each in vitro proliferation experiment, DCs were matched according to these maturation markers. To obtain responder T cells, T cells from TCR transgenic mice were isolated from splenocytes by negative selection using appropriate combinations of anti-B220, anti-CD8, or anti-CD4 Dynabeads (Dynal) according to the manufacturer’s instructions, followed by CFSE labeling. On average, more than 85% pure TCR transgenic T cells were recovered and used for in vitro proliferation. In the coculture system, 1 × 105 CFSE-labeled T cells were mixed with 1 × 104 DCs in round-bottom, 96-well tissue culture plates for the times indicated in Figure 5 (65).
Cell transfer experiments.
CD8+ and CD4+ spleen cells from OT-I, OT-II, or DO11.10 TCR transgenic mice were purified by negative selection applying magnetic cell sorting (MACS; Miltenyi Biotec), according to the manufacturer’s instructions. Usually, purity of isolated naive transgenic T cells was greater than 90% (CD62Lhigh). CFSE-labeled TCR transgenic T cells were adoptively transferred i.v. in Ebag9+/+ or Ebag9–/– recipient animals. Alternatively, OT-I/Ebag9+/+ and OT-I/Ebag9–/– CD8+Vα2 T cells (CD45.2 congenic mouse strain) were injected i.v. into congenic C57BL/6/LY-5.1 (CD45.1 congenic mouse strain) recipients. One day after adoptive transfer, mice were immunized i.v. or s.c. with OVA protein and CpGs, as specified in the figure legends. To determine proliferation of adoptively transferred T cells, mice were sacrificed 2–3 days after immunization. Splenocytes or inguinal lymph node cells were prepared, and cells were stained with anti-clonotypic TCR or anti-CD45.2, -CD4, or -CD8 antibodies. CFSE dilution was determined by flow cytometry using a FACSCalibur or a FACSCanto II cytometer with CellQuest or FACSDiva and FloJo software, respectively (BD Biosciences). Figures were assembled in FreeHand 10 software (Adobe).
H-2Kb:Ig dimer staining, intracellular IFN-γ staining and flow cytometric analysis.
Single-cell suspensions of spleen cells were prepared and restimulated for 4 days at 2 × 106 cells/ml with 1 μg/ml Tag epitope IV peptide in complete RPMI medium supplemented with 10 U/ml IL-2, essentially as previously described (32). For dimer staining, cells were incubated for 1 hour with Tag peptide–loaded H-2Kb:Ig dimers (BD Biosciences) and PE-labeled anti-mouse IgG1 secondary antibody according to the manufacturer’s instructions. Cells were stained simultaneously with APC-conjugated anti-CD8a mAb. For intracellular IFN-γ staining, cells were restimulated for 4 hours with 1 μg/ml Tag peptide in the presence of Golgi-Plug (1:1,000; BD Biosciences). Then, cells were prepared and stained as previously described (67). Analysis was performed by flow cytometry.
L. monocytogenes infection.
Mice were infected with the L. monocytogenes strain EGD essentially as previously described (67). Briefly, 5 × 103 bacteria in a volume of 200 μl PBS were injected into a lateral tail vein. For determination of bacterial burdens in liver, mice were killed, livers were homogenized in PBS, serial dilutions of homogenates were plated on PALCAM agar supplemented with selective antibiotics (Merck), and colonies were counted after 48 hours of incubation at 30°C.
Yeast 2-hybrid screen.
A full-length human EBAG9 cDNA was used for screening a human lymph node cDNA library (BD Biosciences) expressed in AH109 yeast cells according to the MATCHMAKER GAL4 2-hybrid system 3 (BD Biosciences), essentially as previously described (12).
Plasmids, GST pulldown assays, coimmunoprecipitation, SDS-PAGE, and immunoblotting.
