

Abstract

A de novo preparation of α-keto-imides via ynamide oxidation is described. With a number of alkyne oxidation conditions screened, a highly efficient RuO2-NaIO4 mediated oxidation and a DMDO oxidation have been identified to tolerate a wide range of ynamide types. In addition to accessing a wide variety of α-keto-imides, the RuO2-NaIO4 protocol provides a novel entry to the vicinal tricarbonyl motif via oxidation of push-pull ynamides, and imido acylsilanes from silyl-substituted ynamides. Chemoselective oxidation of ynamides containing olefins can be achieved using DMDO, while the RuO2-NaIO4 protocol is not effective. These studies provide further support for the synthetic utility of ynamides.
Introduction
In our efforts to explore the reactivity of ynamides and to establish their utility as versatile synthons, 1–4 we arrived at α-keto-imides5 [see 1a in Scheme 1], a hitherto underrepresented chemical entity. Literature precedents on the engagement of α-keto-imides towards diastereoselective outcomes,6 as well as a significant body of literature surrounding the structurally related α-keto-amides [1b] and esters [1c],7–11 prompted us to pursue an optimized entry to these molecules. Ynamide preparation via copper-mediated amidation of bromoalkynes2c–i along with the recently reported amidations of terminal acetylenes2b have tolerated a variety of amide types and alkyne functionality,2e,2g,3,12 providing access to a wide variety of ynamides. Building on this versatile synthon, we pursued a highly efficient oxidative protocol transforming a range of ynamide types to access a structurally diverse array of α-keto-imides.
Scheme 1.

Ynamide-Derived α-Keto-Imides.
The limited literature involving α-keto-imides confirms their utility as synthons with the potential of incorporating elements of stereocontrol. Some examples are successes in hetero-Diels Alder reactions,6a an intriguing divergent diastereoselective allylation,6b diastereoselective cyanation,6c and Grignard additions.6d In these reports, preparations of α-keto-imides were accomplished by amidation of α-keto-acids, or by ozonolytic or osmium mediated oxidative cleavage of acrylimides. Although there is a compelling parallel between α-keto-imides 1a and the related α-keto-amides [1b] and esters [1c], the latter has attracted much more synthetic interest as evident from an array of elegant solutions to their construction,8 including seminal work on oxidations of ynamines7a–d that is related to the efforts to be described herein as well as advances in their applications as synthons,9 and a greater understanding of their role as pharmacophores.10
The underlying reactivity of α-keto-amides [1b] and esters [1c] stems from the enhanced electrophilicity of their respective keto carbonyl group. Transformations of these compounds have involved the addition of nucleophiles9a with particular interest placed on attaining stereochemical control through a chirality-inducing element substituted on the oxygen or nitrogen atom. It is noteworthy that α-keto-amides 1b have also been engaged in pinacol-type couplings9b as well as photocyclizations leading to β-lactams.9c In the biological setting, this reactivity is implicated in the engagement of key cysteine and lysine residues important to protease,10a–d lipase,10e and histone deacetylase activity.10f Facile hydrate formation of the α–keto carbonyl, which serves as a transition state mimic of the tetrahedral intermediates associated with amide and ester hydrolysis, has also been associated with enzyme inhibitory activity.10b Given the close analogy between α-keto-imides 1a and α-keto-amides or esters [1b or 1c], access to α-keto-imides should prove to be of significance in organic synthesis and medicinal chemistry. We wish to report here highly efficient oxidative transformations of ynamides to novel α-keto-imides.
Results and Discussion
Our first experiences with α–keto-imides arose from studies aimed at the preparation of benzofurans via a Rh(I)-catalyzed demethylation-cyclization of o-anisole-substituted ynamides such as 2 [Scheme 2].4c While not highly reproducible, α–keto-imide 35 could be obtained in 45% yield by exposure of ynamide 2 to the action of Wilkinson’s catalyst and AgBF4, and an X-ray structure of α–keto-imide 35 was also attained [see Supporting Information]. Although we have not identified the stoichiometric oxidant involved, α–keto-imide formation has been correlated with the use of contaminated/decomposed samples of Wilkinson’s catalyst containing triphenylphosphine oxide.
