

Abstract
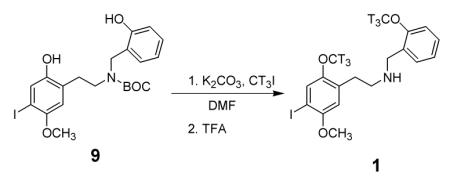
The title compound ([3H]INBMeO) was prepared by an O,O-dimethylation reaction of a t-BOC protected diphenolic precursor using no carrier added tritiated iodomethane in DMF with K2CO3. Removal of the t-BOC protecting group and purification by HPLC afforded an overall yield of 43%, with a radiochemical purity of 99% and specific activity of 164 Ci/mmol. The new radioligand was suitable for labeling human 5-HT2A receptors in two heterologous cell lines and had about 20-fold higher affinity than [3H]ketanserin.
1. Introduction
The serotonin 2A (5-HT2A) receptor is a member of the G protein-coupled receptor (GPCR) superfamily. Of the seven different subtypes of serotonin receptors, the 5-HT2A receptor has been closely linked to complex behaviors. It is expressed at high density within the frontal cortex, but at lower levels in many other brain areas.1-3 It has been implicated in working memory, cognitive processes, affective disorders such as schizophrenia, and mediates the primary effects of hallucinogenic drugs.4-7 In addition, many peripheral tissues also express the 5-HT2A receptor. For example, 5-HT2A receptors are involved in platelet function, and induce contraction within the vasculature.8,9 Thus, radiolabeled agonist and antagonist ligands are of high importance in the pharmacology community to measure density and changes in density of these receptors.
Unfortunately, few ligands are highly specific for 5-HT2A receptors, and those that are, such as M100907,10 are antagonists. It is desirable, therefore, to have available a radiolabeled agonist ligand. Furthermore, it is typical that agonist ligands label only a high-affinity subset of the total receptor population, so any useful radioligand should have high specific activity. One ligand that is widely used for this purpose is [125I]-2,5-dimethoxy-4-iodoamphetamine (([125I]DOI).11,12 Although this ligand does not discriminate between the 5-HT2A, 5-HT2B, and 5-HT2C receptors,13 the high specific activity of 125I makes it very useful for rapid assays of low density receptor populations. This radioligand is presently commercially available, but major drawbacks to the use of [125I]DOI are its short half-life and high cost. In our own laboratory, for example, experiments have often been postponed until the need for results is sufficiently compelling, or until we have delayed a sufficient number of experiments to justify the cost of [125I]DOI. Therefore, a high-specific activity radioligand with a longer half-life is very desirable. Recently, we reported on a series of high affinity N-benzyl phenethylamine ligands for the 5-HT2A receptor.14 The most potent of the ones reported, N-(2-methoxybenzyl)-2,5-dimethoxy-4-iodophenethylamine (INBMeO, 1), had exceptionally high affinity for the 5-HT2A receptor, and was obviously suitable for the introduction of 3-9 tritium atoms by O-methylation. Based on preliminary work, we had concluded that six tritium atoms should provide sufficient radioactivity for a useful ligand. This report describes the synthesis and preliminary characterization of N-(2-[3H-OCH3]-benzyl)-2-[3H-OCH3]-5-methoxy-4-iodophenyl)ethylamine (1).
In order to prepare 1 with six tritium atoms it was planned to bis-methylate the diphenolic precursor 9 using carrier-free tritiated iodomethane. The overall synthesis of this precursor was carried out as shown in Scheme 1.
Scheme 1.
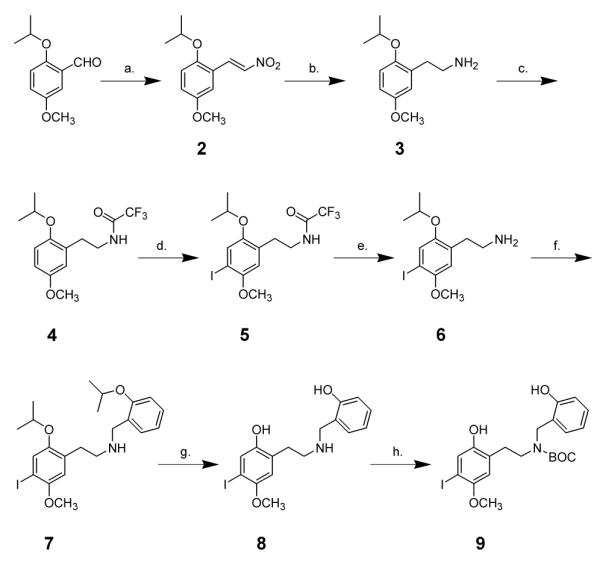
Reagents and conditions: (a) EtNO2, AcOH, reflux; (b) LiAlH4; (c) CF3COOEt, heat; (d) ICl, AcOH; (e) HCl; (f) o-isopropyloxybenzaldehyde, NaCNBH3; (g) BCl3; (h) (tert-BOC)2O.
Preliminary studies of the methylation reaction, utilizing unlabeled iodomethane, demonstrated that treatment of the N-tert-BOC protected bisphenol 9 with sodium hydride in DMF, followed by the addition of a slight excess of iodomethane, afforded intermediate 10a (Scheme 2). Partially methylated intermediates were likely by-products, so HPLC conditions were developed to distinguish between 1, and 9-14. Table 1 shows the retention parameters for the desired product 1, the N-Boc protected bisphenol 9, the O,O-dimethylated-N-Boc-protected intermediate 10, the partially O-methylated N-Boc-protected potential byproducts 11 and 12, and the deprotected analogs 13 and 14.
