

Abstract
The importance of the active site region aspartyl residues 25 and 29 of the mature HIV-1 protease (PR) for the binding of five clinical and three experimental protease inhibitors (symmetric cyclic urea inhibitor DMP323, non-hydrolysable substrate analog (RPB) and the generic aspartic protease inhibitor acetyl-pepstatin (Ac-PEP)) was assessed by differential scanning calorimetry. ΔTm values, defined as the difference in Tm for a given protein in the presence and absence of inhibitor, for PR with DRV, ATV, SQV, RTV, APV, DMP323, RPB and Ac-PEP are 22.4, 20.8, 19.3, 15.6, 14.3, 14.7, 8.7, and 6.5 °C, respectively. Binding of APV and Ac-PEP is most sensitive to the D25N mutation, as shown by ΔTm ratios [ΔTm(PR)/ΔTm(PRD25N)] of 35.8 and 16.3, respectively, whereas binding of DMP323 and RPB (ΔTm ratios of 1-2) is least affected. Binding of the substrate-like inhibitors RPB and Ac-PEP is nearly abolished (ΔTm(PR)/ΔTm(PRD29N) ≥ 44) by the D29N mutation, whereas this mutation only moderately affects binding of the smaller inhibitors (ΔTm ratios of 1.4-2.2). Of the 9 FDA approved clinical HIV-1 protease inhibitors screened, APV, RTV and DRV competitively inhibit porcine pepsin with Ki values of 0.3, 0.6 and 2.14 μM, respectively. DSC results were consistent with this relatively weak binding of APV (ΔTm 2.7 °C) compared with the tight binding of AcPEP (ΔTm ≥17 °C). Comparison of superimposed structures of the PR/APV complex with those of PR/Ac-PEP and pepsin/pepstatin A complexes suggests a role for Asp215, Asp32 and Ser219 in pepsin, equivalent to Asp25, Asp25′ and Asp29 in PR, in the binding and stabilization of the pepsin/APV complex.
Keywords: Retroviral protease, aspartic protease, protein structure, calorimetry, kinetics
INTRODUCTION
Aspartic proteases of the stomach, lysosomes, kidney, yeast granules and fungi are all single chain pepsin-like proteases with similar molecular sizes (∼330 residues)1. They are strikingly similar both in their sequence and tertiary fold (e.g. active-site, location of the disulfide bridges etc.) and exhibit a twofold symmetry in the conformation throughout the molecule2,3. The active site of these aspartic proteases is formed by two aspartic acid residues (positions 32 and 215 in pepsin)2 located in a conserved triad AspThrGly that is preserved throughout evolution4,5. It is speculated that eukaryotic aspartic proteases evolved from a primordial aspartic protease which was about half the size of pepsin and was active as a homodimer1,6. Retroviral proteases fulfill these criteria. They are small (100-125 amino acids), active as homodimers and bear a striking resemblance to pepsin both in their tertiary fold and catalytic mechanism6,7. Therefore, gene duplication and fusion may have led to the emergence of single chain aspartic proteases with an overall fold that is conserved in all organisms despite significant divergence of their primary sequences6.
In HIV-1, a single copy of the protease, composed of 99 amino acids, is synthesized as part of the ∼160 kDa Gag-Pol precursor flanked by the 58 amino acid long transframe region (TFR) and the reverse transcriptase at its N- and C-termini, respectively7,8. The Gag-Pol consists of the structural proteins (matrix, capsid, P2, nucleocapsid) followed by the TFR and the functional proteins (protease, reverse transcriptase, integrase). Dimer formation of the protease domains while in the form of a precursor is essential to form an active protease that catalyzes the cleavage of the peptide bonds at its termini, which is essential for its release, and the processing of the Gag and Pol polyproteins to produce mature structural and functional proteins required for the maturation and propagation of virus8,9. The role of the native TFR sequences is not fully understood but they may function similarly to the proregion found in zymogen forms of cellular aspartic proteases2,8. It has been shown that cleavage at the N-terminus of the protease, which precedes the C-terminal cleavage, is concomitant with the appearance of mature-like catalytic activity and stable dimer structure formation10,11.
Because of its crucial role, the protease has been one of the primary targets for drug development and treatment of HIV infection. Currently, nine FDA-approved potent protease inhibitors (PI) are used in treating patients with HIV/AIDS and successfully curb the progression of the disease (HIV Drug Resistance Database: hivdbstanford.edu/index.html). In spite of the new treatment regimens including the highly active antiretroviral therapy (HAART), rapid emergence of viral variants because of the decrease in drug potency is evident (HIV Drug Resistance Database). Extensive studies have been performed to understand the mechanism of HIV-1 protease drug resistance8,9. The drug-resistance mutations can map either to the active-site or the dimer interface or distally to these regions. These mutations can decrease inhibitor affinity by direct or indirect perturbations of the binding site, and influence the kinetic parameters and the stability of the dimeric fold. In spite of these changes, these mutant enzymes are still capable of catalyzing cleavage at the required junctions in the Gag and Gag-Pol polyproteins required for virus maturation and generation of progeny virus. New potent second generation inhibitors, such as DRV and ATV, have been designed to combat drug resistance. The binding of DRV to the mature protease is enthalpically driven unlike the binding of earlier generation of drugs12,13. In addition to the hydrogen bond interactions mostly to the main chain atoms, which cannot be directly altered by mutations, the binding of DRV is also characterized by extra polar interactions with Asp29 and Asp30 residues of the protease9. In spite of the higher affinity of DRV and ATV to the protease, clinical evidence for reduced virological response as a result of selection of drug-resistance mutations has been reported (HIV Drug Resistance Database).