Human γ2-adaptin–HA plasmid was provided by K. Nakayama (University of Tsukuba, Tsukuba, Japan) (7), and the GST-fusion constructs of γ1- and γ2-adaptin (68) were a donation from J.S. Bonifacino (National Institute of Child Health and Human Development, Bethesda, Maryland, USA). For GST fusion protein pulldown assays, cell extracts were prepared from transfected Jurkat or MDA-MB435 cells by using Triton X-100–containing lysis buffer (0.5% Triton X-100, 50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1 mM MgCl2). Then, cell lysates from 2 × 106 cells were incubated with 8 μg of GST fusion protein immobilized on glutathione–Sepharose 4B beads. Beads were washed extensively with the same buffer, and bound protein was analyzed by SDS-PAGE. For coimmunoprecipitation studies, stably transfected MDA-MB435 or transiently transfected Jurkat cells (1 × 107 cells) were solubilized in Triton X-100–containing lysis buffer (see above). Immunoprecipitation, gel electrophoresis, and immunoblotting were performed essentially as previously described (12). CTL lysates from equal number of cells for quantitation of SNARE molecules were generated in RIPA buffer (total number of mice was 9 per group).
Subcellular fractionation.
To resolve the distribution of endosomal-lysosomal SNARE molecules, 8 × 107 to 1 × 108 CTLs (days 6–7) were resuspended in 1.5 ml of homogenization buffer (10 mM Tris-HCl, pH 7.4, 250 mM sucrose, 1 mM EDTA, 0.2 mM PMSF, Roche protease inhibitor tablets) and homogenized on ice with 20 strokes of a Dounce homogenizer with a tight-fitting pestle. A postnuclear supernatant was centrifuged for 45 minutes at 100,000 g to sediment the microsomal fraction. Pellets were resolubilized in homogenization buffer and loaded on a continuous preformed 10%–30% OptiPrep gradient (Axis-Shield). The gradient was centrifuged in a SW-40 rotor at 185,000 g for 3 hours. Fractions were taken from the top, and protein was precipitated with TCA and further analyzed by immunoblotting.
Metabolic labeling, subcellular fractionation and pulse-chase analysis of cathepsin D.
Metabolic labeling of day 6–7 CTLs (8 × 107 to 1 × 108) was done as previously described (12). Cells were suspended in 1.5 ml of homogenization buffer (10 mM Tris-HCl, pH 7.4, 250 mM sucrose, 1 mM EDTA, 0.2 mM PMSF, Roche protease inhibitor tablets) and homogenized on ice with 20 strokes of a Dounce homogenizer with a tight-fitting pestle. A postnuclear supernatant was centrifuged for 20 minutes at 20,000 g to enrich a light mitochondrial and lysosomal fraction. Pellets were resolubilized in homogenization buffer and loaded on a continuous preformed 10%–30% OptiPrep gradient. The gradient was centrifuged in a SW-40 rotor at 52,000 g for 14 hours. Fractions were taken from the top, and an equal volume of 2× NP-40 lysis buffer was added. Distribution of radioactivity was determined using liquid spectrometry after TCA precipitation.
Secretion assay.
CTLs (1 × 105 cells/well; 100 μl/well) derived from MLR were cultured in round-bottom 96-well plates coated with anti-CD3ε mAb (5 μg/ml), or in plates left uncoated. After 4 hours, plates were centrifuged and cell supernatant was assayed for granzyme A activity as described previously (38).
Immunological synapse formation.
Polybead carboxylate latex microspheres (6 μM) were coated with 5 μg anti-CD3 mAb according to the manufacturer’s instructions (Polysciences). In some experiments, Dynabeads (4.5 μm) precoated with CD3/CD28 antibodies (Invitrogen) were used instead (Supplemental Figures 6 and 7). CTLs were mixed with the anti-CD3–coated latex beads, centrifuged at low speed (380 g), and incubated for 5 minutes at 37°C in RPMI without FCS, allowing the formation of conjugates. Subsequently, cells were resuspended in complete RPMI medium and incubated for the indicated times at 37°C. Next, conjugates were plated on poly-L-lysine–coated coverslips, fixed in either acetone or 4% PFA, permeabilized, and stained with a biotinylated anti-perforin antibody and streptavidin–Alexa Fluor 568 conjugate (Invitrogen).