Scheme 2.

Methylation and deprotection of precursor 9, to produce 1, as well as partially methylated byproducts 13 and 14.
Table 1.
HPLC Retention times for 1 and byproducts obtained in the radiochemical synthesis
| Compound | 9 | 10 | 11 | 12 | 13 | 14 | INBMeO |
|---|---|---|---|---|---|---|---|
|
Retention time (min) |
26.3 | 31.7 | 27.7 | 27.7 | 14.7 | 15.5 | 17.5 |
Conditions for the deprotection of 10 and for isolation of pure INBMeO (1) were also developed. Thus, treatment of a mixture comprised of 10 and small amounts of 11 and 12, with trifluoroacetic acid (TFA) for 15 min provided 1, along with 13 and 14 (derived from 11 and 12). Base treatment failed to remove the phenols 13 and 14. Surprisingly, INBMeO was found to decompose in the presence of 1 N sodium hydroxide.
Unfortunately, when the O-methylation reaction conditions were applied on the scale that would be used in the radiosynthesis, only minute amounts of 10a, 11a, and 12a were produced. Further experiments revealed that the starting material 9 and the monomethylated intermediate 11a decomposed when a solution of 9-12a in DMF was treated with sodium hydride for 15 h at room temperature. A similar experiment using potassium carbonate was more promising, showing the disappearance of intermediate 11a only. Therefore, the methylation reaction using potassium carbonate to replace sodium hydride was examined. These conditions gave 10a, accompanied by small amounts of 11a, 12a, and unreacted 9. Deprotection gave INBMeO (1b) in 54% yield.
A similar reaction mixture was produced in the master radiosynthesis. Thus, treatment of a DMF solution of the bisphenol 9 with an 11-fold excess of iodomethane (100 mCi, specific activity 80 Ci/mmol)) in the presence of potassium carbonate gave primarily 10b (10.6 mCi), accompanied by small amounts of 9, 11b, and 12b. Because we had found that 10a was more stable than 1a, only a portion of 10b (3.7 mCi) was deprotected. The product was purified by preparative HPLC to afford 1.8 mCi (49% yield) of [3H]INBMeO (1b) of nominal specific activity 160 Ci/mmol. A portion of 10b was used to determine experimentally the specific activity, which was found to be 164 ± 1 Ci/mmol. Because high specific activity compounds are known to undergo radiolytically-promoted decomposition, the purity of [3H]INBMeO was monitored over a nine-month period. As shown in Figure 1, [3H]INBMeO is usable for a minimum of 60 days (∼95% purity) and we have carried out successful experiments beyond 150 days (∼90% purity).
Figure 1.
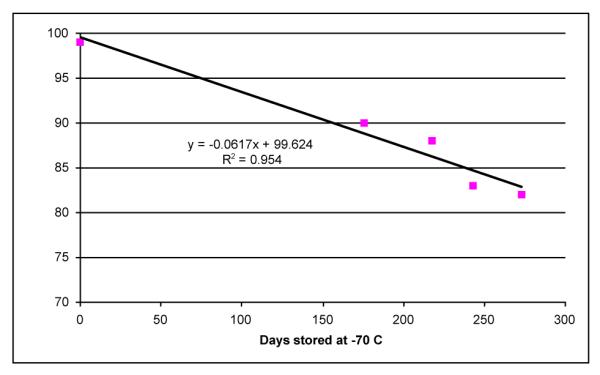
Stability of [3H]INBMeO when stored at -70 °C in EtOH.
We then used the new radioligand [3H]INBMeO 1b to carry out receptor saturation isotherm experiments in two cells lines stably expressing human 5-HT2A receptors. For a comparison, we used [3H]ketanserin (67 Ci/mmol, PerkinElmer). The results of that comparison are presented in Figure 2 and Table 2. The affinity of [3H]INBMeO was about 20-fold higher than that of ketanserin, and it labeled about 60% of the sites labeled by ketanserin. Use of [3H]INBMeO as the radioligand in competition binding assays with four ligands known to have high affinity for the 5-HT2A receptor demonstrated that all of the ligands had comparable affinity, whether [3H]ketanserin or [3H]INBMeO was used (Table 3). Curiously, although INBMeO is an agonist, it labeled approximately 60% of the sites that were labeled by ketanserin.
Figure 2.

Representative Receptor Isotherm using A. [3H]ketanserin and, B. [3H]INBMeo to label 5-HT2A receptors in Hh2A (HEK) and A20 (A549) cells.
Table 2.
A comparison of receptor saturation isotherm data for [3H]ketanserin and [3H]INBMeO
| [3H]ketanserin | [3H]INBMeO | |||
|---|---|---|---|---|
| Cells | Kd (nM) | Bmax (fmol/mg) |
Kd (nM) | Bmax (fmol/mg) |
| A20 | 2.2 ± 0.3 | 890 ± 160 | 0.097 ± 0.02 | 480 ± 70 |
| Hh2A | 2.7 ± 0.2 | 3260 ± 360 | 0.15 ± 0.01 | 2210 ± 100 |
(A20 cells n = 10, Hh2A cells n = 4)
Table 3.