While the signature triplet Asp-Thr-Gly (residues 25-27 in HIV protease) critical for the active-site geometry and catalytic function is common to all aspartic proteases, the second conserved triplet Gly86-Arg87-Asn/Asp88 (in HIV) in the sole α-helix of each subunit is unique to retroviral proteases4,7,14. The Asp residue in position 29 is also highly conserved in retroviral proteases. Residue Asp29 forms vital intra-monomer and inter-monomer hydrogen bonds with Arg87 and Arg8′ (prime denotes the second subunit) residues, respectively15. Subtle mutation of the Asp29 or Arg87 residue increases the dimer dissociation constant (Kd), R87K mutation exerting a dramatic effect (> 104) on Kd compared with a D29N (> 102) mutation16. In very recent studies we have also shown that similar to the Asp29 residue, the Asp25 residue, in addition to its catalytic function, plays a role in stabilizing both the dimer fold and substrate/inhibitor binding17. For example, the D25N mutation increases the ligand dissociation constant of PRD25N/DRV by a factor of ∼106 relative to PR/DRV. Conversely, Arg8 and Asp30 mutations that are selected upon patient therapy have only small effects on the kinetic parameters and stability of the mature protease11,18.
Because of the crucial structural and functional roles exhibited by the two conserved aspartyl residues Asp25 and Asp29 of HIV-1 protease, we have now systematically examined the effect of individual substitution mutations (D25N or D29N) on the binding of selected PIs (DRV, ATV, SQV, RTV, APV) based on DSC analyses and compared them to the binding of one tightbinding symmetric inhibitor (DMP323) and 2 substrate mimetics (RPB and Ac-PEP). PIs currently used in the treatment of HIV-infected patients have also been shown to inhibit the secreted aspartyl proteases of the fungus, Candida albicans19. Furthermore, in vitro growth of Pneumocystis was also shown to be significantly inhibited in the presence of clinically achievable concentration of several PIs, possibly by inhibiting aspartyl proteases of this organism20. Because of the conservation of the active site region in all aspartyl proteases, Asp25 and Asp29 of HIV represented by the conserved Asp32/215 and Ser36/219 residues, respectively, in single chain pepsin-like aspartic proteases, we also examined the inhibition and stabilization of pepsin, using the PIs designed for HIV-1, by kinetics and calorimetry. The most potent of these inhibitors with pepsin was APV. Predicted interactions of this inhibitor with the active site of pepsin are discussed.
MATERIALS AND METHODS
Substrate and inhibitors
Acetyl pepstatin (Ac-PEP), RPB, and pepsin substrate (H-Pro-Thr-Glu-Phe-[p-NO2-Phe]-Arg-Leu-OH) were purchased from Bachem Bioscience (King of Prussia, PA) and substrate IV (Lys-Ala-Arg-Val-Nle-[p-NO2-Phe]-Glu-Ala-Nle-NH2) from California Peptide Research (Napa, CA). DMP323 was obtained from the DuPont Merck Pharmaceutical Company (Wilmington, DE). Clinical inhibitors of HIV-1 protease, APV, ATV, DRV, INV, LPV, NFV, RTV, SQV and TPV were obtained from the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Stock solutions of the inhibitors (100 mM) were prepared in DMSO. Secondary stock solutions (typically 160 μM) were prepared by dilution of the DMSO stock solution into 5 mM sodium acetate buffer, pH 6, and diluted as required with the same buffer before use in the quench protocol for protease folding as described previously21. Because of limited solubility, the secondary stock solution of TPV was prepared at 80 μM.
PR and pepsin purification
The mature HIV-1 protease optimized for kinetic and structural studies (pseudo wild-type (PR11) and its mutants PRD25N22 and PRD29N23 were purified from inclusion bodies using an established protocol as described previously involving size-exclusion chromatography under denaturing conditions followed by reverse-phase HPLC11,24. Proteins were folded according to described protocols17,21.
Porcine pepsin (∼30 mg) from Calbiochem (Cat. No. 516360) was dissolved in column buffer (CB: 20 mM sodium phosphate buffer, pH 6, 50 mM NaCl). The sample was spun at full speed in an Eppendorf microcentrifuge for 5 min and the supernatant was subjected to size exclusion column chromatography (Superdex-75, 16 × 60 cm, GE Healthcare) equilibrated in CB at a flow rate of 1.4 ml/min at room temperature. Peak fractions were combined and dialyzed in the appropriate buffer. Protein concentration (mg/ml) was determined spectrophotometrically using ε (0.1%) = 1.36 at 280 nm. Identity and purity were confirmed by MALDI-TOF and active-site titration.
