

Abstract
The MEI-1/MEI-2 microtubule-severing complex, katanin, is required for oocyte meiotic spindle formation and function in C. elegans, but the microtubule-severing activity must be quickly downregulated so that it does not interfere with formation of the first mitotic spindle. Post-meiotic MEI-1 inactivation is accomplished by two parallel protein degradation pathways, one of which requires MEL-26, the substrate-specific adaptor that recruits MEI-1 to a CUL-3 based ubiquitin ligase. Here we address the question of how MEL-26 mediated MEI-1 degradation is triggered only after the completion of MEI-1’s meiotic function. We find that MEL-26 is present only at low levels until the completion of meiosis, after which protein levels increase substantially, likely increasing the post-meiotic degradation of MEI-1. During meiosis, MEL-26 levels are kept low by the action of another type of ubiquitin ligase, which contains CUL-2. However, we find that the low levels of meiotic MEL-26 have a subtle function, acting to moderate MEI-1 activity during meiosis. We also show that MEI-1 is the only essential target for MEL-26, and possibly for the E3 ubiquitin ligase CUL-3, but the upstream ubiquitin ligase activating enzyme RFL-1 has additional essential targets.
Keywords: Meiosis, Protein degradation, Spindle, C. elegans
Introduction
Fertilization triggers a cascade of effects on the oocyte, including maturation, cytoskeletal rearrangements and completion of meiosis (Harris et al., 2006; McCarter et al., 1999; McNally and McNally, 2005). A dramatic change occurs a short time later during the transition from meiosis to mitosis. Meiotic and mitotic spindles form sequentially in the same cytoplasm, but differ considerably in size, morphology, cellular location and the presence of centrosomes only in the mitotic spindle (Albertson, 1984; Schatten, 1994). This transition from one type of spindle to the other necessitates strict regulation of the gene activities unique to each type of spindle: meiotic-specific spindle components must be quickly inactivated prior to mitotic spindle formation and mitotic-specific components must remain inactive until after the meiotic divisions are completed. This problem is particularly acute in the newly fertilized C. elegans embryo where the mitotic spindle forms only about 15 min after the completion of the second meiotic division (Kemphues et al., 1986; Yang et al., 2003). Recently, it has become apparent that rapid turnover of many maternally-supplied proteins is critical during the oocyte to embryo transition [reviewed in Bowerman and Kurz (2006) and DeRenzo and Seydoux (2004)].
The MEI-1/MEI-2 katanin microtubule-severing complex is an example of a protein that is required strictly for meiotic spindle formation in C. elegans. While essential for meiosis, MEI-1/MEI-2 must be inactivated prior to mitosis (Clark-Maguire and Mains, 1994a). During meiosis, MEI-1/MEI-2 are required to generate microtubule fragments that seed microtubule nucleation and later contribute to meiotic spindle shortening (McNally et al., 2006; Srayko et al., 2006) and these proteins are required for translocation of the meiotic spindle to the oocyte cortex (Yang et al., 2003). MEI-1 and MEI-2 localize to the meiotic spindle but disappear prior to mitosis (Clark-Maguire and Mains, 1994a; Srayko et al., 2000). mel-26 encodes an ubiquitin E3 ligase substrate-specific adaptor essential for post-meiotic MEI-1 degradation (Dow and Mains, 1998; Furukawa et al., 2003; Pintard et al., 2003b; Xu et al., 2003). If MEI-1 degradation is blocked, e.g., in the absence of MEL-26 or in the presence of a gain-of-function (gf) mei-1 mutation that prevents MEI-1 binding to MEL-26, MEI-1 microtubule-severing activity is then ectopically expressed during mitosis, leading to small and mis-positioned spindles (Clark-Maguire and Mains, 1994a). The question of why MEL-26 mediated MEI-1 degradation does not begin until the completion of meiosis is the subject of this paper.
Synthesis and degradation of proteins requires precise regulation to control the concentrations of active molecules present in cells at critical times. In eukaryotes, the bulk of protein degradation is performed by the 26S proteasome. Ubiquitination marks proteins for degradation by the proteasome, and ubiquitin addition involves a multienzyme pathway (Glickman and Ciechanover, 2002; Kerscher et al., 2006). The ubiquitin-activating enzyme (E1) forms a high energy thioester bond with ubiquitin, which is then passed to the ubiquitin-conjugating enzyme (E2). Ubiquitin is then transferred to the substrate by an ubiquitin-protein ligase (E3). The Cullin (CUL) family of E3 ubiquitin ligases bind to ubiquitin bound-E2 using the common Rbx subunit while substrate-specificity is provided by variable subunits, which include F-box family members for CUL-1/SCF E3 ligases and BC-box family members for CUL-2/VHL E3 ligases (Kipreos, 2005; Petroski and Deshaies, 2005). CUL-3 based E3-ubiquitin ligases use BTB family members for substrate-specificity, of which MEL-26 was a founding member (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003b;Xu et al., 2003).
There are several levels of regulation for E3 ubiquitin ligase induced protein degradation. The subunits conferring substrate-specificity to CUL-based E3 ligases are themselves subject to autodegradation, and this has been demonstrated for MEL-26 (Luke-Glaser et al., 2005; Pintard et al., 2003b). The association of the F-box substrate adaptors of CUL-1-based E3 ubiquitin ligases with their target substrates stabilizes the F-box against autoubiqui-tination and degradation (Galan and Peter, 1999; Wirbelauer et al., 2000). In addition, CUL-based ubiquitin E3 ligases are themselves conjugated to the NEDD8 type of ubiquitin-like molecule, which activates ubiquitin ligase activity (Parry and Estelle, 2004; Petroski and Deshaies, 2005). Cyclical neddylation/deneddylation is required for activity of C. elegans CUL-3/MEL-26 (Pintard et al., 2003a). Another level of control includes different types of ubiquitin E3 ligases regulating one another (Bashir et al., 2004; Vodermaier, 2004; Wei et al., 2004).
Substrates are often phosphorylated prior to ubiquitination (Glickman and Ciechanover, 2002; Hunter, 2007; Petroski and Deshaies, 2005), and phosphorylation of MEI-1 by the DYRK minibrain kinase MBK-2 is required for timely degradation of MEI-1 (Ming Pang et al., 2004; Pellettieri et al., 2003; Quintin et al., 2003; Stitzel et al., 2006). MBK-2 has additional targets in the oocyte, including the OMA-1/OMA-2 and MEX-5/MEX-6 proteins and components of the germ plasm (DeRenzo et al., 2003; Feng et al., 1999; Nishi and Lin, 2005; Shirayama et al., 2006; Stitzel et al., 2006). However, we recently found that MBK-2 activity is not necessary for MEL-26 induced MEI-1 degradation, and instead MBK-2 and MEL-26 act in parallel pathways that are both required for MEI-1 degradation (Lu and Mains, 2007). MBK-2 mediated degradation appears to be coupled with meiotic exit after which MEL-26 then completes the process of MEI-1 degradation.
The parallel MEL-26 and MBK-2 systems explain how MEI-1 microtubule-severing activity is degraded during mitosis, but this leads to the question of how MEI-1 degradation is delayed until the completion of meiosis. Stitzel et al. (2007) recently showed that the timing of the MBK-2 branch of the MEI-1 degradation pathway is regulated by the degradation of the cortical protein EGG-3 by the anaphase promoting complex (APC) as meiosis proceeds. EGG-3 degradation releases MBK-2 from the cortex, giving it access to cytoplasmic substrates. In this paper, we characterize the temporal regulation of MEL-26/CUL-3 mediated MEI-1 degradation. Immuno-localization indicates that MEL-26 protein does not accumulate to high levels until after meiosis has been completed and accumulation of high levels of MEL-26 is prevented during meiosis by another type of E3 ubiquitin ligase, one that includes CUL-2. However, the low levels of MEL-26 present during meiosis are active and function to moderate MEI-1 during that time, although ectopic high levels of MEL-26, like premature release of cortical MBK-2, are not sufficient to block MEI-1 function. Finally, we show that MEI-1 is the major target of MEL-26, and probably also of CUL-3, while RFL-1, an enzyme in the neddylation pathway that activates ubiquitin ligases, has additional essential targets.