Scheme 2.

α-Keto-Imide Formation RhCl(PPh3)3-AgBF4.
A more consistent entry to α–keto-imides from ynamides became apparent during our exploration of the dimethyldioxirane (DMDO) oxidation of ynamides 4 [Scheme 3].5 We were interested in probing the possibility of arriving at push-pull carbenes 5 derived from the oxidation of 1 through the rearrangement of presumed oxirenes A. This event was confirmed by the isolation of push-pull carbene-derived cyclopropanes 6. The formation of α–keto-imides 7 was often a competing outcome of these reactions, presumably resulting from a second oxidation of the carbenes 5, although oxidation of oxirenes A to 1,3-dioxabicyclobutanes 8 followed by rearrangement to α–keto-imides 7 cannot be ruled out.
Scheme 3.

DMDO Oxidation of Ynamides.
Persuing the purposeful preparation of α-keto-imides, we examining a number of alkyne oxidation conditions of ynamide 9 [Table 1]. In addition to DMDO oxidation,5,13 α–keto-imide 10 formation could be achieved by the action of ozone,7b–d m-CPBA,14 and RuO4 generated in situ from either RuO2 or RuCl3.7a,15 We also examined the action of I2 in DMSO,16 as well as CuCl2 in DMSO17 at elevated temperatures (150 °C), however, no evidence of α–keto-imide 10 was found, with complete consumption of the starting ynamide 9 [entries 6 and 7]. Oxidation by DMDO18 provided the α–keto-imide in 86% isolated yield [entry 1] with ~5% yield of what appears to be the corresponding α–keto-carboxylic acid accompanied by the free Evans’ oxazolidinone auxiliary. The stability of α–keto-imides in wet acetone suggests that the formation of α–keto-carboxylic acid does not occur by a simple event of hydrolyzing the respective imide motif.5 We are currently still investigating this mechanistic issue. m-CPBA oxidation provided only trace amounts of 10 [entry 3].19 The RuO4 mediated oxidation quickly became the method of choice, yielding 10 in quantitative yields [entries 4 and 5].
Table 1.
Conditions for α-Keto-Imide Formation.
 | ||
|---|---|---|
| entry | conditions | yield [%]a |
| 1 | DMDO, acetone, rt, 2.5 h | 86% |
| 2 | 1) O3, CH2Cl2, −78 °C to rt, 2 h; 2) DMS | 70% |
| 3 | m-CPBA, CH2Cl2, rt, 2.5 h | trace |
| 4 | RuO2•H2O, NaIO4 CH2Cl2, CH3CN, H2O, rt, 4 h | 96% |
| 5 | RuCl3•H2O, NaIO4, CH2Cl2, CH3CN, H2O, rt, 4 h | 99% |
| 6 | I2, DMSO, 150°C, 1 h | ndb |
| 7 | CuCl2, DMSO, 150°C, 24 h | nd |
Isolated yields.
Not detected.
We proceeded to examine the scope of RuO2-NaIO4 mediated oxidation varying ynamide electronic properties [Table 2]. The ynamides examined varied in the nature of the electron withdrawing group on nitrogen, as well as the electron withdrawing or donating ability of the alkyne substituent [entries 4–6]. All oxidations proceeded in moderate to high yield, and the resulting α-keto-imides tolerated routine laboratory handling such as purification and storage. In addition to facile preparation of a variety of α-keto-imides, this method provides ready access to the vicinal tricarbonyl motif as in 20 and 21 [entries 4 and 5] with long standing chemical and biological intrigue,20 and these preparations also showcase the synthetic utility of so called push-pull ynamides 14 and 15. Imido acylsilanes such as 22 [entry 6] should be poised for umpolung chemistry elegantly demonstrated by Johnson’s tandem alkylation-aldolizations of silylglyoxylates.11
Table 2.