Differential scanning calorimetry (DSC)
The experimental protocol for PR sample preparation was as described previously21 and gave a final protein concentration of ∼0.3 mg/mL (13-14 μM as dimer) in 50 mM sodium acetate buffer, pH 4.8-5.0. The inhibitors were present at a final concentration of 28 μM (approximately twice the concentration of the dimeric proteins). Thermal denaturation scans were carried out in a MicroCal VP-DSC microcalorimeter, and data analysis was performed as described17. DSC scans of the inhibitor complexes with PR constructs are provided in supporting information Fig. S1. The measured Tm values (Table 1) were reproducible and provided a useful indication of relative thermal stability. Apparent ΔH values for the unliganded proteins, PR, PRD29N and PRD25N, were estimated by curve fitting with the Origin software provided by MicroCal, and agreed well with the baseline-corrected peak areas determined by integration. The relatively weak transitions in the absence of inhibitors did not permit reliably estimating the ΔCp. A literature value25,26 of ΔCp = 3 kcal/mol·deg for PR was used in calculations for all three constructs. Ligand-binding constants for the inhibitor complexes with PR were estimated at Tm and extrapolated to 25 °C by the methods of Brandts and Lin27 (equations 6 and 9; note that in these authors' notation, the binding constant KL is the reciprocal of our ligand-dissociation constants KL in Table 1). Literature values12,26,28-30 were used for ΔH and Cp of binding. Extrapolation was not possible in the absence of thermodynamic data for binding of the inhibitors to PRD25N and PRD29N.
Table 1.
DSC data for inhibitor binding to PR, PRD25N and PRD29N
| Inhibitor | Tm (°C) | ΔTm (°C) | KL at Tm (μM)a | KL at 25 °C (nM) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PR | PRD25N | PRD29N | PR | PRD25N | PRD29N | PR | PRD25N | PRD29N | PR (lit.) |
PR (calc.)a |
|
| none | 65.7 | 58.4 | 59.0 | - | - | - | - | - | - | - | - |
| DRV | 88.1 | 61.5 | 72.7 | 22.4 | 3.1 | 13.7 | 0.018 | 16.6 | 1.34 |
0.005b; 0.010c |
0.014 |
| ATV | 86.5 | 61.4 | 71.7 | 20.8 | 3.0 | 12.7 | 0.036 | 17.4 | 1.80 | 0.035 c | 0.186 |
| SQV | 85.0 | 62.6 | 72.3 | 19.3 | 4.2 | 13.3 | 0.066 | 10.0 | 1.51 | 0.28d,e | 8.14 |
| RTV | 81.3 | 61.1 | 66.1 | 15.6 | 2.7 | 7.1 | 0.27 | 20.5 | 9.3 | 0.10d,e | 9.72 |
| APV | 80.0 | 58.7 | 67.5 | 14.3 | 0.3 | 8.5 | 0.44 | 274 | 6.2 | 0.2d,e | 3.46 |
| DMP323 | 81.0 | 67.3 | 69.2 | 14.7 | 8.9 | 10.2 | 0.30 | 1.6 | 3.7 | 3.8 f | - |
| RPB | 74.4 | 64.6 | 59.2 | 8.7 | 6.2 | 0.2 | 2.9 | 4.4 | 1178 | 50 g | - |
| Ac-PEP | 72.2 | 58.8 | 58.3 | 6.5 | 0.4 | - | 6.1 | 202 | - | 440 e | 2956 |
Ligand dissociation constants calculated by the method of Brandts and Lin27 with ΔH = 43.3, 53.1 and 26.4 kcal/mol (this work) for PR, PRD25N and PRD29N, respectively. ΔCP = 3 kcal/mol·degree as measured25,26 for PR was used in calculations for all three constructs. KL values for PR at Tm (this work) and at 25 °C are italicized for ease of comparison.
Cited from Refs.
DSC studies of pepsin were carried out in 50 mM sodium formate buffer, pH 3.0-3.2 (the same buffer used for kinetics studies; see below), at pepsin concentrations of 14-21 μM without added inhibitor or in the presence of a 1.4- or 2-fold molar excess of inhibitors APV or Ac-PEP, respectively.
Isothermal titration calorimetry (ITC)
Measurements were performed at 28 °C using a high-precision VP-ITC titration calorimeter (MicroCal LLC) equipped with a ∼1.43 ml cell. Purified pepsin was dialyzed against 50 mM sodium formate, pH 3.0. The DRV and APV solutions were prepared at a final concentration of 160 and 152 μM, respectively, and contained a final concentration of 0.5% DMSO derived from the stock solutions of the inhibitors. DMSO was added to the enzyme solution to give the same 0.5% concentration, in order to eliminate possible thermal effects on dilution of the organic solvent from the titrant. For titration with APV (152 μM), the pepsin solution (21 μM) was titrated with 5-μl aliquots of the inhibitor solution. For titration with DRV (160 μM), the pepsin solution (14.4 μM) was titrated with 6-μl portions of the inhibitor. Analysis of the data was performed using the Origin software provided with the instrument. Attempted ITC of pepsin (14.3 μM) with RTV (240 μM) under the same conditions was unsuccessful because no significant thermal change was observed on titration at 28 °C.