Materials and methods
Strains and culture conditions
C. elegans strains were cultured at 15 °C as described by Brenner (1974). L4 stage animals were upshifted to the appropriate temperatures, and complete broods were scored for hatching as described in Mains et al. (1990b). Between 500 and 5000 embryos from at least four hermaphrodites are reported for each strain. The following alleles were used: LG I, mei-1(ct46 and ct46ct101), mei-2(sb39, ct98 and ct102), mel-26(ct61 and ct61sb4), unc-29(e1072); LG II, emb-27(g48); LG III, tbb-2(sb26), rft-1(or198), cul-2(ek1), pod-1(ye11 and or548), lon-1(e185), dpy-18(e364); LG IV, mbk-2(dd5), dpy-20(e1280). Mutations were often linked to morphological mutations listed above to ease strain construction, and the chromosomal translocation hT2(I;III) (Zetka and Rose, 1992) was used to balance cul-2 and mel-26.
Antibody production
Fragments of mel-26 cDNA were amplified by RT-PCR using Superscript II (Invitrogen) on gravid hermaphrodite poly(A)+ RNA isolated with the FasTrack mRNA Isolation System (Invitrogen). A 366 bp fragment was then PCR amplified with Herculase (Stratagene) and forward (gggatccccgctttgctcattgctgccgat) and reverse (ctgaatt-cattacagtccagatggaggtgg) primers. A 230 bp region was amplified by substituting gggatcctgagaaaacgagctgttacg for the former forward primer. Both fragments were cloned into pGEX-3X (Pharmacia) and transformed into BL21 CodonPlus (DE3)-RP to produce glutathione S-transferase (GST) fusion proteins. Recombinant protein was isolated using a glutathione Sepharose 4B column (Pharmacia Biotech) with EDTA-free protease inhibitor cocktail (Roche) added to the re-suspended bacterial pellet according to the GST Gene Fusion System (Pharmacia Biotech). The larger fusion protein was excised from an SDS-PAGE gel and injected into rats or rabbits (Southern Alberta Cancer Research Institute and the Animal Resource Centre, University of Calgary). The smaller MEL-26 protein was coupled to cyanogen bromide (CNBr)-activated Sepharose (Pharmacia Biotech). Antiserum was affinity purified by passage through an immobilized E. coli lysate affinity column (Pierce) and then through the column containing the smaller MEL-26 fragment before use.
Microscopy
Temperature sensitive (ts) strains were upshifted 18–24 h prior to fixation, except for stains with emb-27, which were upshifted for 2 h to avoid degeneration of embryos. Embryos were fixed with methanol-acetone (Miller and Shakes, 1995) and blocked with either goat and/or donkey 20% normal serum. Rat anti-MEL-26 antibodies (1:50) and mouse anti-α-tubulin monoclonal antibody (Sigma clone M1A, 1:200) were applied for 1 h at 37 °C, indocarbocyanine (Cy3; Jackson ImmunoResearch) donkey anti-rat and fluorescein (FITC, TAGO Inc.) goat anti-mouse (both at 1:100) were incubated at room temperature for 1 h. DNA was then stained with 1 μg/ml 4′,6-diamidine-2′-phenylindole dichloride (DAPI, Roche) for 20–30 s, and specimens were mounted in Slowfade (Molecular Probes). Images were collected by a Hamamatsu Orca ER digital camera using Axiovision 3.0. Z-stacks (1 μm per slice) were taken from a Zeiss Axioplan 2. Images were processed by digital deconvolution using the Axiovision 3.0 constrained iterative algorithm and processed with Adobe Photoshop. Where a uniform black background was desirable, the non-staining regions outside of the embryo have been digitally removed. Meiotic stages were judged based upon the number of DAPI stained polar bodies.
In utero spindle imaging
Wild-type and mei-1(ct46) worms expressing GFP-tubulin were anesthetised, mounted and subjected to time-lapse fluorescence microscopy as described previously (McNally et al., 2006). Images were captured at 15 second intervals with a Photometrics Quantix CCD controlled by IP Lab software using shuttered, attenuated Hg-arc illumination and a Nikon Microphot SA wide field microscope with a 60X Plan Apo 1.4 objective. Spindle length was determined from individual time-lapse images using the Measure Length function of IP Lab Spectrum.
RNAi
Feeding RNAi was done using the library described by Kamath et al. (2003). L4 animals were grown on bacteria induced to produce the appropriate double stranded RNA for 24 h, and were transfered to fresh plates spread with RNA producing bacteria. Broods from the second set of plates were reported.
Results
MEL-26 does not accumulate to high levels until the completion of meiosis
mel-26 encodes a maternally required protein whose mRNA is present predominantly in the gonad of hermaphrodites and in fertilized embryos (Dow and Mains, 1998). MEL-26 acts as the substrate-specific adaptor for the CUL-3 based E3 ubiquitin ligase to down-regulate MEI-1 microtubule-severing activity when meiosis is completed (Clark-Maguire and Mains, 1994a; Furukawa et al., 2003; Pintard et al., 2003b; Xu et al., 2003). However, MEL-26 mediated degradation of MEI-1 must not occur until MEI-1 fulfills its meiotic functions. To address the timing of MEL-26 function, we generated a polyclonal antibody against the C-terminal region of MEL-26 to determine when and where it is present in the newly fertilized embryo.
MEL-26 was detected in a punctate cytoplasmic pattern in the newly fertilized embryo. Levels are low during meiosis I and II (Figs. 1A–D), consistent with the requirement for MEL-26 for viability only following completion of meiosis (Mains et al., 1990b). MEL-26 levels immediately increase when the pronuclei form after meiosis (Figs. 1E, F and 2A), and high levels of MEL-26 were present throughout the early mitotic divisions (Figs. 1G–N). As shown in Fig. 2G, among embryos in meiosis I or meiosis II, we found that a minority of embryos (5/17 and 3/17, respectively), had levels of MEL-26 noticeably higher than that shown in Figs. 1A–D, and none had levels comparable to post-meiotic embryos. In contrast, after completion of meiosis, 28/30 embryos had high levels of MEL-26 staining (Fig. 2G). In multicellular embryos, MEL-26 is enriched at the membranes between cells (Figs. 1K–N and Luke-Glaser et al., 2005). MEL-26 expression remained strong at the 16-cell stage, but decreased to low levels by the 64-cell stage (data not shown).
Fig. 1.

Time course of MEL-26 accumulation in wild-type embryos. The left column shows deconvolved indirect immunofluorescence images of embryos stained with anti-MEL-26 (red) while the right column shows merged images of the same embryos stained with anti-MEL-26 (red), anti-tubulin (green) and DAPI (blue). (A, B) meiosis I. (C, D) Meiosis II, note the first polar body (arrow). (E, F) Pronuclear formation, levels of MEL-26 have increased. (G, H) Pronuclear fusion. (I, J) Anaphase of the first mitotic division. (K, L) Late two cell stage, and (M, N) four cell stage. Note that MEL-26 is enriched at the membrane between cells of multicellular embryos. (O, P) Two cell mel-26(ct61sb4); tbb-2(sb26) shows no MEL-26 staining. The mutation encoded by the allele ct61sb4 results in a truncation N-terminal to the region used to raise the antisera. tbb-2(sb26) suppresses ct61sb4 lethality, restoring normal embryo morphology. Note that the low levels of MEL-26 present at meiosis I and II (A–D) are above the background seen in (O, P).
Fig. 2.