Generation of retroviral vectors and transduction of T cells.
To generate pMP71-mEG, the murine EBAG9 cDNA coding sequence was amplified by PCR from total murine cDNA and cloned into the single Not-I site of pMP71GPRE, thereby creating a mEBAG9-GFP fusion protein. Ecotropic packaging cells GP+E86 were cotransfected with the retroviral vector plasmid pMP71-mEG and pWLneo (Stratagene) to generate ecotropic vector particle–producing cells. Single-cell suspensions of spleens from OVA-specific TCR transgenic (OT-1) C57BL/6 mice were prepared and cultured (2 × 106/ml) for 24 hours in the presence of 1 μg/ml chicken OVA257–264 (SIINFEKL) peptide (Biosyntan) and 10 U/ml IL-2. Stimulated cells were transduced on 2 consecutive days in RetroNectin-coated plates (Takara) (69).
Electron microscopy.
CTLs were labeled overnight with 2 mg/ml horseradish peroxidase (Serva) in complete RPMI medium. Cells were washed 3 times with PBS and fixed in 2% formaldehyde/1% glutaraldehyde for 60 minutes on ice, followed by DAB staining. Lymphocytes were refixed with 2% formaldehyde/1.5% glutaraldehyde in 0.1 M cacodylate buffer for 2 hours and treated with 1% osmium tetroxide/1.5% potassium ferrocyanide in 0.1 M cacodylate buffer for 2 hours. Samples were dehydrated through a graded series of ethanols and embedded in Poly/Bed 812 (Polysciences). Ultrathin sections were stained with uranyl acetate for 3 minutes and examined with a Zeiss EM 910. Digital images were taken with a 1kx1k high-speed slow-scan CCD camera (Proscan) and analySIS 3.2 software.
Confocal image acquisition and correlation analysis.
Activated T cells were grown for at least 4 hours on fibronectin-coated coverslips (Sigma-Aldrich). For activation of the TCR, coverslips were either left untreated or additionally coated with anti-CD3ε mAb (5 μg/ml) and anti-CD28 mAb (3 μg/ml). Cells were rinsed in PBS and fixed in –20°C acetone for 5 minutes, followed by permeabilization with PBS containing 0.05% Triton X-100 for 5 minutes. Next, cells were stained for 2 hours with the antibodies indicated in Figures 7, 8, and 14 (1:100 in PBS with 5% BSA). Bound antibodies were detected with Alexa Fluor 568– or Cy5-conjugated secondary antibodies. Images were acquired with a Zeiss LSM510Meta confocal setup on an Axiovert 200 M inverted microscope equipped with a 3-line laser. Digital images were collected using a ×63 phase contrast plan–apochromat oil objective NA 1.4 or a ×100 DIC oil objective. For all acquisition settings, the main beam splitter was HFT UV/488/453/633, with following parameters for each fluorochrome: FITC and GFP, 488 nm (light wavelength excitation [λexc]) and BP500–530 nm (light wavelength emission [λem]); Alexa Fluor 568, 543 nm (λexc) and BP565–615IR (λem); Cy5 and Alexa Fluor 647, 633 nm (λexc) and LP650 nm (λem). The image acquisition was done sequentially (multi-track) to minimize potential cross-talk between the fluorophores, and images were processed using LSM Examiner 3.2 and LSM Browser software (Zeiss). To better visualize colocalization of proteins, the Cy5/Alexa Fluor 647 were reassigned to green or red colors.