Kinetic inhibition studies
Kinetics of hydrolysis of the chromogenic pepsin substrate, (HPro-Thr-Glu-Phe-[p-NO2-Phe]-Arg-Leu-OH)31, were measured in 120 μl cuvettes at 310 nm in 50 mM sodium formate buffer, pH 3.0-3.2, at 28 °C. For rapid screening of inhibitors, assays were carried out with a final substrate concentration of 175 μM and inhibitor concentrations of 16 μM, with the exception of DMP323, where 24 μM inhibitor was used. Inhibitors that exhibited >50% inhibition under these conditions were selected for further study. Kinetic experiments for determination of Ki contained pepsin at a final concentration of 4.6 μg/ml (0.13 μM). Substrate concentrations were determined from the UV spectrum of the substrate stock solutions (ε28012,000 M−1cm−1). Initial rates (vi) in absorbance at 310 nm (A310) per second, typically observed over the first 10-20% of reaction, were converted to μM/s using a molar absorbance difference between substrate and products of 1800, as reported by Dunn and coworkers31. Linear Dixon plots of 1/vi vs inhibitor concentration were determined in the presence of four substrate concentrations in the range of 60-350 μM, and their slopes were replotted vs 1/[S]. The slopes of these secondary plots are equal to Km/VmaxKi; Ki for each inhibitor was calculated from this relationship by use of values for Km = 93 μM and Vmax = 2.1 μM/s determined in the absence of inhibitor (supporting information Fig. S2).
RESULTS
DSC Analysis of wild-type (PR) and active-site mutants of the mature protease in the absence and presence of different inhibitors
As described in previous studies17,21 we have used a facile method for preparing folded PR in the presence and absence of inhibitors at concentrations suitable for calorimetric studies, in order to assess conveniently the effects of eight diverse inhibitors (Fig. 1) on the thermal stability of mature wild-type protease (PR) as well as the two mutant constructs PRD25N and PRD29N, in which essential Asp residues were replaced with Asn. Three different structural types were examined (Fig. 1): five tight-binding inhibitors that are drugs in current clinical use; a symmetrical inhibitor, DMP323; and the extended, substrate-like inhibitors RPB and Ac-PEP. All inhibitors were present in ∼twofold molar excess over the protein and at concentrations well above their dissociation constants for PR. Thermograms for each of these protein/inhibitor combinations are given in supporting information Fig. S1. Observed Tm values in the presence and absence of the inhibitors are listed in Table 1, as are calculated27 values of the ligand dissociation constants (KL) at the respective Tm's of the complexes. These values were extrapolated to 25 °C for the clinical inhibitors and Ac-PEP by use of literature values for ΔH and Cp of binding for ATV26, SQV30, RTV30, APV28 and Ac-PEP29. For DRV, a ΔH of −12.1 kcal/mol12 was used, with a Cp value of −414 kcal/mol•degree as estimated from an average of known values for clinical inhibitors26. KL values calculated by this method differed significantly from those determined directly by ITC; however there was a consistent free-energy relationship between the ITC values and the present extrapolated values as shown by a logarithmic plot (supporting information Fig. S3). The point for ATV fell far from the correlation line when the positive value of Cp = +470 cal/mol•degree reported by Yanchunas et al.26 was used in the calculation; however when Cp = −470 cal/mol•degree (consistent with all other Cp values for the inhibitors, which were negative) was used for ATV, a reasonable agreement with the rest of the data (Fig. S3) was obtained.
Fig. 1. Structures of the PR inhibitors.

The structures are oriented so that the motif corresponding to P1, that is common to all the clinical inhibitors, as well as analogous substructures in the other inhibitors, are overlaid. Absolute configurations (shown only for the two carbon atoms in this common motif) in the acyclic inhibitors are S for the carbon atoms marked with an asterisk, and otherwise are as indicated. Note that in RPB the hydroxymethylene moiety is replaced by a methylene group (reduced peptide bond) as shown by the arrow.
Effects of the inhibitors on the thermal stability of PRD25N and PRD29N are summarized in relation to PR in Figure 2. With the exception of APV, the clinical inhibitors exhibited very similar patterns of relative thermal stabilization of the three proteins examined (as measured by ΔTm), in particular, stabilization was greatest for PR, least for PRD25N and intermediate for PRD29N. As indicated by Fig. 2, when compared to the clinical inhibitors, DMP323 stabilized PRD29N and PRD25N to about the same extent, and the two extended, peptide-like inhibitors RPB and Ac-PEP, had virtually no effect on the thermal stability of PRD29N.
Fig. 2. Effect of PR inhibitors on ΔTm values for PR, PRD25N and PRD29N.