A comparison of competition binding for selected 5-HT2A ligands using [3H]ketanserin and [3H]INBMeO. Ki values given are nM ± SEM.
| Displacing Liganda |
A20 Cells | Hh2A cells | ||
|---|---|---|---|---|
| [3H]ketb | [3H]INBc | [3H]ketb | [3H]INBc | |
| 5-HT | 120 ± 21 | 160 ± 17 | 130 ± 65 | 150 ± 20 |
| Cinanserin | 17 ± 2.8 | 24 ± 2.4 | 13 ± 5.1 | 22 ± 5.9 |
| DOI | 29 ± 5.2 | 28 ± 8 | 13 ± 2.0 | 14 ± 1.2 |
| LSD | 2.6 ± 0.9 | 4.0 ± 0.5 | 3.1 ± 0.7 | 3.5 ± 0.4 |
(n = 3)
[3H]ketanserin
[3H]INBMeO
4. Conclusions
We have prepared a new high specific activity tritiated ligand for the 5-HT2A/2C receptors. It appears to have utility similar to ketanserin, but has about 20-fold higher affinity for 5-HT2A/2C receptors. It may be an economical alternative to the use of [125I]DOI as a radioligand for these receptors.
5. Experimental
5.1 Chemistry — General
The chemicals, reagents and solvents used in the synthesis were inspected and released for use based on visual inspection and conformance of the individual lot with the manufacturer’s Certificate of Analysis. Nuclear magnetic resonance (NMR) spectra were determined on a Bruker Avance 300 MHz NMR spectrometer. HPLC analyses were performed on a dual pump system (Ranin HPXL solvent delivery system) equipped with a Rheodyne injector, a Varian ProStar 325 UV-Vis detector, β-RAM radio-detector, (IN/US and a Systems, Inc.) connected after the UV detector, controlled by Varian Star Workstation software. The columns and solvent systems are given below. The water used in HPLC solvent systems was obtained from a Millipore Milli-Q Plus Ultra-Pure Water System.
Radioactive samples were counted on Tri-Carb Liquid Scintillation Analyzer (Packard Bioscience Company, model: 2100TR) using IN-FLOW 3 as liquid scintillation counting cocktail (IN/US Systems, Inc.).
HPLC conditions for analysis: Phenomenex Synergy Fusion-RP column (3×150 mm, 4 μm), solvent A: [20 mM NaH2PO4-H2O], solvent B: CH3CN, gradient, 0-30 min, solvent B: 20-90%; 30-40 min, solvent B: 90%; flow rate: 0.6 ml/min, UV detector wavelength: 210 nm and 215 nm, β-RAM detector (for analyzing radio-chemical purity of samples).
HPLC conditions for purification of the final radioligand: Phenomenex Synergy Polar-RP column (3×150 mm, 4 μm), solvent A: 80% [20 mM NaH2PO4-H2O]/20% EtOH, solvent B: EtOH, gradient, 0-30 min, solvent B: 30-80%; 30-40 min, solvent B: 80%; flow rate: 0.4 ml/min, UV detector wavelength: 210 nm and 215 nm.
5.1.1 2-(2-Isopropoxy-5-methoxyphenyl)nitroethene 2
A solution of 2-isopropoxy-5-methoxybenzaldehyde15,16 (15.0 g, 72 mmol) and n-butylamine (6.84g, 94 mmol) in 200 mL of toluene was heated at reflux for 45 min with H2O removal using a Dean-Stark apparatus. The solvents were removed by rotary evaporation and the residue was dissolved in nitromethane (75 mL). This solution was stirred efficiently while glacial acetic acid (13.0 g, 216 mmol) was added dropwise. After stirring at room temperature for 12 h, the solvents were removed by rotary evaporation. The residue was dissolved in dichloromethane, washed with H2O (2 × 100 mL), brine (100 mL), and dried over MgSO4. After filtration to remove the drying agent, the filtrate was concentrated by rotary evaporation to afford a yellow solid that was recrystallized from methanol to provide 2-(2-isopropoxy-5-methoxyphenyl)-nitroethane (14.22 g, 83.3%) as bright yellow crystals, mp 65 °C. 1H NMR (300 MHz, CDCl3, δ ppm): 1.41 (s, 6H), 3.81 (s, 3H), 4.60 (m, 1H), 6.94 (m, 3H), 7.86 (d, J = 4.29 Hz, 1H), 8.14 (d, J = 4.29 Hz, 1H); CIMS m/z 238 (M+1), Anal. (C12H15NO4) C, H, N.