Colocalization was quantified by calculating the Pearson’s correlation coefficient (r), according to the following formula (Ch, channel; values are pixel intensities):
(Equation 1)
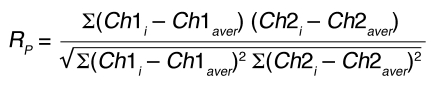
Individual dots were marked as regions of interest using the LSM software, essentially as previously described (57). For each region of interest the software calculated r. Maximal r values (rmax) were calculated for EBAG9-GFP and anti-GFP staining, followed by detection with a Cy5-conjugated secondary antibody. Minimal r values (rmin) were determined for GM130 and CD8a staining. In all experiments the intensity of signals was kept equal. r was plotted against the frequency of vesicles counted for 1 class of r values. The Pearson’s correlation coefficient takes into consideration similarities between shapes, while ignoring the intensities of signals, and thus is applicable when fluorescence intensities are equal (70). Thus, for each experiment comparing WT and gene-deleted mice, average signal intensities between fluorescence detection channels were calculated and showed no significant differences. In addition, in each individual confocal microscopy analysis, average pixel intensities (channel 1 or 2) obtained for Ebag9+/+ and Ebag9–/– CTLs showed indistinguishable values. In agreement with Zinchuk et al. (71) and based on the rmax and rmin values determined, r values greater than 0.5 were considered as good colocalization. All figures were assembled with Corel Draw (Corel).
Statistics.
Results are expressed as mean ± SD or SEM. Data were considered statistically significant for P ≤ 0.05, which was determined using the 2-tailed unpaired Student’s t test or the Mann-Whitney U test, where appropriate. The 1-way ANOVA test was used for the calculation of frequency distributions in electron microscopy images.
Supplementary Material
Acknowledgments
We would like to thank G. Willimsky, E. Vasyutina, and C. Birchmeier (Max-Delbrück-Center) for technical advice. We thank Kerstin Gerlach and Katja Scholz for expert technical assistance and T. Kammertöns, T. Schüler, and T. Blankenstein for useful comments on the manuscript. This work was funded by grants from the Deutsche Krebshilfe, the DFG, and a MDC-Charité clinical cooperation project to U.E. Höpken and A. Rehm. J. Wolf is a fellow of the Max-Delbrück-Center PhD graduate program.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: AP1, adaptor protein complex 1; BLOC-1, biogenesis of lysosome-related organelles complex-1; BLOS2, BLOC-1 subunit 2; EBAG9, estrogen receptor–binding fragment-associated antigen 9; HBV, hepatitis B virus; MLR, mixed lymphocyte reaction; MVB, multivesicular body; SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor; Tag, T antigen; TGN, trans-Golgi network.
Citation for this article: J. Clin. Invest. 119:2184–2203 (2009). doi:10.1172/JCI37760
See the related Commentary beginning on page 2136.
References
- 1.Russell J.H., Ley T.J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 2.Stinchcombe J., Bossi G., Griffiths G.M. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004;305:55–59. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 3.Stinchcombe J.C., Griffiths G.M. Normal and abnormal secretion by haemopoietic cells. Immunology. 2001;103:10–16. doi: 10.1046/j.1365-2567.2001.01225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voskoboinik I., Smyth M.J., Trapani J.A. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh P., Dahms N.M., Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. Mol. Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 6.Robinson M.S., Bonifacino J.S. Adaptor-related proteins. Curr. Opin. Cell. Biol. 2001;13:444–453. doi: 10.1016/S0955-0674(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 7.Takatsu H., Sakurai M., Shin H.W., Murakami K., Nakayama K. Identification and characterization of novel clathrin adaptor-related proteins. J. Biol. Chem. 1998;273:24693–24700. doi: 10.1074/jbc.273.38.24693. [DOI] [PubMed] [Google Scholar]
- 8.Lewin D.A., et al. Cloning, expression, and localization of a novel gamma-adaptin-like molecule. FEBS Lett. 1998;435:263–268. doi: 10.1016/S0014-5793(98)01083-7. [DOI] [PubMed] [Google Scholar]