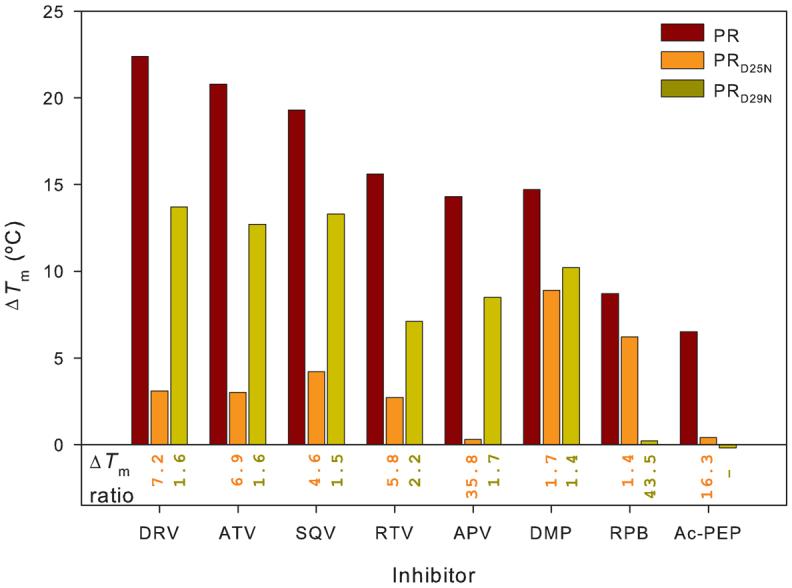
The bar graphs (ΔTm) represent the increase in Tm observed on binding of each of the inhibitors to each of the PR constructs (PR, red; PRD25N, orange; PRD29N, dark yellow). Numbers under the bar graphs are the ratios of ΔTm(PR)/ΔTm(PRD25N) (orange) and ΔTm(PR)/ΔTm(PRD29N) (dark yellow) for each of the inhibitors; large numbers for this ratio indicate that the mutated residue makes a large contribution to inhibitor binding. Data for PR and PRD25N in the absence of inhibitor and in the presence of a twofold molar excess of DRV or RPB are taken from ref. 17.
Assessment of the inhibition of pepsin by HIV-1 protease inhibitors
In light of the marked sensitivity of most of the clinical inhibitors to the highly conserved Asp25 and Asp29 residues, as well as observations that some of these drugs also inhibit the aspartyl proteases of other pathogens such as Candida albicans32,33 and Plasmodium falciparum34, we were interested in investigating the possible interactions of these drugs with another well characterized, monomeric aspartyl protease whose active site bears significant homology with that of PR. Porcine pepsin was chosen as the prototypical monomeric aspartyl protease because of its (1) high homology to human pepsin and (2) commercial availability in sufficient quantities to permit diverse experiments. Even though inhibition of human pepsin by RTV35 and SQV36 has been alluded to, to our knowledge, quantitative data have not been reported.
In order to ensure that the preparation of pepsin used was of sufficient purity and homogeneity for crystallization attempts as well as for the present physical studies, the commercial product was purified by size-exclusion chromatography, which removed significant UV-absorbing, non-protein impurities eluting as a broad peak (retention volume of 80-120 ml) just after the symmetric pepsin peak (retention volume of 62-70 ml) to provide a yield of ∼18% pure pepsin (see Materials and Methods). SDS-polyacrylamide gel analysis and coomassie blue G-250 staining showed the presence of a single band corresponding to the correct molecular size (34.5 kDa). A set of inhibitors consisting of the drugs described above with the addition of INV, LPV, NFV and TPV, as well as DMP323 and RPB, was screened for pepsin inhibition (see Materials and Methods for details). Inhibition was significant (75-95% under the screening conditions) only with DRV, APV and RTV, marginal (≤ 50%) with TPV, LPV, SQV and NFV, and undetected with ATV, INV, DMP323 and RPB.
Binding and inhibition of pepsin by the three most effective inhibitors was further examined kinetically. Fig. 3A shows Dixon plots of the reciprocal of the initial rate of cleavage of the pepsin substrate as a function of DRV concentration at pH 3 and 28 °C in the presence of four substrate concentrations. For competitive inhibitors, the linear plots intersect at a common point with x = –Ki and y = 1/Vmax. Replots of the slopes of these lines against 1/[S] (Fig. 3B) gave lines of slope Km/VmaxKi for each inhibitor, from which Ki values were determined for DRV, RTV and APV (Table 2). Dissociation constants, KL, of 1.54 and 0.18 μM for pepsin/DRV and pepsin/APV, respectively, (Fig. 4) determined by ITC at the same pH and temperature as the kinetic studies, were consistent with the inhibition data (Table 2). Titration of pepsin with RTV at 28 °C produced thermal changes that were virtually identical to those observed upon titration of buffer alone; thus, binding of RTV could not be measured by ITC at this temperature.
Fig. 3. Kinetic inhibition of pepsin by DRV, APV and RTV at pH 3.

For experimental details see text. A. Dixon plots showing the concentration dependence of inhibition by DRV at varying substrate concentrations. The lines intersect at −Ki as required for competitive inhibition. B. Replots of the slopes of the Dixon plots vs. 1/[substrate] for the data shown in A as well as for APV and RTV. The slopes of these secondary plots are equal to Km/VmaxKi for each inhibitor; thus, steeper slopes correspond to smaller values of Ki.