RuO2-NaIO4 Mediated Oxidation.
| entry | ynamides | α-keto-imidesa | yield [%]b |
|---|---|---|---|
| 1 |
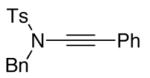 11 11
|
 17 17
|
91 |
| 2 |
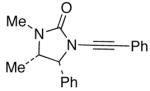 12 12
|
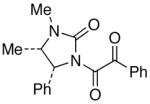 18 18
|
77 |
| 3 |
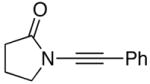 13 13
|
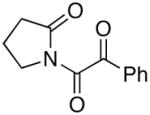 19 19
|
95 |
| 4 |
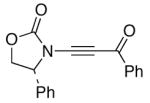 14 14
|
 20 20
|
71 |
| 5 |
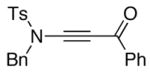 15 15
|
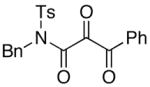 21 21
|
79 |
| 6 |
 16 16
|
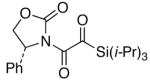 22 22
|
99 |
5 mol% RuO2•H2O, 3 equiv NaIO4, CH2Cl2/CH3CN/H2O, rt, 4 h.
Isolated yields.
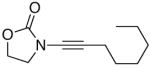
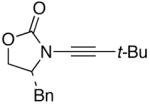
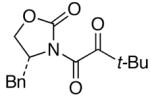
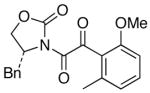
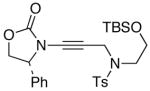
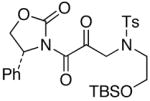
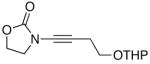
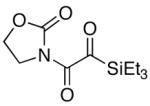
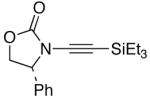
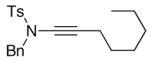
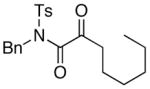
We then examined the preparation of α-keto-imides from ynamides with varied alkyne substitution and compared DMDO and RuO2-NaIO4 conditions [Table 3]. Throughout this series, the RuO2-NaIO4 mediated oxidation provided higher yields of the doubly oxidized products. Increasing steric bulk surrounding the alkyne [from entries 1–4] was well tolerated by both methods, and is accompanied by increased yields under DMDO oxidation conditions (remainder of the material is hydrolyzed). Both the TBS-silyl ether and THP acetal protecting groups, as well as the N-tosyl group are tolerated under reaction conditions [see 27→37 and 28→38 in respective entries 5 and 6]. The yield of α-keto-imide 38 suffers from elimination of the O-THP group [entry 6].21 High yields of triethylsilyl imido acylsilanes 39 and 40 were obtained employing the RuO2-NaIO4 conditions [entries 7 and 8]. The preparation of these less hindered silanes [relative to tri-isopropylsilane 22 in Table 2] was pursued due to their added susceptibility towards engagement of the acylsilane.11 In contrast, the DMDO oxidation of silylated ynamides 29 and 30 did not provide any trace of imido acylsilanes 39 and 40.22 DMDO oxidation of both N-sulfonyl-substituted ynamides 31 and 32 also did not yield the respective α-ketoimides 4123 and 42.22
Table 3.
RuO2-NaIO4 versus DMDO Oxidation.
| entry | ynamides | α-keto-imides | yield [%]a:DMDOb | RuO4c |
|---|---|---|---|---|
| 1 |
 23 23
|
 33 33
|
42d | 81 |
| 2 |
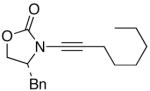 24 24
|
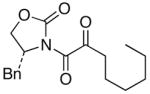 34 34
|
41 | 85 |
| 3 |
 25 25
|
 35 35
|
95 | 95 |
| 4 |
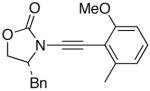 26 26
|
 36 36
|
82 | 79 |
| 5 |
 27 27
|
 37 37
|
69 | 85 |
| 6 |
 28 28
|
 38 38
|
52 | 68 |
| 7 |
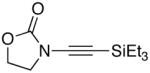 29 29
|
 39 39
|
nde | 90 |
| 8 |
 30 30
|
 40 40
|
nd | 86 |
| 9 |
 31 31
|
 41 41
|
nd | 95 |
| 10 |
 32 32
|
 42 42
|
nd | 87 |
Isolated yields.
4 equiv DMDO, acetone, rt, 2.5 h.