- 9.Zizioli D., Meyer C., Guhde G., Saftig P., von Figura K., Schu P. Early embryonic death of mice deficient in gamma-adaptin. J. Biol. Chem. 1999;274:5385–5390. doi: 10.1074/jbc.274.9.5385. [DOI] [PubMed] [Google Scholar]
- 10.Hartmann-Stuhler C., Prange R. Hepatitis B virus large envelope protein interacts with gamma2-adaptin, a clathrin adaptor-related protein. J. Virol. 2001;75:5343–5351. doi: 10.1128/JVI.75.11.5343-5351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rost M., et al. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 2006;281:29297–29308. doi: 10.1074/jbc.M603517200. [DOI] [PubMed] [Google Scholar]
- 12.Rüder C., et al. EBAG9 adds a new layer of control on large dense-core vesicle exocytosis via interaction with snapin. Mol. Biol. Cell. 2005;16:1245–1257. doi: 10.1091/mbc.E04-09-0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ilardi J.M., Mochida S., Sheng Z.H. Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat. Neurosci. 1999;2:119–124. doi: 10.1038/5673. [DOI] [PubMed] [Google Scholar]
- 14.Jahn R., Lang T., Südhof T.C. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/S0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 15.Hong W. Cytotoxic T lymphocyte exocytosis: bring on the SNAREs! Trends Cell Biol. 2005;15:644–650. doi: 10.1016/j.tcb.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Das V., et al. Activation-induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity. 2004;20:577–588. doi: 10.1016/S1074-7613(04)00106-2. [DOI] [PubMed] [Google Scholar]
- 17.Starcevic M., Dell’Angelica E.C. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 2004;279:28393–28401. doi: 10.1074/jbc.M402513200. [DOI] [PubMed] [Google Scholar]
- 18.Tsuneizumi M., et al. Overrepresentation of the EBAG9 gene at 8q23 associated with early-stage breast cancers. Clin. Cancer Res. 2001;7:3526–3532. [PubMed] [Google Scholar]
- 19.Takahashi S., et al. EBAG9/RCAS1 expression and its prognostic significance in prostatic cancer. Int. J. Cancer. 2003;106:310–315. doi: 10.1002/ijc.11205. [DOI] [PubMed] [Google Scholar]
- 20.Ogushi T., et al. Estrogen receptor-binding fragment-associated antigen 9 is a tumor-promoting and prognostic factor for renal cell carcinoma. Cancer Res. 2005;65:3700–3706. doi: 10.1158/0008-5472.CAN-04-3497. [DOI] [PubMed] [Google Scholar]
- 21.Nakashima M., Sonoda K., Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat. Med. 1999;5:938–942. doi: 10.1038/11383. [DOI] [PubMed] [Google Scholar]
- 22.Park K.J., Krishnan V., O’Malley B.W., Yamamoto Y., Gaynor R.B. Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchiya F., et al. Molecular cloning and characterization of mouse EBAG9, homolog of a human cancer associated surface antigen: expression and regulation by estrogen. Biochem. Biophys. Res. Commun. 2001;284:2–10. doi: 10.1006/bbrc.2001.4892. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe T., et al. Isolation of estrogen-responsive genes with a CpG island library. Mol. Cell. Biol. 1998;18:442–449. doi: 10.1128/mcb.18.1.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsson S., et al. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 26.Pauklin S., Sernandez I.V., Bachmann G., Ramiro A.R., Petersen-Mahrt S.K. Estrogen directly activates AID transcription and function. J. Exp. Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyubchenko T.A., Wurth G.A., Zweifach A. Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity. 2001;15:847–859. doi: 10.1016/S1074-7613(01)00233-3. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima K., Nakashima M., Sonoda K., Kikuchi M., Watanabe T. Expression of RCAS1 and FasL in human trophoblasts and uterine glands during pregnancy: the possible role in immune privilege. Clin. Exp. Immunol. 2001;123:481–486. doi: 10.1046/j.1365-2249.2001.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsushima T., et al. Receptor binding cancer antigen expressed on SiSo cells, a novel regulator of apoptosis of erythroid progenitor cells. Blood. 2001;98:313–321. doi: 10.1182/blood.V98.2.313. [DOI] [PubMed] [Google Scholar]