Table 2.
ITC and kinetic data for inhibitor binding to PR and pepsin.
| Enzyme | Ligand | Binding constant (Ka) (M−1) |
ΔH (kcal/mol) |
−TΔS (kcal/mol) |
KLa (nM) |
Kib (nM) |
|---|---|---|---|---|---|---|
| PRc | APVd | (5.0 ± 0.3) × 109 | −6.9 | −6.3 | 0.2 | - |
| RTVd | (1.0 ± 0.1) × 1010 | −4.3 | −9.4 | 0.1 | - | |
| DRVe | 2.2 × 1011 | −12.1 | −3.1 | 0.005 | - | |
| Pepsinf | APV | (5.45 ± 0.5) × 106 | −4.2 ± 0.3 | −5.1 | 180 | 300 |
| RTV | NDg | ND | ND | ND | 600 | |
| DRV | (6.5 ± 0.6) × 105 | −6.0 ± 0.1 | −2.0 | 1540 | 2140 | |
Fig. 4. Calorimetric titration of pepsin with DRV and APV.
The titrations were performed at 28 °C in 50 mM sodium formate buffer, pH 3. Pepsin concentration in the cell was 14.4 μM for the titration with DRV and 21 μM for the titration with APV. Panels A and B are experimental traces of the thermal response on titration with successive aliquots of the inhibitor solutions; panels C and D are integrated data after processing by the instrument's Origin software. The data in panel D were corrected for a baseline of 1.1 kcal/mol per injection of APV. Curve fitting of the integrated data gave a binding constant (KL) of (6.5 ± 0.6) × 105 M and a stoichiometry of 0.90 ± 0.01 for DRV; and a binding constant of (5.5 ± 0.5) × 106 and a stoichiometry of 0.97 ± 0.004 for APV.
On DSC (Fig. 5, upper panel) at pH 3, under the conditions of the ITC and kinetics experiments, unliganded pepsin exhibited a single transition with a Tm of 74.2 °C. This Tm was significantly higher than that reported (62.2 °C) for the major, high temperature transition of native pepsin at pH 5.3, whereas the ΔH (184 kcal/mole) under the present conditions was similar to the value of 176 kcal/mol at pH 5.337. APV, the most effective of the clinical PIs towards pepsin, stabilizes the pepsin/APV complex only slightly (ΔTm 2.7 °C) (Fig. 5, upper panel). In contrast, the tight binding pepsin inhibitor Ac-PEP increased the Tm of pepsin by ≥17 °C.
Fig. 5. DSC thermograms of pepsin at pH 3.0 (upper panel) and PR at pH 4.8 (lower panel) with APV and Ac-PEP.

For conditions see Materials and Methods. The numbers correspond to the apparent Tm of each protein (maximum of the transition) in °C, and ΔTm values on inhibitor binding are shown above the plots. Data are baseline corrected and normalized to the appropriate PR (dimer) or pepsin (monomer) concentrations. Data for PR in the absence of inhibitor are taken from ref. 17. The Tm for pepsin/Ac-PEP represents a lower limit as a result of denaturation and probable aggregation of the protein at the high temperature required for dissociation of the inhibitor, as indicated by the sharp downward trend of the DSC trace.
The substrate specificity of PR is reflected in its natural cleavage sites in Gag-Pol, which generally have an aromatic or hydrophobic residue at P1 and a hydrophobic or proline residue at P1′. For pepsin, both P1 and P1′ are almost always aromatic or non-polar residues. Thus the substrates preferred by these two enzymes have some similarity at the cleavage position, and given the sensitivity of pepsin to PR inhibitors, it was of interest to determine whether there was any cross reactivity between these two enzymes and the substrates designed to be specific for each protease. Incubation of substrate IV with pepsin under the assay conditions described at pH 3 produced an absorbance change at 310 nm, which surprisingly was an increase rather than the decrease observed upon cleavage of the Nle-[p-NO2Phe] bond in this substrate by PR. The reaction was allowed to proceed essentially to completion (∼15 min) and the products were analyzed by MALDI-MS. The most intense peak observed (M/z 778) corresponded to the N-terminal peptide Lys-Ala-Arg-Val-Nle-[p-NO2-Phe] derived from cleavage at the [p-NO2-Phe]-Glu bond of substrate IV. A less intense peak (M/z 586) corresponded to Lys-Ala-Arg-Val-Nle, from cleavage at the normal Nle-[pNO2Phe] site. In contrast, no absorbance change was detected upon incubation of PR with the synthetic pepsin substrate at either pH 3 (pepsin assay conditions) or at pH 5 in the presence of 250 mM NaCl (PR assay conditions).
DISCUSSION
Role of conserved Asp25 and Asp29 residues in inhibitor binding
Previously we examined the role played by the catalytic Asp25 residues of HIV-1 PR in stabilizing the dimeric fold in the presence of inhibitors. When Asp25 is replaced by Asn (PRD25N), the dissociation constant (KL) for DRV, a potent second generation PI, is increased by approximately 6 orders of magnitude. This large change in binding is also reflected by the observed Tm values of PR and PRD25N in the presence of DRV. The Tm for PRD25N is only slightly affected by a twofold molar excess of DRV, whereas that of PR is increased by 22° under similar conditions. We concluded that Asp25, in addition to its catalytic function, makes a major and specific contribution to the binding of DRV. In the present study we have extended these observations, and include the role of a second essential Asp residue, Asp29, in binding of DRV and four other clinically significant inhibitors as well as three other inhibitors of different structural types.