5.1.2 2-(2-Isopropoxy-5-methoxyphenyl)ethylamine 3
To lithium aluminum hydride (9.07 g, 238 mmol) in a flame-dried 2 L reaction vessel, was added 600 mL of anhydrous tetrahydrofuran. A solution of 2-(2-isopropoxy-5-methoxyphenyl)-nitroethane (20.0 g, 84 mmol) in 150 mL of THF was then added dropwise over 45 min. The mixture was heated at 65 °C for 4 h, after which TLC analysis showed absence of starting material. After cooling to room temperature the reaction was quenched by the stepwise addition of 50% THF/H2O (10 mL), 15% NaOH solution (10 mL), and H2O (30 mL). The precipitated solids were removed by vacuum filtration and the filtrate was reduced under vacuum to afford a colorless oil. The oil was dissolved in Et2O, washed with H2O (2 × 100 mL), and then extracted into 1M HCl (2 × 200 mL). The acidic extract was washed with Et2O (2 × 75 mL), and then made strongly basic with 5M NaOH. The basic solution was extracted with Et2O (3 × 100 mL), the ether extract was washed with H2O (2 × 75mL), brine (75 mL), dried over MgSO4, and filtered. The filtrate was concentrated by rotary evaporation and the residue was distilled at 97 °C and 0.17 torr, providing 13.32 g (75.5%) of the product as a clear oil. 1H NMR (300 MHz, CDCl3, δ ppm): 1.28 (s, 3H), 1.32 (s, 3H), 1.70 (bs, 2H), 2.73 (t, J = 6.67 Hz, 2H), 2.92 (t, J = 6.67 Hz, 2H), 3.75 (s, 3H), 4.42 (m, 1H), 6.74 (m, 3H); CIMS m/z 210 (M+1), Anal. (C12H19NO2) C,H,N.
5.1.3 N-(2,2,2-Trifluoroacetyl)-2-(2-isopropoxy-5-methoxyphenyl)ethylamine 4
To a solution of the amine 3 (20.0 g, 96 mmol) in anhydrous THF (100 mL) and triethylamine (9.65 g, 96 mmol) in a dried 250 mL round bottomed flask was added ethyl trifluoroacetate (17.73 g, 125 mmol), and the solution was stirred efficiently at room temp overnight. TLC showed complete consumption of the starting material. The solvents were then removed by rotary evaporation and the residue was dissolved in 150 mL of CH2Cl2. The organic solution was washed with H2O (2 × 100 mL), brine (100 mL), dried over MgSO4, and filtered to remove the drying agent. The filtrate was concentrated by rotary evaporation to afford 23.42 g (80.3%) of amide 4 as a waxy white solid. This material could be recrystallized from methanol, distilled (155 °C at 0.2 torr), or, most advantageously, sublimed (30 °C at 0.2 torr), resulting in a crystalline product with a sharp melting point of 35 °C. 1H NMR (300 MHz, CDCl3, δ ppm): 1.32 (s, 3H), 1.34 (s, 3H), 2.87 (t, J = 6.15, 2H), 3.56 (q, J = 8.40, 2H), 3.71 (s, 3H), 4.46(m, 1H), 6.74 (m, 2H), 6.87 (d, 1H), 7.32 (bs, 1H), CIMS m/z 306 (M+1), Anal. (C14H18F3NO3) C,H,N.
5.1.4 N-(2,2,2-Trifluoroacetyl)-2-(4-iodo-2-isopropoxy-5-methoxyphenyl)ethylamine 5
Acetic anhydride (25 mL) was added to a solution of trifluoroacetamide 4 (5.0 g, 16.4 mmol) dissolved in glacial acetic acid (25 mL), and the reaction was cooled to 10 °C. A solution of iodine monochloride (2.92 g, 18 mmol) in acetic acid (5 mL) was added dropwise over 30 min to the vigorously stirred solution. The reaction was stirred for 6 h, and then poured onto ice. Excess saturated aqueous Na2SO3 was added to neutralize any free halogen. A white precipitate was collected from the aqueous mixture by vacuum filtration and washed on the filter with H2O. The aqueous filtrate was extracted with CH2Cl2 (3 × 75 mL). The collected solid was dissolved in CH2Cl2 (125 mL), combined with the organic extracts, washed with H2O (2 × 75mL), brine (75 mL) and dried over MgSO4. After filtration, the solvent was removed by rotary evaporation and the residue was recrystallized from methanol to provide the iodinated trifluoroacetamide 5 (5.59 g, 79.1%) as a white crystalline solid; mp 93 °C. 1H NMR (300 MHz, CDCl3, δ): 1.32 (s, 3H), 1.34 (s, 3H), 2.86 (t, J = 5.89 Hz, 2H), 3.56 (q, J = 9.06 Hz, 2H), 3.85 (s, 3H), 4.49(m, 1H), 6.60 (s, 1H), 7.06 (bs, 1H), 7.33 (s, 1H, ArH), CIMS m/s 432 (M+1), Anal. (C14H17F3INO3) C,H,N.
5.1.5 2-(4-Iodo-2-isopropoxy-5-methoxyphenyl)ethylamine 6
A solution of the trifluoroacetamide 5 (2.50 g, 58 mmol) in EtOH (50 mL) was cooled to 10 °C and aqueous 5M NaOH (10 mL) was added. After stirring overnight at RT H2O (50 mL) was added and the solution was extracted with Et2O (3 × 75 mL). The organic extracts were washed with H20 (3 × 75 mL), brine (75 mL), dried with MgSO4., and filtered. The filtrate was concentrated by rotary evaporation and the resulting golden oil (1.74 g, 89.3%) was used without further purification. An analytical sample was purified by radial TLC (Chromatotron, silica plate, CH2Cl2 as eluent, with nitrogen bubbled through conc NH4OH as the atmosphere). 1H NMR (300 MHz, CDCl3, δ ppm): 1.29 (s, 3H), 1.31 (s, 3H), 1.65 (bs, 2H), 2.72 (t, J = 7.16 Hz, 1H), 2.92 (t, J = 7.16 Hz, 1H), 3.71 (q, J = 10.27 Hz, 2H), 3.82 (s, 3H), 4.42 (m, 1H), 6.65 (s, 1H), 7.25 (s, 1H), CIMS m/z 336(M+1), Anal. (C12H18INO2) C,H,N.