Mutations affecting MEL-26 temporal expression. Merged images are of MEL-26 (red), tubulin (green) and DAPI (blue). (A) Low levels of MEL-26 are apparent in the meiotic stage wild-type embryo (i) compared to the pronuclear fusion stage wild-type embryo (ii) in the same field. cul-2 (B) and rfl-1 (C) embryos show increased MEL-26 staining at meiosis compared to wild type. (D) emb-27 embryo arrested at meiosis I shows low levels of MEL-26. (E) zyg-11 embryos have a prolonged meiosis (as do cul-2 embryos), but levels of meiotic MEL-26 do not increase as they do in cul-2. Prolonged meiosis may result in very low levels of MEL-26 in (D) and (E). (F) The increased levels of meiotic MEL-26 characteristic of cul-2 are epistatic to the low levels seen in emb-27 in the cul-2; emb-27 double mutant. (G) CUL-2 prevents premature accumulation of MEL-26. The histogram shows the percent of embryos showing very low (white), moderate (grey) and high (black) levels of MEL-26 at the stages indicated for each genotype. Wild-type embryos show an abrupt increase in MEL-26 after meiosis. cul-2 embryos are strongly positive at meiosis I and increase slightly thereafter. An APC subunit (emb-27) mutant arrests at meiosis I and is similar to wild type at that stage, but cul-2 is epistatic to emb-27 as doubly mutant embryos are similar to cul-2. The numbers of embryos scored are as follows. Meiosis I: wild type (17), cul-2 (29), emb-27 (19), cul-2; emb-27 (25). Meiosis II: wild type (17), cul-2 (16). Prometaphase: wild type (30), cul-2 (20).
To demonstrate antibody-specificity, we used the genetic null allele mel-26(ct61sb4) (Lu and Mains, 2007). This mutation encodes a truncated protein missing the terminal 75 amino acids (Dow and Mains, 1998), which contains the region used to affinity purify the antisera (Materials and Methods). As shown in Figs. 10, P, no MEL-26 staining was apparent in embryos with this null mutation. This demonstrates that the low meiotic levels of MEL-26 seen in Figs. 1A–D and 2A represent a true signal. We conclude that the timing of the post-meiotic disappearance of MEI-1 corresponds to the period when MEL-26, as measured by immunostaining, accumulates to higher levels (C. elegans meiotic stage embryos cannot be isolated for Western analysis to independently quantify MEL-26 levels).
Timing of MEL-26 accumulation is not regulated by autodegradation
We next sought to identify the factors that regulate the accumulation of MEL-26 during meiosis. Many ubiquitin substrate targeting subunits are themselves regulated by autoubiquitination and degradation, and this has been demonstrated by accumulation of higher levels of MEL-26 in cul-3(RNAi) embryos (Luke-Glaser et al., 2005; Pintard et al., 2003b). While we also observed higher levels of MEL-26 in post-meiotic cul-3(RNAi) embryos consistent with previous reports, we did not observe higher levels of MEL-26 during meiosis (data not shown). Thus autodegradation affects the levels, but not the timing, of post-meiotic MEL-26 accumulation. Likewise, the presence of the substrate can stabilize ubiquitin ligase substrate adaptors (Galan and Peter, 1999; Wirbelauer et al., 2000); however, we found MEL-26 accumulation was not altered in the background of the null allele mei-1(ct46ct101) (data not shown).
The CUL-2 ubiquitin ligase is required to regulate meiotic levels of MEL-26
cul-2 encodes another E3 ligase implicated in many processes in the newly fertilized C. elegans embryo, including meiotic progression (Feng et al., 1999; Liu et al., 2004; Sonneville and Gonczy, 2004; Vasudevan et al., 2007; Zhong et al., 2003). While a cul-2 null allele had no obvious effect on post-meiotic levels of MEL-26, we found that this mutation resulted in higher MEL-26 levels in 16/24 embryos in meiosis I (Fig. 2B). As cul-2 embryos proceed from meiosis I to meiosis II, there was an increase in the proportion of embryos with high levels of MEL-26 staining compared to wild type (Fig. 2G).
rfl-1 is part of the heterodimeric E1 activating enzyme in the ubiquitin-like neddylation pathway that activates ubiquitin E3 ligases. During mitosis, RFL-1 activates the E3 ligase containing CUL-3 and MEL-26 to degrade MEI-1 (Kurz et al., 2002; Pintard et al., 2003a). rfl-1 mutants, like cul-2 mutants, had increased accumulation of MEL-26 during meiosis (Fig. 2C). The increase in MEL-26 accumulation seen in both rfl-1 and cul-2 mutant embryos suggests that during meiosis rfl-1 might regulate CUL-2 based E3 ligases.
Two other genes previously implicated in MEL-26 function were also examined. MBK-2 has been shown to act in parallel with MEL-26 for MEI-1 degradation (Lu and Mains, 2007) and is also required for the coordinated regulation of several maternally-provided proteins (Ming Pang et al., 2004; Nishi and Lin, 2005; Pellettieri et al., 2003; Quintin et al., 2003). mbk-2 mutant embryos had no effect on the timing of MEL-26 expression (data not shown), consistent with our previous observation that mbk-2 and mel-26 act independently of one another (Lu and Mains, 2007). MEL-26 membrane localization requires POD-1(Luke-Glaser et al., 2005), an actin-binding protein (Rappleye et al., 1999). However, pod-1 mutants did not affect the temporal pattern of MEL-26 expression (data not shown).
In summary, MEL-26 levels are kept low during meiosis through the action of a CUL-2 based ubiquitin ligase, which in turn may be regulated by RFL-1 mediated neddylation.
CUL-2 regulates the accumulation of MEL-26 during meiosis
We next asked if the increased levels of MEL-26 seen in rfl-1 and cul-2 mutants requires progression through meiosis, or if fertilization per se triggers increased MEL-26 accumulation. Embryos mutant for emb-27, which encodes the APC subunit APC-6, arrest at metaphase of meiosis I (Golden et al., 2000). These embryos expressed low levels of MEL-26 (Fig. 2D), indicating that induction of higher MEL-26 levels requires at least passage through the first meiotic division.
Mutations in cul-2 result in a prolonged meiosis II (Liu et al., 2004; Sonneville and Gonczy, 2004), and so it is possible that the increased levels of meiotic MEL-26 in cul-2 mutants are misleading. Many mutant cul-2 meiotic embryos could simply be chronologically older than the corresponding wild-type meiotic embryos. Similarly, we have found that mutant rfl-1 embryos also appear to have an extended meiosis (J. Johnson and E. Raharjo, unpublished), which might explain the ectopic meiotic MEL-26 observed in rfl-1 mutants. To test whether increased chronological age leads to the premature meiotic increase in MEL-26 accumulation, we examined zgy-11 mutants. The zgy-11 gene encodes the substrate adaptor that acts with CUL-2 to control meiotic progression (Vasudevan et al., 2007). Although zyg-11 mutants also display a prolonged meiosis II (Liu et al., 2004; Sonneville and Gonczy, 2004), these mutant embryos did not show increased meiotic MEL-26 levels (Fig. 2E). This result argues that CUL-2 and RFL-1 may play a direct role in MEL-26 regulation, and additionally that ZYG-11 is not the substrate adaptor for MEL-26.
Another way to test the possibility that CUL-2 is involved in regulating MEL-26 other than by altering meiotic timing is to examine emb-27; cul-2 double mutants. If CUL-2 is actively involved in MEL-26 regulation prior to passage through meiosis II, then the high levels of meiotic MEL-26 in cul-2 mutants would be epistatic to the low levels seen in emb-27 APC mutants blocked at meiosis I. As shown in Figs. 2B, D, F, G, we found that cul-2(+) function is required to prevent MEL-26 accumulation in emb-27 mutant embryos. Thus CUL-2 is responsible for keeping MEL-26 levels low during meiosis, through a mechanism independent of its role in meiotic progression.