5 mol %RuO2•H2O, 3 equiv NaIO4, CH2Cl2/CH3CN/H2O, rt, 4h.
See reference 5.
Not detected by TLC or 1HNMR.
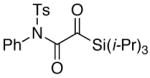
The last class of ynamides examined were those containing a tethered olefin [Table 4]. Ynamide oxidation employing RuO2-NaIO4 led to the formation of a large number of higher polarity products [SiO2 TLC analysis], attributed to alkene dihydroxylation and further cleavage reactions,24 as well as the potential for ketal and hemi-ketalization of the resulting dihydroxy-keto-imides. In these cases, DMDO oxidation proceeded with chemoselective oxidation of the ynamide moeity. All the olefinic motifs in these substrates were stable to DMDO oxidation except for the relatively more electron rich styryl group in 45 [entry 3], which suffered from competitive epoxidation. In addition, for entries 1–4 and 6, we observed a noticeable amount of the free oxazolidinone auxiliary, thereyby suggesting the formation of the corresponding α–keto-carboxylic acid.5 Intramolecular cyclopropanation through intermediate push-pull carbenes 55 [Scheme 3] was not oberved in any of these reactions.
Table 4.
Chemoselectivity in Oxidatively Sensitive Ynamides.
| entry | ynamides | α-keto-imidesa | yield [%]b |
|---|---|---|---|
| 1 |
 43 43
|
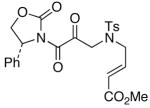 50 50
|
46 |
| 2 |
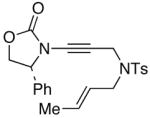 44 44
|
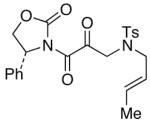 51 51
|
35 |
| 3 |
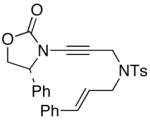 45 45
|
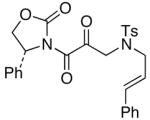 52 52
|
15c |
| 4 |
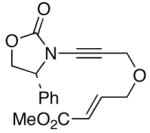 46 46
|
 53 53
|
40 |
| 5 |
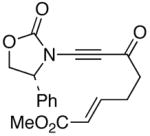 47 47
|
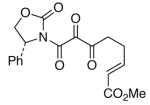 54 54
|
65 |
| 6 |
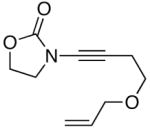 48 48
|
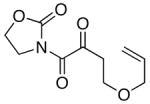 55 55
|
39 |
| 7 |
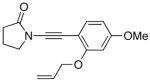 49 49
|
 56 56
|
88 |
4 equiv DMDO, acetone, it, 2.5 h.
Isolated yields.
Also isolated~33% yield of the epoxidized α-keto-imide.
CONCLUSION
We have described here efficient preparations of α-keto-imides through oxidations of ynamides. Both RuO2-NaIO4 and DMDO oxidations tolerate a wide range of ynamide types and substituents. In addition to facile preparation of a variety of α-keto-imides, the RuO2-NaIO4 mediated oxidation provides ready access to the vicinal tricarbonyl motif via the oxidation of push-pull ynamides as well as imido acylsilanes via the oxidation of silylated ynamides. The RuO2-NaIO4 protocol does however lead to complex mixtures during the oxidation of olefin containing ynamides. In these cases, the chemoselective oxidation of such ynamides can be achieved employing DMDO. We believe these protocols provide practical access to a class of building blocks that will be significant in synthesis.