ΔTm is defined as the difference in Tm for a given protein in the presence and absence of inhibitor; thus a large ΔTm correlates with strong inhibitor binding and stabilization of the dimer-inhibitor complex, whereas a small or zero ΔTm correlates with weak or non-existent inhibitor binding. The ΔTm values reported here for PR are in good agreement with, and follow a trend similar to, those previously determined for ATV, SQV, RTV, APV and Ac-PEP by use of a fluorescence method26. Dissociation constants derived from several sources12,17,26,38-40 revealed a qualitatively consistent relationship between small values of KL (high affinity) and large ΔTm's for PR with the entire set of eight inhibitors in the present study, despite the differences in their structure (Table 1).
The selected set of inhibitors can be divided into three types: the tight-binding, non-peptide drugs DRV, ATV, SQV, RTV and APV; the symmetrical inhibitor DMP323; and the substrate-like inhibitors RPB and Ac-PEP. The data of Fig. 2 show that all the inhibitors examined bind more strongly to PR than to either PRD25N or PRD29N, indicative of significant contributions of both Asp25 and Asp29 to the stability of the PR-inhibitor complexes. However different patterns of relative stabilization are observed for the three types of inhibitors. For the five tight-binding drugs, substitution of Asp25 by Asn has a large destabilizing effect on binding, such that these inhibitors increase the Tm of PRD25N by only 0.4-4.2 °C, as compared to 15-22° for PR. The D29N substitution has smaller but significant effects on ΔTm for these inhibitors. In contrast, binding of the cyclic, symmetrical inhibitor DMP323 was less affected by the D25N mutation. Binding of the extended, substrate-like molecules RPB and Ac-PEP was almost completely abolished by the D29N mutation, such that these inhibitors, which increase the Tm of PR by ∼9 and 6.5 °C, respectively, had virtually no effect on the Tm of PRD29N. A major difference between the effects of these latter two inhibitors is the effect of the D25N mutation, such that the binding of Ac-PEP to PRD25N (ΔTm < 1 °C) was virtually nil, whereas the binding of RPB increased its Tm by ∼6 °C.
The ratios ΔTm(PR)/ΔTm(PRD25N) and ΔTm(PR)/ΔTm(PRD29N) (“ΔTm ratio”) for each inhibitor (shown under the bars in Fig. 2) provide a convenient indication of the importance of Asp25 and Asp29, respectively, to the binding of a given inhibitor. Thus all the drugs, with the notable exception of APV, exhibit similar sensitivities to the D25N mutation (ΔTm ratios in the range of 5-7). The much larger value of the ΔTm ratio for APV suggests that Asp25 is relatively more important for the binding of APV than for that of the other drugs. The comparison with DRV, which differs only by a second, fused tetrahydrofuran ring (Fig. 1), is particularly interesting. This additional ring permits new hydrogen bonding interactions with Asp29 that are not possible with APV, and subtle changes in geometry of the PR/DRV complex also likely result in stronger hydrogen bonds with Asp30, Gly27 and Asp30′12. We speculate that these enhanced interactions with DRV may help to compensate for the substitution of Asp25 in the D25N mutant, whereas the binding of APV depends much more heavily on interactions mediated by the catalytic Asp25 residues.
Binding of the five clinical inhibitors examined is less affected (smaller ΔTm ratio) by the Asp29 mutation than Asp25, whereas DMP323 exhibits an equal sensitivity to both mutations. This relative lack of sensitivity to the D25N mutation is quite surprising, but may result from differences between the geometry of the symmetrical pair of hydroxyl groups in DMP323 compared to the single hydroxyl that is present in the other inhibitors (Fig. 1). An interaction between the flap residues Ile50/50′ and the carbonyl oxygen of DMP323, which replaces a structural water in the enzyme-inhibitor complex41, may also provide strong, additional stabilization that can compensate for the loss of interaction with Asp25.
Finally we compare the two substrate-like inhibitors, RPB and Ac-PEP. Both are highly sensitive to the D29N substitution. This is likely because of the extended nature of these substrates that brings their termini close to Asp29. In the PR/Ac-PEP complex, a hydrogen bond between the Val carbonyl and the carboxyl group of Asp29, as well as several backbone hydrogen bonds of both Asp29 and Asp29′ with Val and Ala of the inhibitor, support a role for Asp29. In the PR/RPB complex, hydrogen bonds are possible between the carbonyl group of Thr in the P3 position of RPB and a backbone amino group of Asp29, and between the terminal amide nitrogen of RPB and the backbone carbonyl of Asp29′. RPB binding is much less affected by the D25N mutation (ΔTm ratio 1.4) than Ac-PEP (ΔTm ratio 16.2) because RPB lacks the hydroxyl (shown in red for all the other inhibitors in Fig. 1) that is crucial for interaction with the catalytic Asp carboxyl groups.