The premature increase in meiotic MEL-26 in cul-2 mutants could lead to a decrease in MEI-1 levels during meiosis. By scoring levels of cytoplasmic anti-MEI-1 staining in meiotic embryos we determined that meiotic MEI-1 staining in cul-2 mutant embryos did not decrease. Compared to later mitotic stage embryos, cul-2 meiotic embryos (9/11) showed higher levels of MEI-1 (Fig. 3B). This was similar to wild type, where 21/26 meiotic embryos displayed higher cytoplasmic MEI-1 staining compared to later-stage embryos (Fig. 3A). Interestingly, both meiotic and mitotic MEI-1 levels appeared higher in cul-2 mutants than corresponding wild-type embryos, consistent with the previous report that cul-2(+) is required for efficient MEI-1 degradation (Stitzel et al., 2006). Thus any increase in MEI-1 degradation in cul-2 mutants due to ectopic MEL-26 may be offset by loss of CUL-2 dependent MEI-1 degradation. In this scenario, CUL-2 rather than MEL-26 is limiting in cul-2 mutants.
Fig. 3.

Meiotic cytoplasmic MEI-1 levels are not altered in cul-2 embryos. (A–D) Embryos stained with anti-MEI. (A′–D′) Anti-tubulin staining of the same embryos. In each panel, levels in meiotic embryos are compared to the mitotic embryo present in the same field. (A, A′) Wild-type cytoplasmic MEI-1 staining is higher in the meiotic embryo than a later-stage embryo. (B, B′) cul-2 has higher levels of cytoplasmic MEI-1 at meiosis than mitosis, and levels at both divisions are higher than corresponding wild-type embryos. (C, C′) egg-3 (RNAi) embryos show similar cytoplasmic MEI-1 levels at meiosis and mitosis, indicative of premature MEI-1 degradation. (D, D′) cul-2; egg-3(RNAi) embryos have similar levels of cytoplasmic MEI-1 at meiosis and mitosis, like egg-3 alone, but cytoplasmic levels at both divisions are increased, as seen with cul-2 single mutants. Note that in all panels meiotic spindles (arrows) are present and contain MEI-1.
Other factors also might limit meiotic MEI-1 degradation when MEL-26 is ectopically expressed in cul-2 mutants. One candidate is MBK-2, a component of the pathway that degrades MEI-1 in parallel to MEL-26. Ectopic MBK-2 expression in egg-3 mutants leads to decreased levels of cytoplasmic MEI-1:GFP during meiosis, resulting in meiotic embryos with the same level of cytoplasmic MEI-1 as later-stage mitotic embryos (Stitzel et al., 2007). As was observed with MEI-1:GFP, we found that cytoplasmic anti-MEI-1 staining was reduced in egg-3(RNAi) in 14/20 meiotic embryos (Fig. 3C). However, it should be noted that meiotic spindles still formed in egg-3(RNAi) embryos indicating that ectopic MBK-2 alone is not sufficient to block MEI-1 function [Fig. 3C and Maruyama et al. (2007) and Stitzel et al. (2007)]. To test the possibility that both the MBK-2 and MEL-26 pathways are required to prevent MEI-1 dependent meiotic spindle formation, we examined egg-3(RNAi); cul-2. These embryos resembled both of the egg-3(RNAi) and cul-2 single mutants. 7/10 meiotic embryos showed decreased levels of cytoplasmic MEI-1 during meiosis similar to those seen in mitosis, like egg-3 alone, indicating premature loss of cytoplasmic MEI-1. MEI-1 levels in egg-3 (RNAi); cul-2 embryos were increased during both meiotic and mitotic divisions, as seen in cul-2 single mutants. However, MEI-1 levels were still sufficient for meiotic spindle formation (Fig. 3D). Therefore factors, which may include CUL-2, appear to be required in addition to MBK-2 and MEL-26 to eliminate sufficient MEI-1 to prevent meiotic spindle formation.
MEL-26 has a meiotic function
The low levels of MEL-26 present at meiosis are clearly above the background seen in a mel-26 null mutant (Fig. 1, A–D compared to O–P). Therefore, we asked if MEL-26 has a meiotic function, perhaps in moderating MEI-1 activity. Any meiotic function of MEL-26 examined by loss of function mutations would be obscured by the subsequent mitotic lethality caused by ectopic MEI-1 expression, therefore we examined double mutants of a null mel-26 allele with tbb-2(sb26). This mutant β-tubulin rescues lethality by partially inhibiting ectopic MEI-1 severing of mitotic microtubules (Lu et al., 2004). Among the survivors of this strain, the appearance of males is a measure of their meiotic abnormalities: while autosomal meiotic nondisjunction leads to lethality, nondisjunction of the X chromosome instead results in viable males. We have previously used the high incidence of males (Him) phenotype as a sensitive metric of mei-1 function (Clandinin and Mains, 1993; Clark-Maguire and Mains, 1994a; Lu and Mains, 2005). Among self progeny of wild-type hermaphrodites (Hodgkin et al.,1979) and tbb-2(sb26) (Lu et al., 2004), males are rare, ~0.2% of the progeny. However, we found approximately a 6-fold increase to 1.3% among the surviving mel-26(null); tbb-2(sb26) progeny. Similar results (1.4% male) were seen for mei-1(ct46); tbb-2(sb26). ct46 is an allele that encodes a protein resistant to MEL-26 degradation (Clark-Maguire and Mains, 1994a; Pintard et al., 2003b; Xu et al., 2003), suggesting that MEI-1 is the relevant MEL-26 target leading to the high incidence of males.
To determine if the meiotic abnormalities seen in the absence of mel-26 resulted in increased MEI-1 activity during meiosis, we measured the length of meiotic spindles in live mei-1(ct46) embryos that have the form of MEI-1 that appears completely resistant to MEL-26 action (Lu and Mains, 2007). The genetic behavior of mei-1(ct46) is identical to loss of mel-26 (Dow and Mains, 1998; Mains et al., 1990a). Since partial loss of MEI-1 activity results in metaphase meiotic spindles that are longer than wild type (McNally et al., 2006), excess MEI-1 activity due to loss of MEL-26 inhibition in mei-1(ct46) should result in meiotic spindles that are shorter than wild type. Indeed, time-lapse imaging of GFP-tubulin in mei-1(ct46) embryos revealed meiotic metaphase spindles that were significantly shorter (p b0.005) than wild type (Fig. 4). Following completion of metaphase, C. elegans meiotic spindles undergo a period of shortening and rotation, and by this time mutant and wild-type spindles were of comparable sizes.
Fig. 4.
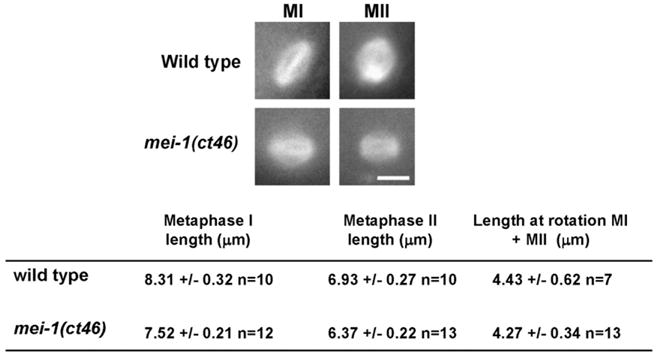
Decreased MEI-1 degradation during meiosis leads to shorter meiotic metaphase spindles. C. elegans meiotic spindles initially maintain a constant length during metaphase I and metaphase II (Yang et al., 2003). However, just prior to anaphase they shorten and rotate from parallel to perpendicular to the cortex. The constant lengths of metaphase I and metaphase II spindles prior to shortening and their lengths at rotation were determined from time-lapse sequences of GFP: tubulin in both wild-type and mei-1(ct46) embryos in utero. Representative images of metaphase spindles (scale bar, 5 μm) and average metaphase spindle lengths are shown and tabular data are presented +/− the standard deviation. n =the number of individual spindles used to determine each average.