5.1.6 N-(2-isopropoxybenzyl)-2-(4-iodo-2-isopropoxy-5-methoxyphenyl)ethylamine 7
To a solution of the crude amine 6 obtained above (0.99 g, 2.95 mmol) in toluene (30 mL) in a 100 mL round bottomed flask was added 0.629 g (3.83 mmol) of 2-isopropoxy-benzaldehyde,17 and the reaction was heated at reflux for one hour with H2O removal using a Dean-Stark trap. When H2O production ceased, the toluene was removed by rotary evaporation and the residue was dissolved in anhydrous EtOH (50 mL). Sodium borohydride (0.56 g, 15 mmol) was added portion-wise and the reaction was stirred at room temperature for 4 h. The excess NaBH4 was decomposed by transferring the reaction to a 250 mL flask and adding methanol (50 mL). The solution was then heated at reflux until gas evolution ceased. The solvent was removed by rotary evaporation and the residue was dissolved in Et2O (50 mL). The ether solution was extracted with 1M HCl (3 × 75mL). The acidic aqueous extract was washed with Et2O (3 × 25mL), followed by basification with 2M NaOH. The liberated free base was extracted into Et2O (3 × 75 mL), the organic layer was washed with H2O (2 × 50mL), brine (50 mL), dried over MgSO4, and filtered. The filtrate was reduced to dryness by rotary evaporation and the resulting golden oil was purified using radial TLC (Chromatotron, 4 mm silica plate, CH2Cl2 as eluent, and nitrogen bubbled through conc. NH4OH as the atmosphere), providing 0.86 g (60.1%) of the pure amine as a clear oil. 1H NMR (300 MHz, CDCl3, δ ppm): 1.24 (s, 3H), 1.25 (s, 3H), 1.26 (s, 3H), 1.27 (s, 3H), 1.80 (bs, 1H), 2.80 (m, 4H), 3.73 (s, 2H), 3.80 (s, 3H), 6.65 (s, 1H), 6.67-6.90 (m, 3H), 7.13-7.24 (m, 2H), HRMS (ESI): calculated for C22H30INO3: 484.1349. Found: 484.1346
5.1.7 N-(2-hydroxybenzyl)-2-(4-iodo-2-hydroxy-5-methoxyphenyl)ethylamine 8
A solution of amine 7 obtained in the previous step (0.75 g, 1.55 mmol) in anhydrous CH2Cl2 (30 mL) in a 100 mL single neck round bottomed flask was cooled to -78 °C and a solution of 1M BCl3 in CH2Cl2 (4.65 mL, 4.65 mmol) was added dropwise using a syringe. The reaction was allowed to warm to room temperature and was stirred overnight. The flask was cooled in an ice bath and methanol (20 mL) was added dropwise. The solvent was then removed by rotary evaporation. Methanol (30 mL) was added to the residue, the solvent was removed by rotary evaporation, and the procedure was repeated using 25 mL of toluene. The resulting salt was basified with excess 1M NH4OH, and the aqueous solution was extracted with CH2Cl2 (3 × 25mL). The combined organic extract was washed with H2O (25 mL), brine (25 mL), dried with MgSO4, filtered, and the solvent was evaporated under vacuum. The residual free base was purified by radial TLC (Chromatotron, 4 mm silica plate, CH2Cl2 as eluent, with nitrogen bubbled through NH4OH as the atmosphere) to provide 0.442 g (71.4%) of the O,O-dealkylated amine 8 as a golden oil. 1H NMR (300 MHz, CDCl3, δ ppm): 2.80 (t, J = 8.4 Hz, 2H), 2.95 (t, J = 8.4 Hz, 2H), 3.80 (s, 3H), 3.95 (s, 2H), 6.55 (s, 1H), 6.60-6.90 (m, 3H), 7.00-7.25 (m, 2H), HRMS (ESI): Calculated for C16H18INO3: 400.0410. Found: 400.0412.
5.1.8 N-(tert-butoxycarbonyl)-N-(2-hydroxybenzyl)-2-(4-iodo-2-hydroxy-5-methoxyphenyl)ethylamine 9
A solution of amine 8 prepared in the previous step (1.56 g, 3.59 mmol) in 10 mL of dry THF was cooled to 0 °C and a solution of di-tert-butyl dicarbonate (0.784 g, 3.59 mmol) in dry THF (2 mL) was added dropwise using a syringe. The reaction was allowed to warm to room temperature and was stirred for a total of 2 h. The THF was removed by rotary evaporation and the residue was dissolved in 20 mL CH2Cl2. This organic solution was washed with H2O (2 × 25 mL), brine (25 mL), dried with MgSO4, and filtered. The filtrate was concentrated by rotary evaporation, and the residual dark brown oil was purified using radial TLC (Chromatotron, 4 mm silica plate, CH2Cl2 as eluent, and nitrogen bubbled through NH4OH as the atmosphere) to provide 0.984 g (54.9%) of the t-Boc protected amine 9 as a light golden oil. After drying under high vacuum the oil solidified to an easily handled glass. 1H NMR (300 MHz, CDCl3, δ ppm): 1.33 (s, 9H), 2.67 (t, J = 7.05 Hz, 2H), 3.38 (m, 2H), 3.70 (s, 3H), 4.25 (s, 2H), 4.45 (bs, 1H), 6.30 (s, 1H), 6.65-6.90 (m, 2H), 7.00-7.22 (m, 3H), 9.25 (bs, 1H), CIMS m/z 500 (M+1), Anal. C21H26INO5 C,H,N.