EXPERIMENTAL SECTION
General Procedure For RuO2-NaIO4 Mediated Oxidation of Ynamides
To a solution of ynamide 29 (377.0 mg, 1.670 mmol, 1 equiv) in CH2Cl2 (5 mL) and CH3CN (5 mL) was added NaIO4 (1.07 g, 5.02 mmol, 3 equiv) in H2O (7.5 mL), and then RuO2•H2O (11.2 mg, 0.0840 mmol, 5 mol%). The reaction mixture was stirred vigorously at rt, and the reaction progress was followed by thin layer chromatography. The sides of the reaction flask were rinsed with CH3CN (~1 mL) at 2 h, and the reaction stirred for another 2 h. The reaction mixture was then filtered through a plug of SiO2 rinsing with CH2Cl2. Further purification was accomplished by silica gel flash column chromatography [gradient elution: 17–50% EtOAc in hexanes] to afford α–keto-imide 39 as a bright yellow oil (386.0 mg, 90% yield). Rf = 0.41 [50% EtOAc in hexanes]; 1H NMR (500 MHz, CDCl3) δ 0.83 (q, 6H, J = 8.0 Hz), 1.03 (t, 9H, J = 8.0 Hz), 4.02 (m, 2H), and 4.59 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 2.3, 7.1, 41.1, 64.7, 154.2, 171.5, and 232.8; IR (neat) cm−1 2958w, 2914w, 2879w, 1780s, 1681s, 1642m, 1478w, 1390s, 1361m, 1335m, 1226s, 1117m, 1028m, and 967m; mass spectrum (APCI): m/e (% relative intensity) 258 (10) (M+1)+, 202 (90), 172 (100), and 128 (25); HRMS (ESI) m/e calcd for C11H20NO4Si+ (M+H+) 258.1156, found 258.1146.
General Procedure For DMDO Oxidation of Ynamides
To a solution of ynamide 49 (45.4 mg, 0.167 mmol) in acetone (12 mL) was added DMDO (6.0 mL, 0.11 M in acetone, 4 equiv)18,25 at rt. The resulting reaction mixture was stirred for 2.5 h before it was filtered through Celite™ rinsing with CH2Cl2 and concentrated in vacuo. The crude residue was purification by silica gel flash column chromatography [gradient elution: 17–67% EtOAc in hexanes] to provide α–keto-imide 56 as a yellow crystalline solid (44.6 mg, 88% yield). Rf = 0.33 [67% EtOAc in hexanes]; mp = 152.0 – 153.0 °C; 1H NMR (500 MHz, CDCl3) δ 2.17 (m, 2H), 2.52 (br s, 2H), 3.83 (t, 2H, J = 7.5 Hz), 3.85 (s, 3H), 4.49 (ddd, 2H, J = 6.0, 1.0, 1.0 Hz), 5.33 (ddt, 1H, J = 10.5, 1.0, 1.0 Hz), 5.39 (ddt, 1H, J = 17.5, 1.5, 1.5 Hz), 5.96 (ddt, 1H, J = 17.5, 10.5, 6.0 Hz), 6.39 (d, 1H, J = 2.0 Hz), 6.63 (dd, 1H, J = 9.0, 2.5 Hz), and 8.04 (d, 1H, J = 9.0 Hz); 13C NMR (125 MHz, CDCl3) δ 18.5, 32.1, 43.6, 55.9, 70.1, 99.2, 107.1, 116.4, 119.7, 132.3, 132.8, 161.1, 166.5, 168.1, 175.7, and 186.1; IR (film) cm−1 3079w, 2938w, 2899w, 2852w, 1738m, 1673m, 1656m, 1595s, 1575m, 1504m, 1447m, 1421m, 1363s, 1286m, 1251s, 1231s, 1204s, 1175m, 1115m, 993s, 909m, and 837m; mass spectrum (APCI): m/e (% relative intensity) 304 (65) (M+1)+, and 191 (100); HRMS (MALDI) m/e calcd for C16H17NO5Na+ (M+Na+) 326.0999, found 326.0998.
Supplementary Material
Acknowledgments
Authors thank the NIH [GM066055] and University of Wisconsin for support. We thank Dr. Haibing Song at Nankai University for solving single-crystal X-ray structure.
Footnotes
SUPPORTING INFORMATION AVAILABLE. Experimental procedures, characterization data for all new compounds, 1H NMR, 13C NMR spectra, and X-ray structural data are available free of charge via Internet at http://pubs.acs.org.