As an independent test of MEL-26 regulation of meiotic MEI-1, we examined genetic interactions between these genes. If low levels of MEL-26 do indeed antagonize MEI-1 function in meiotic spindles, then decreasing MEL-26 activity should compensate for mutations with sub-optimal levels of microtubule-severing activity. To investigate this possibility, we made use of partial loss-of-function alleles of mei-2 (Mains et al., 1990a; Srayko et al., 2000). Since MEI-1 and MEI-2 are co-dependent on one another for localization (Clark-Maguire and Mains, 1994a; Srayko et al., 2000) and MEI-1 requires MEI-2 for microtubule-severing activity (McNally et al., 2006), we reasoned that decreasing the level of MEI-1 degradation with a mel-26 mutant might lead to increased levels of MEI-2, thereby rescuing the meiotic lethality caused by mei-2 partial loss-of-function mutations. Indeed, when we partially lowered the level of mel-26 using a strain heterozygous for the dominant-negative (dn) allele mel-26(ct61) (Mains et al., 1990b), we increased the hatching of the temperature-sensitive (ts) allele mei-2(sb39) at all temperatures tested (Table 1, lines 1–3). We further lowered microtubule-severing activity using a strong mei-2 allele in combination with mei-2(sb39) and again observed rescue with the addition of mel-26(dn)/+ (lines 4–5). Finally, to guard against possible background effects in the strains, we substituted a less ts mei-2 allele, ct98, for sb39 and the mei-1(ct46) allele that is refractory to MEL-26 regulation in place of mel-26(dn). We again saw a rescue of the meiotic lethality (lines 6–8). We conclude that the low levels of MEL-26 in meiosis appear to antagonize MEI-1/MEI-2 microtubule-severing activity in the meiotic spindle. [As previously observed (Mains et al., 1990a), the genetic interaction was reciprocal, decreasing the level of mei-2 suppressed the mitotic lethality caused by mel-26 and mei-1(gf) (Table 1)].
Table 1.
Decreased levels of mel-26 compensates for partial loss of mei-2.
| Genotypea | Percent hatching |
|||
|---|---|---|---|---|
| 15 °C | 20 °C | 24 °C | 25 °C | |
| mel-26(dn)/+ | 95 | 79 | 8.9 | 7.8 |
| mei-2(sb39) | 84 | 74 | 13 | 1.1 |
| mei-2(sb39) mel-26(dn)/mei-2(sb39) + | –b | 92 | 29 | 4.4 |
| mei-2(sb39)/mei-2(0) | 52 | 39 | 3.1 | – |
| mei-2(sb39) mel-26(dn)/mei-2(0) + | 76 | 72 | 10 | – |
| mei-2(ct98)/mei-2(0) | – | – | – | 28 |
| mei-1(gf)/+ | – | – | – | 0 |
| mei-2(ct98) mei-1(gf)/mei-2(0) | – | – | – | 45 |
| mbk-2(ts) | 92 | 62 | 2.6 | – |
| mei-2(sb39); mbk-2(ts) | 85 | 69 | 17 | – |
| mei-2(sb39)/mei-2(0); mbk-2(ts) | 58 | 35 | 1.8 | – |
mel-26(dn)=ct61, mei-2(0) =ct102, which is a strong allele. mei-2(sb39) is hypomorphic, with less activity than mei-2(ct98). mei-1(gf)=ct46, an allele refractory to MEL-26 degradation, mbk-2(ts) =dd5.
Not determined.
Thus, based on increased levels of nondisjunction, altered spindle morphology and genetic interactions, we conclude that MEL-26 moderates MEI-1 activity during meiosis.
We next asked whether the MBK-2 pathway of MEI-1 degradation is likewise active during meiosis. Using a similar strategy to that described above, we asked whether a ts allele of mbk-2 can rescue mei-2 hypomorphs. At 24 °C, where ts alleles of both genes were strongly compromised, the hatching rate of the double mutant increased from 2.6% to 17% (Table 1, lines 9–10) clearly demonstrating rescue of mbk-2 lethality. This is expected because mbk-2 lethality results in part due to ectopic mitotic MEI-1, and by lowering mei-2 hatching increases (Quintin et al., 2003). Here we are concerned with the reciprocal genetic interaction, asking if lowering mbk-2 in turn increases the viability of mei-2 hypomorphs. The levels of hatching for mei-2(ts) (Table 1, lines 2 and 10) and mei-2(ts)/mei-2(0) (lines 6 and 11) were similar with or without mbk-2(ts) at all temperatures tested. Unless one assumes that mei-2 rescued 100% of the lethality due to mbk-2 (which seems unlikely, especially given MBK-2’s many other targets), then these values indicate that the reciprocal suppression of mei-2 meiotic lethality by mbk-2 was occurring, preventing increased lethality in the double mutant. For example, if decreased mei-2 suppressed half of the mbk-2 lethality (so ~50% hatching is then the theoretic maximum for hatching due to lethality caused by other MBK-2 targets), the observed hatching frequency of mei-2; mbk-2 indicates an approximately two-fold suppression of the lethality seen in the mei-2(ts) single mutant. However, because we do not know the extent of suppression of mbk-2 by mei-2, we cannot come to a firm conclusion as to whether MBK-2, like MEL-26, also moderates MEI-1/MEI-2 activity during meiosis.
MEI-1 is the major target of MEL-26 and CUL-3, but RFL-1 has additional targets
MEL-26 has an essential role in regulating MEI-1 (Clark-Maguire and Mains, 1994a) but MEL-26 also interacts with the actin-binding protein POD-1 and the katanin-related protein FIGL-1 (Luke-Glaser et al., 2005; Luke-Glaser et al., 2007). However, only the interaction between MEI-1 and MEL-26 appears to be essential, leading to complete lethality when this interaction is lost. Data in Table 2 (lines 1 and 2) shows that mel-26 lethality is substantially rescued by tbb-2 (sb26), a tubulin mutation that partly inhibits MEI-1 microtubule severing (Lu et al., 2004). mei-1(ct46), which encodes a product that appears to be completely refractory to MEL-26 regulation (Lu and Mains, 2007), is suppressed by tbb-2(sb26) to a greater extent than the mel-26 null (lines 3 and 4), indicating that misregulation of other MEL-26 targets may contribute some incompletely penetrant lethality. However, we previously reported that a series of mei-2 and mei-1(dn) alleles that suppress mei-1(ct46) also suppress two different mel-26 mutations to a similar extent (Dow and Mains, 1998; Mains et al., 1990a). Thus, MEL-26 regulation of targets other than MEI-1 is not essential for viability.
Table 2.
Genetic interactions of tbb-2 with mel-26, rfl-1 and cul-3.
| Genotype | Percent hatching |
|
|---|---|---|
| 15 °C | 25 °C | |
| mel-26(ct61sb4) | 18 | 0 |
| mel-26(ct61sb4); tbb-2(sb26) | 60 | 40 |
| mei-1(ct46) | 23 | 0 |
| mei-1(ct46); tbb-2(sb26) | 96 | 66 |
| rft-1(or198) | –a | 0 |
| rft-1(or198) tbb-2(sb26) | – | 0 |
| rft-1(or198); mei-2(ct98) | – | 0 |
| cul-3(RNAi) | 19 | 11 |
| cul-3(RNAi); tbb-2(sb26) | 83 | 37 |
Not determined.
Loss of cul-3, rfl-1 and mel-26 all leads to similar mitotic defects due to ectopic MEI-1 expression during mitosis (Clark-Maguire and Mains, 1994a; Kurz et al., 2002). We asked whether cul-3 and rfl-1 have essential targets in addition to MEI-1. As shown in Table 2 (lines 5 and 6), rfl-1 lethality was not rescued by tbb-2(sb26), the suppressor of ectopic MEI-1 activity. Similarly, the mei-2 hypomorph ct98, which ef3ciently suppresses mel-26(dn) (Mains et al., 1990a), also failed to rescue rfl-1 (line 7). Thus RFL-1 must have essential targets in addition to MEI-1. A mutation is not available for cul-3, but a substantial level of cul-3(RNAi) lethality was rescued by tbb-2(sb26) (lines 8 and 9), demonstrating that MEI-1 is indeed the major target of CUL-3. However, because cul-3(RNAi) did not lead to complete lethality, there could be other essential targets that would only be revealed in situations where cul-3 is null. We conclude that MEL-26, and likely CUL-3, have MEI-1 as their only essential target while RFL-1 must regulate essential pathways in addition to MEL-26/MEI-1.