5.1.9 N-(tert-butoxycarbonyl)-N-(2-methoxybenzyl)-2-(2,5-dimethoxy-4-iodophenyl)ethylamine 10a
A mixture of the t-Boc-protected amine 9 (123 mg, 0.246 mmol) in anhyd DMF (20 mL), and NaH (60% in mineral oil, 275 mg, 6.89 mmol) was stirred under argon for 45 min. A solution of CH3I (87 mg, 0.617 mmol) in toluene (4 mL) was added and the reaction was stirred overnight. TLC analysis showed total consumption of the starting material. The reaction was carefully quenched with H2O and the solvent was removed by rotary evaporation under high vacuum. The resulting syrupy residue was dissolved in Et2O (50 mL), washed with H2O (2 × 20 mL), brine (20 mL), dried over MgSO4, and filtered. The filtrate was concentrated by rotary evaporation and the residue was purified by radial TLC (Chromatotron, 2 mm silica plate, CH2Cl2 as eluent, with nitrogen bubbled through conc NH4OH as the atmosphere) to provide 81 mg (63.0%) of chromatographically pure O,O-dimethylated t-Boc protected amine 10a as a clear oil. 1H NMR (300 MHz, CDCl3, δ ppm): 1.33 (s, 9H), 2.66 (t, J = 6.93 Hz, 1H), 2.76 (t, J = 6.93 Hz, 1H), 3.25 (t, J = 6.93 Hz, 1H), 3.35 (t, J = 6.93 Hz, 1H), 3.65 (s, 3H), 3.72 (s, 3H), 3.74 (s, 3H), 4.28 (1, 1H), 4.40 (s, 1H), 6.44, 6.60 (2s, 1H), 6.77 (d, 1H), 6.83 (t, 3H), 7.00-7.20 (m, 3H). HRMS (ESI): Calculated for C23H30INO5: 550.1066 (M + Na). Found: 550.1075.
5.1.10 N-(2-methoxybenzyl)-2-(2,5-dimethoxy-4-iodophenyl)ethylamine hydrochloride 1a
The t-BOC protected amine 10a from the previous step (50 mg, 94.8 mmol) was added to 2M HCl in methanol (3 mL), followed by stirring at room temperature for 12 h, a procedure that completely removed the t-BOC protecting group. The solvent was removed by rotary evaporation and the resulting white powder was recrystallized from acetonitrile to provide 43 mg (98%) of the desired deprotected amine (1a) hydrochloride; melting point 167 °C. 1H NMR (300 MHz, CDCl3, δ ppm): 3.11 (bs, 4H), 3.63 (s, 3H), 3.76 (s, 3H), 3.84 (s, 3H), 4.16 (t, J = 4.90 Hz, 2H), 6.79 (s, 1H), 6.84 (d, 1H), 6.95 (t, 1H), 7.28 (s, 1H), 7.33 (t, 1H), 7.39 (d, 1H), 9.45 (bs, 2H), CIMS m/z 428 (M+1).
5.2 Radiochemical Synthesis
5.2.1 N-(tert-butoxycarbonyl)-N-(2-[3H-OCH3]-benzyl)-2-(2-[3H-OCH3]-5-methoxy-4-iodophenyl)-ethylamine ([10b)
A 0.2 mL aliquot (28.8 μg, 0.0577 μmol) was withdrawn from a solution of 3.5 mg (7.2 mmol) of N-(tert-butoxycarbonyl)-N-(2-hydroxybenzyl)-2-(4-iodo-2-hydroxy-5-methoxyphenyl)ethylamine (9) in anhyd DMF (25 mL). The aliquot was added to DMF (10 mL) containing potassium carbonate (30 mg, 0.22 mmol) in a pear-shaped flask and the solution was stirred under nitrogen for 30 min. The flask was then connected to a vacuum manifold and the mixture was de-gassed three times by freezing/evacuating/thawing. A sealed-tube containing [3H]-methyl iodide (100 mCi, 80 Ci/mmol, 1.25 μmol, 11 eq., PerkinElmer Life Sciences) in toluene (2 mL) was connected to the vacuum manifold using a thermometer adapter that was connected to a double-end female adapter connected to an adapter containing P2O5 (429 mg, 3 mmol) and a cotton plug. The sealed-tube was evacuated for 5 min and the [3H]methyl iodide/toluene solution was transferred to the reaction flask through the P2O5 layer over a 2 hour period. The reaction mixture was stirred at RT for an additional 5 min, cooled with liquid nitrogen, and then thawed. The stirring/freezing/thawing process was repeated three times. Finally, the reaction mixture was stirred at RT for 15 hours. The unreacted [3H]methyl iodide was transferred back to the sealed-tube. H2O (2 mL) was added to the reaction mixture followed by stirring at RT for 30 min. The mixture was transferred to a 15 mL centrifuge tube and extracted with Et2O (5 × 2 mL). The combined ether extract was washed with H2O (3 × 2 mL), dried over Na2SO4, then filtered into a scintillation vial, providing crude intermediate (10b) (10.6 mCi, radiochemical purity: 77%).