References
- 1.For reviews on ynamides, see: Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379.Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei LL. Tetrahedron. 2001;57:7575.For a special issue dedicated to the chemistry of ynamides, see: Tetrahedron-Symposium-In-Print: “Chemistry of Electron-Deficient Ynamines and Ynamides”. Tetrahedron. 2006;62(16)
- 2.For synthesis of ynamides, see: Tracey MR, Hsung RP, Antoline JA, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb Steve M., editor. Chapter 21.4. Georg Thieme Verlag KG; Stuttgart, Germany: 2005. Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833. doi: 10.1021/ja077406x.Sagamanova IK, Kurtz KCM, Hsung RP. Organic Syn. 2007;84:359.Kohnen AL, Dunetz JR, Danheiser RL. Organic Syn. 2007;84:88.Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Tracey MR. J Org Chem. 2006;71:4170. doi: 10.1021/jo060230h.Riddell N, Villeneuve K, Tam W. Org Lett. 2005;7:3681. doi: 10.1021/ol0512841.Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org Lett. 2004;6:1151. doi: 10.1021/ol049827e.Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011. doi: 10.1021/ol035647d.Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J Am Chem Soc. 2003;125:2368. doi: 10.1021/ja021304j.
- 3.For recent references on the chemistry of ynamides, see: Istrate FM, Buzas AK, Jurberg ID, Odabachian Y, Gagosz F. Org Lett. 2008;10:925. doi: 10.1021/ol703077g.Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745.Martínez-Esperón MF, Rodríguez D, Castedo L, Saá C. Tetrahedron. 2008;64:3674.Yavari I, Sabbaghan M, Hosseini N, Hossaini Z. Synlett. 2007;20:3172.Hashimi ASK, Salathe R, Frey W. Synlett. 2007:1763.Rodríguez D, Martínez-Esperón MF, Castedo L, Saá C. Synlett. 2007:1963.Couty S, Meyer C, Cossy J. Synlett. 2007:2819.Tanaka K, Takeishi K. Synthesis. 2007:2920.
- 4.For our recent work on ynamides, see: Yao PY, Zhang Y, Hsung RP, Zhao K. Org Lett. 2008;10 doi: 10.1021/ol801711p. ASAP.Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org Lett. 2008;10:3477. doi: 10.1021/ol801257j.Oppenheimer J, Johnson WL, Tracey MR, Hsung RP, Yao PY, Liu R, Zhao K. Org Lett. 2007;9:2361. doi: 10.1021/ol0707362.Oppenheimer J, Hsung RP, Figueroa R, Johnson WL. Org Lett. 2007;9:3969. doi: 10.1021/ol701692m.You L, Al-Rashid ZF, Figueroa R, Ghosh SK, Li G, Lu T, Hsung RP. Synlett. 2007:1656.Li H, You L, Zhang X, Johnson WL, Figueroa R, Hsung RP. Heterocycles. 2007;74:553.Zhang X, Hsung RP, Li H. Chem Commun. 2007:2420. doi: 10.1039/b701040k.
- 5.For the communication relevant to this manuscript, see: Al-Rashid ZF, Hsung RP. Org Lett. 2008;10:661. doi: 10.1021/ol703083k.
- 6.For preparations and reactions of α-keto-imides, see: Kosior M, Asztemborska M, Jurczak J. Synthesis. 2004;1:87.Chen JH, Venkatesham U, Lee LC, Chen K. Tetrahedron. 2006;62:887. [also see reference 20]Hsu HL, Wu HL, Venkatesham U, Chen KM. J Chinese Chem Soc. 2006;53:449.Raszplewicz K, Sikorska L, Kiegiel K, Jurczak J. Polish J Chem. 2002;76:1595.Kudyba I, Raczko J, Jurczak J. Synthesis. 2004;8:1147. doi: 10.1021/jo0358269.
- 7.For accounts on α-keto-amide formation from RuO4 oxidation of ynamines, see: Müller P, Godoy J. Tetrahedron Lett. 1982;23:3661.For oxidations of ynamines or push-pull ynamines employing ozone and oxygen, see: Schank K, Beck H, Werner F. Helv Chim Acta. 2000;83:1611. this reference includes detailed mechanistic consideration.Schank K, Beck H, Himbert G. Synthesis. 1998:1718.Foote CS, Lin JWP. Tetrahedron Lett. 1968;29:3267.For leading accounts on α-keto-ester formation from oxidation of ynol-ethers, see: Tatlock JH. J Org Chem. 1995;60:6221.Bassignani L, Brandt A, Caciagli V, Re L. J Org Chem. 1978;43:4245.