- 30.Fortier A.H., Nacy C.A., Sitkovsky M.V. Similar molecular requirements for antigen receptor-triggered secretion of interferon and granule enzymes by cytolytic T lymphocytes. Cell. Immunol. 1989;124:64–76. doi: 10.1016/0008-8749(89)90112-3. [DOI] [PubMed] [Google Scholar]
- 31.Catalfamo M., et al. Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TcR stimulation. Immunity. 2004;20:219–230. doi: 10.1016/S1074-7613(04)00027-5. [DOI] [PubMed] [Google Scholar]
- 32.Willimsky G., Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 33.Lanzavecchia A., Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/S0092-8674(01)00455-X. [DOI] [PubMed] [Google Scholar]
- 34.Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 35.Cooper M.A., Fehniger T.A., Fuchs A., Colonna M., Caligiuri M.A. NK cell and DC interactions. Trends Immunol. 2004;25:47–52. doi: 10.1016/j.it.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 36.Bevan M.J. Cross-priming. Nat. Immunol. 2006;7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 37.Reimer T.A., et al. Reevaluation of the 22-1-1 antibody and its putative antigen, EBAG9/RCAS1, as a tumor marker. BMC Cancer. 2005;5:47. doi: 10.1186/1471-2407-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stinchcombe J.C., et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J. Cell Biol. 2001;152:825–834. doi: 10.1083/jcb.152.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riedel D., et al. Rab3D is not required for exocrine exocytosis but for maintenance of normally sized secretory granules. Mol. Cell. Biol. 2002;22:6487–6497. doi: 10.1128/MCB.22.18.6487-6497.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mardones G.A., et al. The trans-Golgi network accessory protein p56 promotes long-range movement of GGA/clathrin-containing transport carriers and lysosomal enzyme sorting. Mol. Biol. Cell. 2007;18:3486–3501. doi: 10.1091/mbc.E07-02-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gieselmann V., Pohlmann R., Hasilik A., Von Figura K. Biosynthesis and transport of cathepsin D in cultured human fibroblasts. J. Cell Biol. 1983;97:1–5. doi: 10.1083/jcb.97.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haddad E.K., Wu X., Hammer J.A., Henkart P.A. Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J. Cell Biol. 2001;152:835–842. doi: 10.1083/jcb.152.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prlic M., Williams M.A., Bevan M.J. Requirements for CD8 T-cell priming, memory generation and maintenance. Curr. Opin. Immunol. 2007;19:315–319. doi: 10.1016/j.coi.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Gourley T.S., Wherry E.J., Masopust D., Ahmed R. Generation and maintenance of immunological memory. Semin. Immunol. 2004;16:323–333. doi: 10.1016/j.smim.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Krzewski K., Strominger J.L. The killer’s kiss: the many functions of NK cell immunological synapses. Curr. Opin. Cell Biol. 2008;20:597–605. doi: 10.1016/j.ceb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shresta S., Pham C.T., Thomas D.A., Graubert T.A., Ley T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998;10:581–587. doi: 10.1016/S0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- 47.Hanna J., et al. Novel APC-like properties of human NK cells directly regulate T cell activation. J. Clin. Invest. 2004;114:1612–1623. doi: 10.1172/JCI22787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Schmidt H., et al. 2-D DIGE analyses of enriched secretory lysosomes reveal heterogeneous profiles of functionally relevant proteins in leukemic and activated human NK cells. Proteomics. 2008;8:2911–2925. doi: 10.1002/pmic.200800170. [DOI] [PubMed] [Google Scholar]
- 49.Haucke V., Wenk M.R., Chapman E.R., Farsad K., De Camilli P. Dual interaction of synaptotagmin with mu2- and alpha-adaptin facilitates clathrin-coated pit nucleation. EMBO J. 2000;19:6011–6019. doi: 10.1093/emboj/19.22.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert C., Doring T., Prange R. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 2007;81:9050–9060. doi: 10.1128/JVI.00479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rost M., Doring T., Prange R. Gamma 2-Adaptin, a ubiquitin-interacting adaptor, is a substrate to coupled ubiquitination by the ubiquitin ligase Nedd4 and functions in the endosomal pathway. J. Biol. Chem. 2008;283:32119–32130. doi: 10.1074/jbc.M802632200. [DOI] [PubMed] [Google Scholar]