5.2.2 Specific Activity Determination
A Beer’s Law calibration curve for 10a was generated by injection of aliquots (2, 3, 4, 5, 10, and 15 μL) of a standard solution (6.23 × 10-6 M) of 10a in CH3OH onto a Supelco Ascentis Express C18 HPLC column (2.1 × 100 mm, 2.7 μm), eluting with a gradient of 55-90% B, over 20 min with solvent A: 0.05% NH4OH-H2O and solvent B: CH3CN, flow rate: 0.4 mL/min, and monitoring at 200 nm. Plotting the amount (μmol, Y) versus the UV response (integrated area, X) gave a straight line (Y = 2.0 × 10-11 X, R2 = 0.9984). To determine the radiochemical purity of 10b the percentage of radioactivity associated with 10b (53.8%) was determined by integration of the counted (β-RAM) peaks and counting (scintillation counter) of the collected total effluent from a separate 8 μCi injection to confirm that no radioactivity remained on the column. To determine the specific activity, three injection of 8.0 μCi each (containing 0.538 × 8μCi = 4.3 μCi) were made. The molar amounts of 10b, determined from the calibration curve, were: 2.65 × 10-5, 2.61 × 10-5, and 2.62 × 10-5 μmol, giving values of 163, 165, and 164 Ci/mmol, respectively, for the specific activity of 10b.
5.2.3 N-(2-[3H-OCH3]-benzyl)-2-(2-[3H-OCH3]-5-methoxy-4-iodophenyl)ethylamine ([3H]INBMeO) (1b)
The crude intermediate 10b (4.8 mCi) was stirred in 1:5 TFA:CH2Cl2 (2 mL) at RT for 1 hour. The solvent was evaporated under a stream of nitrogen and the crude produce (3.8 mCi) was purified by preparative HPLC (sample loaded in 0.5 mL 80% [20 mM NaH2PO4-H2O]/20% EtOH). The pure final product, [3H]INBMeO (1b) (1.8 mCi, radiochemical purity: 99%, specific activity: 160 Ci/mmol) gave a single peak with Rt 18.2 min (UV detector), when coinjected with standard INBMeO 1a.
5.3 Pharmacology
5.3.1 Materials
[3H]ketanserin (67 Ci/mmol) was purchased from Perkin-Elmer (Boston, MA). Cinanserin HCl and ketanserin tartrate were purchased from Tocris Bioscience (Ellisville, MO). DOI and LSD were synthesized in our own laboratory. Serotonin HCl and most other reagents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Cell culture reagents were purchased from Fisher Scientific (Pittsburgh, PA) or Invitrogen (Carlsbad, CA).
5.3.2 Cell Culture
A549 cells, stably expressing the human 5-HT2A receptor and the ICAM-1 luciferase reporter gene (A20), were a generous gift from Dr. Ulrike Weyer-Czernilofsky of the Ernst Boehringer Institut, Bender and Co., Vienna, Austria.18 The creation of the HEK cells stably expressing the human 5-HT2A receptor (Hh2A) has been previously reported.19 Both cell lines were grown in culture plates at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s Medium (DMEM) containing 10% dialyzed fetal bovine serum (dFBS, Hyclone, Logan, UT), 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 300 μg/ml G-418 with 75 μg/ml hygromycin B (A20) or 20 μg/ml Zeocin (Hh2A).
5.3.3 Radioligand Assays
Methods for membrane preparation and radioligand assays were performed as previously described.20 Briefly, cells were grown to confluency in 150 mm culture dishes before being lysed on ice with lysis buffer (1 mM HEPES, 2 mM EDTA, pH 7.4). After 10 min, the cells were scraped and centrifuged for 30 min at 30,000 × g at 4 °C. The supernatant was discarded and the pellets were resuspended in receptor binding buffer (RBB, 50 mM Tris, 4 mM MgCl2, pH 7.4) by mechanical homogenization. This suspension was transferred to microcentrifuge tubes and centrifuged for 10 min at 13,000 × g and 4 °C. The supernatant was discarded and the pellets were frozen at -80 °C until use.
All radioligand binding experiments were performed in the presence of 1% bovine serum albumin in receptor binding buffer, and incubated for 60 minutes at 25 °C. For saturation binding experiments, 0.3-10.0 nM [3H]ketanserin and 0.03-4.0 nM [3H]INBMeo was used. Nonspecific binding was defined by 10 μM cinanserin in a total volume of 500 μL. Competition binding experiments were performed with 1.0 - 3.0 nM [3H]- ketanserin or 0.1 - 0.3 nM [3H]NBMeO in a total volume of 500 μL. Binding experiments were terminated by rapid filtration through FB glass fiber harvest plates with ice cold wash buffer (10mM TRIS, 0.9% NaCl) using a 96-well Packard FilterMate cell harvester. One well in each plate was spotted with 10 μL of radioligand stock solution to quantify total radioactivity, and the plates were dried overnight before 30 μL of Packard Microscint-O scintillation fluid was added to each well. Radioactivity per well was determined using a Packard TopCount scintillation counter. The saturation and competition binding curves were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA).