- 8.For other elegant strategies on α-keto-amide and ester formation, see: Chen J, Cunico RF. J Org Chem. 2004;69:5509. doi: 10.1021/jo040164o.Hua R, Takeda HA, Abe Y, Tanaka M. J Org Chem. 2004;69:974. doi: 10.1021/jo035572r.Chen JJ, Deshpande SV. Tetrahedron Lett. 2003;44:8873.Bolm C, Kasyan A, Heider P, Saladin S, Drauz K, Günther K, Wagner C. Org Lett. 2002;4:2265. doi: 10.1021/ol025911n.Yang Z, Zhang Z, Meanwell NA, Kadow JF, Wang T. Org Lett. 2002;4:1103. doi: 10.1021/ol010297l.Uozumi Y, Arii T, Watanabe T. J Org Chem. 2001;66:5272. doi: 10.1021/jo0156924.Wong MK, Yu CW, Yuen WH, Yang D. J Org Chem. 2001;66:3606. doi: 10.1021/jo0015974.Wasserman HH, Ho WB. J Org Chem. 1994;59:4364.
- 9.For reactions of α-keto-amides and esters, see: Young SW, Kim YH, Hwang JW, Do Y. Chem Commun. 2001:996.Kim SM, Byun IS, Kim YH. Angew Chem Int Ed. 2000;39:728. doi: 10.1002/(sici)1521-3773(20000218)39:4<728::aid-anie728>3.3.co;2-y.Hashizume D, Kogo H, Sekine A, Ohashi Y, Miyamoto H, Toda F. J Chem Soc, Perkin Trans. 1995;2:61.Corey EJ, McCaully RJ, Sachdev HS. J Am Chem Soc. 1970;92:2476. doi: 10.1021/ja00711a045.
- 10.For accounts examining the biological activity of α-keto-amides see: Angelastro MR, Mehdi S, Burkhart JP, Peet NP, Bey P. J Med Chem. 1990;33:11. doi: 10.1021/jm00163a002.Ocain TD, Rich DH. J Med Chem. 1992;35:451. doi: 10.1021/jm00081a005.Li Z, Patil GS, Golubski ZE, Hori H, Tehrani K, Foreman JE, Eveleth DD, Bartus RT, Powers JC. J Med Chem. 1993;36:3472. doi: 10.1021/jm00074a031.Patel DV, Rielly-Gauvin K, Ryono DE, Free CA, Smith SA, Petrillo EW., Jr J Med Chem. 1993;36:2431. doi: 10.1021/jm00069a001.Chiou A, Markidis T, Constantinou-Kokotou V, Verger R, Kokotos G. Org Lett. 2000;2:347. doi: 10.1021/ol991295s.Wada CK, Frey RR, Ji Z, Curtin ML, Garland RB, Holms JH, Li J, Pease LJ, Guo J, Glaser KB, Marcotte PA, Richardson PL, Murphy SS, Bouska JJ, Tapang P, Magoc TJ, Albert DH, Davidsen SK, Michaelides MR. Bioorg Med Chem Lett. 2003;13:3331. doi: 10.1016/s0960-894x(03)00685-1.
- 11.For reactions of silylglyoxylates see: Nicewicz DA, Johnson JS. J Am Chem Soc. 2005;127:6170. doi: 10.1021/ja043884l., also see Supporting Information. Linghu X, Satterfield AD, Johnson JS. J Am Chem Soc. 2006;128:9302. doi: 10.1021/ja062637.
- 12.(a) Kurtz KCM, Hsung RP, Zhang Y. Org Lett. 2006;8:231. doi: 10.1021/ol052487s. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Hsung RP, Zhang X, Huang J, Slafer BW, Davis A. Org Lett. 2005;7:1047. doi: 10.1021/ol0473391. [DOI] [PubMed] [Google Scholar]
- 13.For DMDO-oxidation of alkynes, see: Zeller KP, Kowallik M, Haiss P. Org Biomol Chem. 2005;3:2310. doi: 10.1039/b504296h. this reference includes detailed mechanistic consideration.Murray RW, Singh M. J Org Chem. 1993;58:5076.Curci R, Fiorentino M, Fusco C, Mello R, Ballistreri FP, Failla S, Tommaselli GA. Tetrahedron Lett. 1992;33:7929.