- 52.Mattera R., Ritter B., Sidhu S.S., McPherson P.S., Bonifacino J.S. Definition of the consensus motif recognized by gamma-adaptin ear domains. J. Biol. Chem. 2004;279:8018–8028. doi: 10.1074/jbc.M311873200. [DOI] [PubMed] [Google Scholar]
- 53.Salazar G., et al. BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol. Biol. Cell. 2006;17:4014–4026. doi: 10.1091/mbc.E06-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Pietro S.M., Dell’Angelica E.C. The cell biology of Hermansky-Pudlak syndrome: recent advances. Traffic. 2005;6:525–533. doi: 10.1111/j.1600-0854.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 55.Setty S.R., et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell. 2007;18:768–780. doi: 10.1091/mbc.E06-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krzewski K., Chen X., Strominger J.L. WIP is essential for lytic granule polarization and NK cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2568–2573. doi: 10.1073/pnas.0711593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Menager M.M., et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat. Immunol. 2007;8:257–267. doi: 10.1038/ni1431. [DOI] [PubMed] [Google Scholar]
- 58.Mullock B.M., et al. Syntaxin 7 is localized to late endosome compartments, associates with Vamp 8, and Is required for late endosome-lysosome fusion. Mol. Biol. Cell. 2000;11:3137–3153. doi: 10.1091/mbc.11.9.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pryor P.R., et al. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO Rep. 2004;5:590–595. doi: 10.1038/sj.embor.7400150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peden A.A., Rudge R.E., Lui W.W., Robinson M.S. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J. Cell Biol. 2002;156:327–336. doi: 10.1083/jcb.200107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phiel K.L., Henderson R.A., Adelman S.J., Elloso M.M. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol. Lett. 2005;97:107–113. doi: 10.1016/j.imlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 62.Riethmacher D., Brinkmann V., Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc. Natl. Acad. Sci. U. S. A. 1995;92:855–859. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hogquist K.A., et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 64.Oberg L., et al. Loss or mismatch of MHC class I is sufficient to trigger NK cell-mediated rejection of resting lymphocytes in vivo - role of KARAP/DAP12-dependent and -independent pathways. Eur. J. Immunol. 2004;34:1646–1653. doi: 10.1002/eji.200424913. [DOI] [PubMed] [Google Scholar]
- 65.Hopken U.E., et al. The ratio between dendritic cells and T cells determines the outcome of their encounter: proliferation versus deletion. Eur. J. Immunol. 2005;35:2851–2863. doi: 10.1002/eji.200526298. [DOI] [PubMed] [Google Scholar]
- 66.Schuler T., et al. Generation of tumor-associated cytotoxic T lymphocytes requires interleukin 4 from CD8(+) T cells. J. Exp. Med. 2001;194:1767–1775. doi: 10.1084/jem.194.12.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kursar M., et al. Differential requirements for the chemokine receptor CCR7 in T cell activation during Listeria monocytogenes infection. . J. Exp. Med. 2005;201:1447–1457. doi: 10.1084/jem.20041204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattera R., Arighi C.N., Lodge R., Zerial M., Bonifacino J.S. Divalent interaction of the GGAs with the Rabaptin-5-Rabex-5 complex. EMBO J. 2003;22:78–88. doi: 10.1093/emboj/cdg015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Engels B., et al. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum. Gene Ther. 2003;14:1155–1168. doi: 10.1089/104303403322167993. [DOI] [PubMed] [Google Scholar]
- 70.Zinchuk V., Zinchuk O., Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem. Cytochem. 2007;40:101–111. doi: 10.1267/ahc.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zinchuk, V., and Zinchuk, O. 2008. Quantitative colocalization analysis of confocal fluorescence microscopy images.Curr. Protoc. Cell Biol. Chapter 4, Unit 4.19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.