Discussion
Upon fertilization, the embryo shifts from one developmental program, oogenesis, to another, zygotic development. One of the first events initiated is the completion of meiosis. In the C. elegans embryo, meiosis resumes from its prophase arrest, and the two divisions are rapidly completed (Albertson and Thomson, 1993; Kemphues et al., 1986; Yang et al., 2003). The zygote then transitions to another form of cell division, mitosis. Because the meiosis to mitosis transition is rapid in C. elegans, lasting only about 15 min, the regulation of meiotic-specific components such as the katanin microtubule-severing complex MEI-1/MEI-2 is especially critical. Post-meiotic MEI-1 degradation requires two independent pathways (Lu and Mains, 2007), one involving the MEL-26 substrate adaptor for the CUL-3 based ubiquitin ligase and another that includes the MBK-2 DYRK kinase. While MBK-2 has many other targets (DeRenzo et al., 2003; Feng et al., 1999; Nishi and Lin, 2005; Shirayama et al., 2006; Stitzel et al., 2006), MEL-26 (and likely CUL-3) have MEI-1 as their only essential target in the early embryo (Table 2).
Downregulation of MEI-1 by proteolysis presents the embryo with the problem of maintaining sufficient MEI-1 levels until the completion of meiosis. Recently Stitzel et al. (2007) demonstrated that, at fertilization, MBK-2 is sequestered to the cortex by EGG-3, a protein required for several other aspects of embryo activation (Maruyama et al., 2007). Meiotic APC activity then degrades EGG-3, releasing MBK-2 and giving it access to MEI-1 in the cytoplasm. Here we have described the regulation of the parallel, MEL-26 dependent pathway for MEI-1 degradation (Fig. 5). We find that MEL-26 levels are kept low in meiosis by the activity of another type of ubiquitin ligase that includes CUL-2 as well as by RFL-1, a protein that mediates ubiquitin ligase neddylation and activation. We also show that MEL-26 has a previously undescribed role during meiosis in moderating MEI-1 activity.
Fig. 5.
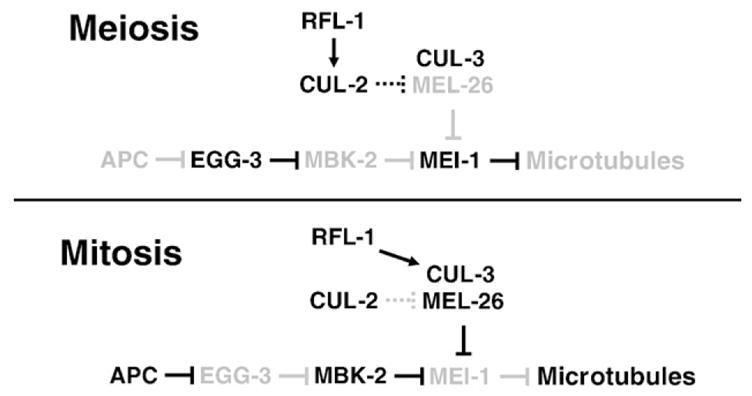
Model of MEI-1 regulation during the meiosis to mitosis transition. Low levels of protein or activity are indicated by lighter shading. During meiosis, CUL-2 (this report) and EGG-3 (Stitzel et al., 2007) keep MEL-26 and MBK-2 activities low, respectively, allowing MEI-1 to accumulate and sever microtubules. It is not known if the interaction between CUL-2 and MEL-26 is direct, and so this interaction is shown with stippled lines. During mitosis the situation is reversed and MEI-1 is degraded, allowing formation of longer microtubules. The kinase that acts in concert with the CUL-3/MEL-26 ubiquitin ligase and the ubiquitin ligase acting with MBK-2 are unknown.
The level of MEL-26, as assessed by antibody staining, is low (but non-zero) during meiosis but then increases dramatically (Figs. 1–3). This dramatic increase likely is required because during mitosis MEL-26 does not have just a subtle role in moderating MEI-1 activity as it did during meiosis, but rather MEL-26 takes part in the total elimination of MEI-1 activity following meiosis. MBK-2 activation occurs during meiosis (Stitzel et al., 2007; Stitzel et al., 2006) while the increase in MEL-26 levels appears to be delayed until meiosis is complete. This is consistent with our previous hypothesis that MBK-2 mediated MEI-1 degradation may link meiotic progression with a first wave of MEI-1 degradation, after which the MEL-26 pathway is required to complete the process (Lu and Mains, 2007).
While substrate adaptors, including MEL-26, are frequently targets of autodegradation, we found that a block to autodegradation alters the level (Luke-Glaser et al., 2005) but not the timing of MEL-26 expression. Instead, maintenance of low MEL-26 levels requires a CUL-2 based ubiquitin ligase: in cul-2 mutants, MEL-26 accumulates to high levels at meiosis (Fig. 2). Since cul-2 mutant embryos have a prolonged meiosis II (Liu et al., 2004; Sonneville and Gonczy, 2004), it was possible that the higher levels of MEL-26 in this background resulted indirectly because embryos still in meiosis were simply chronologically older and so had more time to accumulate MEL-26. We determined that this was not the case because a mutation in zyg-11, the gene that functions with cul-2 to regulate meiotic progression (Vasudevan et al., 2007), did not show premature MEL-26 accumulation. In addition, embryos blocked at meiosis I by the APC mutant emb-27, which by themselves have low MEL-26 levels, require CUL-2 activity to maintain those low levels (Figs. 2 and 3). While the relationship between CUL-2 and MEL-26 could be indirect, the simplest model is that prior to the end of meiosis, CUL-2 directly targets MEL-26 for degradation (Fig. 5). Progression through meiosis results in the inactivation of CUL-2 regulation of MEL-26, allowing accumulation of higher levels of MEL-26 in the embryo. Since CUL-2 is active against many other substrates in the developing embryo (Bowerman and Kurz, 2006; DeRenzo and Seydoux, 2004; Kipreos, 2005), the inhibition of CUL-2 induced degradation of MEL-26 may occur by the inactivation of a CUL-2 substrate-specific adaptor that targets MEL-26 rather than an inactivation of CUL-2 itself. Regulation of one ubiquitin ligase by another, as we see here for CUL-2 and CUL-3, has been described in other systems, such as CUL-1 by the APC ubiquitin ligase (Bashir et al., 2004; Vodermaier, 2004; Wei et al., 2004). During C. elegans sex determination, the FEM proteins provide substrate-specificity for CUL-2 mediated degradation of the terminal sex determination factor for TRA-1 (Starostina et al., 2007), and FEM proteins are themselves the target of a CUL-1 based ubiquitin ligase (Jager et al., 2004).
Meiotic spindle abnormalities might be expected in cul-2 mutants due to increased MEL-26 accumulation, resulting in increased MEI-1 degradation. However, Liu et al. (2004) reported normal meiotic spindles in cul-2 mutants, implying that MEL-26 is not rate limiting at that time, and we did not observe premature MEI-1 degradation (Fig. 3). However, in addition to increasing meiotic MEL-26, loss of cul-2 also delays MEI-1 degradation [Stitzel et al. (2006) and B. Gavinolla and P. Mains, unpublished], and so these opposing effects of cul-2 loss may cancel one another. Alternatively, although mel-26 mutants demonstrate that the protein is necessary for meiotic MEI-1 regulation, an increase of MEL-26 alone might not be sufficient. The same appears to be true for MBK-2, whose ectopic expression during meiosis (alone or in combination with ectopic MEL-26) is also not sufficient to block MEI-1 dependent meiotic spindle formation. Thus other factors (e.g., CUL-2) might be limiting. Finally, MEI-1 might be resistant to MEL-26/MBK-2 degradation when assembled into a spindle and so efficient elimination of MEI-1 may only occur upon spindle disassembly once meiosis is completed.