5.3.4 Screening at Other Receptor Types
INBMeO was submitted to the NIMH-sponsored psychoactive drug screening program. The ligand had low affinity for most receptors, with the following reported Ki values (nM) for receptors where it had significant affinity: 5-HT2C (2); 5HT6 (73 ± 12); μ opiate (82 ± 14); H1 (189 ± 35); 5-HT2B (231 ± 73); kappa opiate (288 ± 50); all other Ki values were greater than 500 nM: 5-HT1A, dopamine D3, histamine H2, 5-HT1D, α1A adrenergic, δ opiate, serotonin uptake transporter, 5-HT5A, 5-HT1B, dopamine D2, 5-HT7, dopamine D1, 5-HT3, 5-HT1E, dopamine D5, muscarinic M1-M5, histamine H3, and the dopamine uptake transporter.
Acknowledgments
This work was supported by NIH grant DA02189 from NIDA and by the NIMH under contract NO1-MH-32005.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Burnet PW, Eastwood SL, Lacey K, Harrison PJ. Brain Res. 1995;676:157–168. doi: 10.1016/0006-8993(95)00104-x. [DOI] [PubMed] [Google Scholar]
- 2.Willins DL, Deutch AY, Roth BL. Synapse. 1997;27:79–82. doi: 10.1002/(SICI)1098-2396(199709)27:1<79::AID-SYN8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 3.Hamada S, Senzaki K, Hamaguchi-Hamada K, Tabuchi K, Yamamoto H, Yamamoto T, Yoshikawa S, Okano H, Okado N. Brain Res. Mol. Brain Res. 1998;54:199–211. doi: 10.1016/s0169-328x(97)00322-7. [DOI] [PubMed] [Google Scholar]
- 4.Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Babler A, Vogel H, Hell D. Neuroreport. 1998;9:3897–3902. doi: 10.1097/00001756-199812010-00024. [DOI] [PubMed] [Google Scholar]
- 5.Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK. J. Pharmacol. Exp. Ther. 2000;292:76–87. [PubMed] [Google Scholar]
- 6.Williams GV, Rao SG, Goldman-Rakic PS. J. Neurosci. 2002;22:2843–2854. doi: 10.1523/JNEUROSCI.22-07-02843.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nichols DE. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Nagatomo T, Rashid M, Abul MH, Komiyama T. Pharmacol Ther. 2004;104:59–81. doi: 10.1016/j.pharmthera.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Cogolludo A, Moreno L, Lodi F, Frazziano G, Cobeno L, Tamargo J, Perez-Vizcaino F. Circ. Res. 2006;98:931–938. doi: 10.1161/01.RES.0000216858.04599.e1. [DOI] [PubMed] [Google Scholar]
- 10.Kehne JH, Baron BM, Carr AA, Chaney SF, Elands J, Feldman DJ, Frank RA, van Giersbergen PL, McCloskey TC, Johnson MP, McCarty DR, Poirot M, Senyah Y, Siegel BW, Widmaier C. J Pharmacol Exp Ther. 1996;277:968–981. [PubMed] [Google Scholar]
- 11.Johnson MP, Hoffman AJ, Nichols DE, Mathis CA. Neuropharmacology. 1987;26:1803–1806. doi: 10.1016/0028-3908(87)90138-9. [DOI] [PubMed] [Google Scholar]
- 12.Glennon RA, Seggel MR, Soine WH, Herrick-Davis K, Lyon RA, Titeler M. J Med Chem. 1988;31:5–7. doi: 10.1021/jm00396a003. [DOI] [PubMed] [Google Scholar]
- 13.Nelson DL, Lucaites VL, Wainscott DB, Glennon RA. Naunyn Schmiedebergs Arch. Pharmacol. 1999;359:1–6. doi: 10.1007/pl00005315. [DOI] [PubMed] [Google Scholar]
- 14.Braden MR, Parrish JC, Naylor JC, Nichols DE. Mol Pharmacol. 2006;70:1956–1964. doi: 10.1124/mol.106.028720. [DOI] [PubMed] [Google Scholar]
- 15.Hofsløkken NU, Skattebøl L. Acta Chem Scand. 1999;53:258–262. [Google Scholar]
- 16.Van Veldhuizen JJ, Gillingham GG, Garber SB, Kataoka O, Hoveyda AH. J. Am. Chem. Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228. [DOI] [PubMed] [Google Scholar]
- 17.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Jr., Hoveyda AH. J. Am. Chem. Soc. 1999;121:791–799. [Google Scholar]
- 18.Weyer U, Schafer R, Himmler A, Mayer SK, Burger E, Czernilofsky AP, Stratowa C. Receptors. Channels. 1993;1:193–200. [PubMed] [Google Scholar]
- 19.Parrish JC, Braden MR, Gundy E, Nichols DE. J Neurochem. 2005;95:1575–1584. doi: 10.1111/j.1471-4159.2005.03477.x. [DOI] [PubMed] [Google Scholar]
- 20.Chambers JJ, Parrish JC, Jensen NH, Kurrasch-Orbaugh DM, Marona-Lewicka D, Nichols DE. J Med Chem. 2003;46:3526–3535. doi: 10.1021/jm030064v. [DOI] [PubMed] [Google Scholar]