- 14.For accounts of peroxyacid oxidation of acetylenes, see: Stille JK, Whitehurst DD. J Am Chem Soc. 1964;86:4871.McDonald RN, Schwab PA. J Am Chem Soc. 1964;86:4866.
- 15.For lead references on ruthenium mediated alkyne oxidation, see: Zibuck R, Seebach D. Helv Chim Acta. 1988;71:237.Pattenden G, Tankard M, Cherry PC. Tetrahedron Lett. 1993;34:2677.For recent applications see: Semmelhack MF, Campagna SR, Federle MJ, Bassler BL. Org Lett. 2005;7:569. doi: 10.1021/ol047695j.Herrera AJ, Rondón M, Suárez E. J Org Chem. 2008;73:3384. doi: 10.1021/jo702663w.For a recent review, see: Plietker B. Synthesis. 2005;15:2453. this review presents the suggested mechanism for RuO4 mediated alkyne oxidation.
- 16.For an account of I2/DMSO mediated alkyne double oxidation, see: Yusybov MSO, Filimonov VD. Synthesis. 1991:131.
- 17.We attempted this conditions because of an intriguing observation: During the preparation of ynamide 13, an extended reaction time of 24 h (rather than 4 h) employing Stahl’s amidation conditions [see reference 2b] with stoichiometric CuCl2 in DMSO under O2 led to the isolation of ~ 7% yield of a mixture of ynamide 13 and α–keto-imide 19 with a ratio of 1.4:1 [see Table 2 for structures].
- 18.DMDO/acetone solutions were prepared following the procedure reported in: Xiong H, Hsung RP, Berry CR, Rameshkumar C. J Am Chem Soc. 2001;123:7174. doi: 10.1021/ja0108638.Aslo see: Murray RW, Singh M, Marron TG, Pfeifer LA, Roush WR. Org Syn. 1997;74:91.
- 19.Further Baeyer-Villiger type oxidation of 1,2-dicarbonyls is known to occur under ozone and peroxyacid oxidation conditions [see references 7b-d and 14]. Although inconclusive, the presence of byproduct benzoyl and phenyl ester-type aromatic proton resonances in the crude 1H-NMR spectra resulting from the oxidation of ynamide 9 under these conditions suggests this course of action.
- 20.For an excellent review on the chemistry of vicinal tricarbonyls and related systems, see: Wasserman HH, Parr J. Acc Chem Res. 2004;37:687. doi: 10.1021/ar0300221.
-
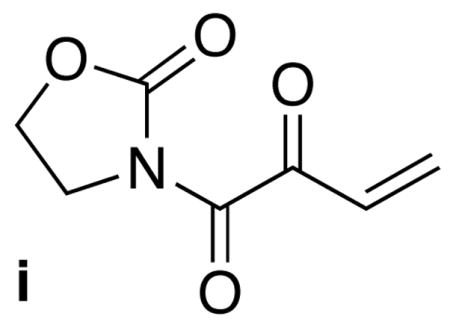
21.Minor amounts of the corresponding enone i apparent by 1H-NMR.
- 22.Silicon d-orbital overlap with the carbene may result a silyl-push-pull carbene [see 5 in Scheme 3 with R = SiR3] that is susceptible to undergo Wolff rearrangement and subsequent transformations through the resulting silyl ketene intermediate. Further studies are underway.
- 23.We observed loss of the N-sulfonyl group during DMDO oxidations of N-sulfonyl-substituted ynamides such as 31 [also observed for 11 shown in Table 2].
- 24.For a lead reference on ruthenium mediated alkene oxidation see: Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936.For a recent application see: Neisius NM, Plietker B. J Org Chem. 2008;73:3218. doi: 10.1021/jo800145x.
- 25.DMDO/acetone concentration determined by 1H-NMR analysis of the crude residue resulting from the reaction (1 h, rt) of a known volume of DMDO/acetone with an excess of thioanisole in Et2O (conc = 0.2 M), comparing the integration values corresponding to the proton peaks belonging to the remaining thioanisole and the resulting methyl phenyl sulfoxide.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.