Neddylation was the only other pathway that we found to regulate MEL-26. RFL-1 is a component of the Nedd-8 E1 complex that leads to ubiquitin ligase activation by coupling the Nedd8 ubiquitin-like molecule to the cullins (Parry and Estelle, 2004; Petroski and Deshaies, 2005). Previously described roles for RFL-1 in C. elegans included CUL-3/MEL-26 activation and early embryo interphase progression (Kurz et al., 2002; Pintard et al., 2003a). Here we find that RFL-1 has an earlier role, likely in activating CUL-2 mediated degradation of MEL-26. Similar to cul-2 mutants, rfl-1 mutants resulted in premature MEL-26 accumulation (Fig. 2), and so the neddylation pathway likely acts on both CUL-2 and CUL-3.
While post-meiotic MEL-26 levels dramatically increase, lower amounts of the protein are clearly present during meiosis (Figs. 1 and 2). We have presented three independent lines of evidence that MEL-26 functions to moderate meiotic MEI-1 activity prior to its degradation of MEI-1 during mitosis. First, partial loss of mel-26 rescues mei-2 hypomorphic mutations, indicating that MEL-26 normally antagonizes meiotic MEI-2 (and by implication, MEI-1) activity (Table 1). Second, the absence of MEL-26 activity towards MEI-1 leads to hyperactive MEI-1 microtubule severing as evidenced by shortened metaphase spindles at both meiosis I and II (Fig. 4). Third, these shortened spindles are not fully functional since they result in higher levels of nondisjunction. MBK-2 may also moderate meiotic MEI-1 activity during meiosis, although the results were less definitive (Table 1). The opposite situation, hypoactive MEI-1, also leads to chromosome segregation defects (Clandinin and Mains, 1993; Clark-Maguire and Mains, 1994b), indicating that the levels of meiotic MEI-1 must be maintained within a precise range.
Why might MEL-26 (and perhaps MBK-2) be necessary to regulate MEI-1/MEI-2 during meiosis and why is ectopic expression of both MEL-26 and MBK-2 not sufficient for large-scale MEI-1 degradation? MEI-1/MEI-2 has multiple functions during meiosis, initially to increase the number of microtubule ends to aid in nucleating the meiotic spindle in the absence of centrosomal-based microtubule organizing centers (McNally et al., 2006; Srayko et al., 2006). Moments later, MEI-1/MEI-2 is required for a different event, spindle shortening and also for translocation of the spindle to the cortex (McNally et al., 2006; Yang et al., 2003). Meiotic spindle shortening is itself a biphasic event (McNally et al., 2006), with only the second stage requiring katanin activity. Perhaps these differing katanin functions require precise and dynamic regulation of microtubule-severing. While MEL-26 regulation of MEI-1/MEI-2 is not essential for completion of meiosis (the level of nondisjunction is relatively low in mel-26 mutants), this regulation would be important in nature as it clearly increases the fidelity of meiotic chromosome segregation.
Acknowledgments
We would like to thank J.D. McGhee, J. Gaudet and D. Hansen and members of their labs for their input during the course of this project. Some of the strains were obtained from the Caenorhabditis Genetics Center, funded by the National Institutes of Health, Center for Research Resources. This work was supported by grants from the Canadian Institute of Health Research and the Alberta Heritage Foundation for Medical Research to P.E.M. and from the NIH (1 R01 GM079421) to F.J.M.
References
- Albertson DG. Formation of the first cleavage spindle in nematode embryos. Dev Biol. 1984;101:61–72. doi: 10.1016/0012-1606(84)90117-9. [DOI] [PubMed] [Google Scholar]
- Albertson DG, Thomson JN. Segregation of holocentric chromosomes at meiosis in the nematode, Caenorhabditis elegans. Chromos Res. 1993;1:15–26. doi: 10.1007/BF00710603. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Kurz T. Degrade to create: developmental requirements for ubiquitin-mediated proteolysis during early C. elegans embryogenesis. Development. 2006;133:773–784. doi: 10.1242/dev.02276. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clandinin TR, Mains PE. Genetic studies of mei-1 gene activity during the transition from meiosis to mitosis in Caenorhabditis elegans. Genetics. 1993;134:199–210. doi: 10.1093/genetics/134.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Maguire S, Mains PE. Localization of the mei-1 gene product of Cae-norhabditis elegans, a meiotic-specific spindle component. J Cell Biol. 1994a;126:199–209. doi: 10.1083/jcb.126.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Maguire S, Mains PE. mei-1, a gene required for meiotic spindle formation in Caenorhabditis elegans, is a member of a family of ATPases. Genetics. 1994b;136:533–546. doi: 10.1093/genetics/136.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRenzo C, Reese KJ, Seydoux G. Exclusion of germ plasm proteins from somatic lineages by cullin-dependent degradation. Nature. 2003;424:685–689. doi: 10.1038/nature01887.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRenzo C, Seydoux G. A clean start: degradation of maternal proteins at the oocyte-to-embryo transition. Trends Cell Biol. 2004;14:420–426. doi: 10.1016/j.tcb.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Dow MR, Mains PE. Genetic and molecular characterization of the Caenor-habditis elegans gene, mel-26, a postmeiotic negative regulator of mei-1, a meiotic-specific spindle component. Genetics. 1998;150:119–128. doi: 10.1093/genetics/150.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Zhong W, Punkosdy G, Gu S, Zhou L, Seabolt EK, Kipreos ET. CUL-2 is required for the G1-to-S-phase transition and mitotic chromosome condensation in Caenorhabditis elegans. Nat Cell Biol. 1999;1:486–492. doi: 10.1038/70272. [DOI] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci U S A. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–790. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Golden A, Sadler PL, Wallenfang MR, Schumacher JM, Hamill DR, Bates G, Bowerman B, Seydoux G, Shakes DC. Metaphase to anaphase (mat) transition-defective mutants in Caenorhabditis elegans. J Cell Biol. 2000;151:1469–1482. doi: 10.1083/jcb.151.7.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JE, Govindan JA, Yamamoto I, Schwartz J, Kaverina I, Greenstein D. Major sperm protein signaling promotes oocyte microtubule reorganization prior to fertilization in Caenorhabditis elegans. Dev Biol. 2006;299:105–121. doi: 10.1016/j.ydbio.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Hodgkin J, Horvitz HR, Brenner S. Nondisjunction Mutants of the Nematode Caenorhabditis elegans. Genetics. 1979;91:67–94. doi: 10.1093/genetics/91.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Jager S, Schwartz HT, Horvitz HR, Conradt B. The Caenorhabditis elegans F-box protein SEL-10 promotes female development and may target FEM-1 and FEM-3 for degradation by the proteasome. Proc Natl Acad Sci U S A. 2004;101:12549–12554. doi: 10.1073/pnas.0405087101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kemphues KJ, Wolf N, Wood WB, Hirsh D. Two loci required for cytoplasmic organization in early embryos of Caenorhabditis elegans. Dev Biol. 1986;113:449–460. doi: 10.1016/0012-1606(86)90180-6. [DOI] [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modi3cation of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Kipreos ET. R TCe., editor. Ubiquitin-mediated pathways C. elegans. Community WormBook. 2005 doi: 10.1895/wormbook.1.36.1. www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Kurz T, Pintard L, Willis JH, Hamill DR, Gonczy P, Peter M, Bowerman B. Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science. 2002;295:1294–1298. doi: 10.1126/science.1067765. [DOI] [PubMed] [Google Scholar]
- Liu J, Vasudevan S, Kipreos ET. CUL-2 and ZYG-11 promote meiotic anaphase II and the proper placement of the anterior–posterior axis in C. elegans. Development. 2004;131:3513–3525. doi: 10.1242/dev.01245. [DOI] [PubMed] [Google Scholar]
- Lu C, Mains PE. Mutations of a redundant {alpha}-tubulin gene affect Caenor-habditis elegans early embryonic cleavage via MEI-1/katanin-dependent and -independent pathways. Genetics. 2005;170:115–126. doi: 10.1534/genetics.104.030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Mains PE. The C. elegans anaphase promoting complex and MBK-2/DYRK kinase act redundantly with CUL-3/MEL-26 ubiquitin ligase to degrade MEI-1 microtubule-severing activity after meiosis. Dev Biol. 2007;302:438–447. doi: 10.1016/j.ydbio.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Lu C, Srayko M, Mains PE. The Caenorhabditis elegans microtubule-severing complex MEI-1/MEI-2 katanin interacts differently with two superficially redundant beta-tubulin isotypes. Mol Biol Cell. 2004;15:142–150. doi: 10.1091/mbc.E03-06-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, Pintard L, Lu C, Mains PE, Peter M. The BTB protein MEL-26 promotes cytokinesis in C. elegans by a CUL-3-independent mechanism. Curr Biol. 2005;15:1605–1615. doi: 10.1016/j.cub.2005.07.068. [DOI] [PubMed] [Google Scholar]
- Luke-Glaser S, Pintard L, Tyers M, Peter M. The AAA-ATPase FIGL-1 controls mitotic progression, and its levels are regulated by the CUL-3MEL-26 E3 ligase in the C. elegans germ line. J Cell Sci. 2007;120:3179–3187. doi: 10.1242/jcs.015883. [DOI] [PubMed] [Google Scholar]
- Mains PE, Kemphues KJ, Sprunger SA, Sulston IA, Wood WB. Mutations affecting the meiotic and mitotic divisions of the early Caenorhabditis elegans embryo. Genetics. 1990a;126:593–605. doi: 10.1093/genetics/126.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mains PE, Sulston IA, Wood WB. Dominant maternal-effect mutations causing embryonic lethality in Caenorhabditis elegans. Genetics. 1990b;125:351–369. doi: 10.1093/genetics/125.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R, Velarde NV, Klancer R, Gordon S, Kadandale P, Parry JM, Hang JS, Rubin J, Stewart-Michaelis A, Schweinsberg P, Grant BD, Piano F, Sugimoto A, Singson A. EGG-3 regulates cell-surface and cortex rearrangements during egg activation in Caenorhabditis elegans. Curr Biol. 2007;17:1555–1560. doi: 10.1016/j.cub.2007.08.011. [DOI] [PubMed] [Google Scholar]
- McCarter J, Bartlett B, Dang T, Schedl T. On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev Biol. 1999;205:111–128. doi: 10.1006/dbio.1998.9109. [DOI] [PubMed] [Google Scholar]
- McNally KL, McNally FJ. Fertilization initiates the transition from anaphase I to metaphase II during female meiosis in C. elegans. Dev Biol. 2005;282:218–230. doi: 10.1016/j.ydbio.2005.03.009. [DOI] [PubMed] [Google Scholar]
- McNally K, Audhya A, Oegema K, McNally FJ. Katanin controls mitotic and meiotic spindle length. J Cell Biol. 2006;175:881–891. doi: 10.1083/jcb.200608117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Shakes DC. Immunofluorescence microscopy. Methods Cell Biol. 1995;48:365–394. [PubMed] [Google Scholar]
- Ming Pang K, Ishidate T, Nakamura K, Shirayama M, Trzepacz C, Schubert CM, Priess JR, Mello CC. The minibrain kinase homolog, mbk-2, is required for spindle positioning and asymmetric cell division in early C. elegans embryos. Dev Biol. 2004;265:127–139. doi: 10.1016/j.ydbio.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Nishi Y, Lin R. DYRK2 and GSK-3 phosphorylate and promote the timely degradation of OMA-1, a key regulator of the oocyte-to-embryo transition in C. elegans. Dev Biol. 2005;288:139–149. doi: 10.1016/j.ydbio.2005.09.053. [DOI] [PubMed] [Google Scholar]
- Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–229. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Pellettieri J, Reinke V, Kim SK, Seydoux G. Coordinate activation of maternal protein degradation during the egg-to-embryo transition in C. elegans. Dev Cell. 2003;5:451–462. doi: 10.1016/s1534-5807(03)00231-4. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B. Neddylation and deneddylation of CUL-3 is required to target MEI-1/katanin for degradation at the meiosis-to-mitosis transition in C. elegans. Curr Biol. 2003a;13:911–921. doi: 10.1016/s0960-9822(03)00336-1. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003b;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- Quintin S, Mains PE, Zinke A, Hyman AA. The mbk-2 kinase is required for inactivation of MEI-1/katanin in the one-cell Caenorhabditis elegans embryo. EMBO Rep. 2003;4:1175–1181. doi: 10.1038/sj.embor.7400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappleye CA, Paredez AR, Smith CW, McDonald KL, Aroian RV. The coronin-like protein POD-1 is required for anterior–posterior axis formation and cellular architecture in the nematode Caenorhabditis elegans. Genes Dev. 1999;13:2838–2851. doi: 10.1101/gad.13.21.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatten G. The centrosome and its mode of inheritance: the reduction of the centrosome during gametogenesis and its restoration during fertilization. Dev Biol. 1994;165:299–335. doi: 10.1006/dbio.1994.1256. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Soto MC, Ishidate T, Kim S, Nakamura K, Bei Y, van den Heuvel S, Mello CC. The conserved kinases CDK-1, GSK-3, KIN-19, and MBK-2 promote OMA-1 destruction to regulate the oocyte-to-embryo transition in C. elegans. Curr Biol. 2006;16:47–55. doi: 10.1016/j.cub.2005.11.070. [DOI] [PubMed] [Google Scholar]
- Sonneville R, Gonczy P. zyg-11 and cul-2 regulate progression through meiosis II and polarity establishment in C. elegans. Development. 2004;131:3527–3543. doi: 10.1242/dev.01244. [DOI] [PubMed] [Google Scholar]
- Srayko M, Buster DW, Bazirgan OA, McNally FJ, Mains PE. MEI-1/MEI-2 katanin-like microtubule severing activity is required for Caenorhabditis elegans meiosis. Genes Dev. 2000;14:1072–1084. [PMC free article] [PubMed] [Google Scholar]
- Srayko M, O’Toole ET, Hyman AA, Muller-Reichert T. Katanin disrupts the microtubule lattice and increases polymer number in C. elegans meiosis. Curr Biol. 2006;16:1944–1949. doi: 10.1016/j.cub.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Starostina NG, Lim JM, Schvarzstein M, Wells L, Spence AM, Kipreos ET. A CUL-2 ubiquitin ligase containing three FEM proteins degrades TRA-1 to regulate C. elegans sex determination. Dev Cell. 2007;13:127–139. doi: 10.1016/j.devcel.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitzel ML, Pellettieri J, Seydoux G. The C. elegans DYRK kinase MBK-2 Marks oocyte proteins for degradation in response to meiotic maturation. Curr Biol. 2006;16:56 –62. doi: 10.1016/j.cub.2005.11.063. [DOI] [PubMed] [Google Scholar]
- Stitzel ML, Cheng KC, Seydoux G. Regulation of MBK-2/Dyrk kinase by dynamic cortical anchoring during the oocyte-to-zygote transition. Curr Biol. 2007;17:1545–1554. doi: 10.1016/j.cub.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Starostina NG, Kipreos ET. The Caenorhabditis elegans cell-cycle regulator ZYG-11 de3nes a conserved family of CUL-2 complex components. EMBO Rep. 2007;8:279–286. doi: 10.1038/sj.embor.7400895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodermaier HC. APC/C and SCF: controlling each other and the cell cycle. Curr Biol. 2004;14:R787–R796. doi: 10.1016/j.cub.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Wirbelauer C, Sutterluty H, Blondel M, Gstaiger M, Peter M, Reymond F, Krek W. The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. Embo J. 2000;19:5362–5375. doi: 10.1093/emboj/19.20.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- Yang H, McNally K, McNally FJ. MEI-1/katanin is required for translocation of the meiosis I spindle to the oocyte cortex in C. elegans. Dev Biol. 2003;260:245–259. doi: 10.1016/s0012-1606(03)00216-1. [DOI] [PubMed] [Google Scholar]
- Zetka MC, Rose AM. The meiotic behavior of an inversion in Caenorhabditis elegans. Genetics. 1992;131:321–332. doi: 10.1093/genetics/131.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. 2003;423:885–889. doi: 10.1038/nature01747. [DOI] [PubMed] [Google Scholar]