The above results of our screening using DSC indicate that, at least for the PIs tested in this study, the interaction(s) of the conserved Asp29 residue of PR is not as critical for the tight binding as is the catalytic Asp25 residue. It would be of general interest to determine if inhibitors that are designed to interact equally very strongly with both these Asp residues exhibit a different drug resistance profile than the current PIs or even curb resistance. Thus, the methodology described in this paper may help to screen and identify future inhibitors with different properties.
Inhibition of porcine pepsin by PR inhibitors
Kinetic evaluation of Ki for the three drugs that were found to inhibit pepsin most effectively, DRV, RTV and APV (Figure 3) indicated a competitive mode of inhibition, with Dixon plots of 1/vi vs inhibitor concentration at different substrate concentrations that intersect at a common point on the abscissa corresponding to −Ki. DRV and APV gave an adequate thermal response to permit determination of the ligand dissociation constant, KL, by ITC. The values obtained (Table 2) were in satisfactory agreement with the kinetically determined values of Ki, as expected for competitive inhibition. Interestingly, for DRV the entropic contributions to the binding constants to PR12 and pepsin (this work) are similar (presumably due to the role of desolvation of the hydrophobic inhibitor); in contrast, the enthalpy for binding of DRV to PR is 12 kcal/mol but only 6 kcal/mol for binding to pepsin. This difference accounts for ∼80% of the 7200 kcal/mol difference in ΔG for binding of DRV to the two enzymes. APV exhibits a similar but less dramatic enthalpic contribution to its selectivity toward PR.
APV is a more effective inhibitor of pepsin than is DRV, but it still has ∼1000-fold lower affinity for pepsin (KL = 180 nM; this work) relative to PR (KL ∼0.2 nM)26,39. Consistent with its weaker binding to pepsin, APV raised the ΔTm of pepsin by only 2.7 °C as compared to 14.3 °C for PR (Fig. 5). Although dimer stabilization concomitant with inhibitor binding could also contribute to the larger ΔTm values for PR/APV in contrast with monomeric pepsin/APV, the large ΔTm is not dependent on the dimeric nature of PR. The prototypical, tight-binding pepsin inhibitor, Ac-PEP (KL ∼50 pM for its isovaleryl analogue, pepstatin A42), raised the Tm of pepsin by ≥17 °C, an effect that is comparable to the ΔTm of 14.3 °C observed for PR/APV (Fig. 5, lower panel). In contrast Ac-PEP had a significantly smaller effect (6.5 °C) on the Tm of PR. The striking reversal of the effects of these two inhibitors on the ΔTm for the two enzymes provides a dramatic demonstration of the effects of tight binding of acetyl pepstatin to pepsin and of APV to PR.
Predicted structure of a pepsin/APV complex
Retroviral proteases are about a third the size of single chain two-domain mammalian (e.g. renin and pepsin) and fungal (e.g. endothiapepsin, penicillopepsin and rhizopuspepsin) aspartic proteases. These monomeric enzymes are proposed to have evolved by gene duplication and fusion of retroviral-like genes6. Crystal structures have been determined for both PR (5HVP)43 and human pepsin (1PSO)44 in the presence of Ac-PEP and pepstatin A, respectively. Despite differences in both size and overall amino acid sequences between monomeric pepsin (Mr 34.5 kDa) and symmetrical, dimeric PR (Mr for the monomer 10.7 kDa), significant homologies exist between their active sites in which the amino acid sequence Asp-Thr-Gly is conserved. In the context of the present study, the Asp25/25′ catalytic pair of PR corresponds to Asp215 and Asp32 in pepsin, whereas the counterparts in pepsin to Asp29/29′ are two Ser residues at positions 219 and 36 (Fig. 5). Some similarities in the overall shape and folding of the two enzymes are also present (Fig. 6).
Fig. 6. Line ribbon representations of the crystal structures of PR co-crystallized with the generic aspartic protease inhibitor, acetyl-pepstatin (5HVP, left) and human pepsin co-crystallized with pepstatin A (1PSO, right).

The two pepstatins differ only in the presence of an acetyl or an isovalyl group at the N-terminus. Only the residues common to both pepstatins are shown (as red sticks). The highly conserved Asp25 (which form part of the active site AspThrGly triad, unique to all aspartic acid proteases) and Asp29 residues in HIV-1 protease that invariably interact with substrates and clinical inhibitors are indicated in red as stick representations. Catalytic Asp32 and Asp215 residues of pepsin, and Ser219 and Ser36 residues that are in equivalent positions to that of Asp29 and Asp29′ of HIV-1 are also indicated in red. The structure of the N-terminal half of pepsin (residues 1-172, middle) mimics that of the single subunit of the HIV-1 protease dimer in its overall structure, substrate binding site and flaps3, 6.
In order to visualize a possible orientation for the bound APV inhibitor and to speculate on its interactions with pepsin in the absence of crystal structure data, we used the pepsin/pepstatin A structure (1PSO) as a starting point and superimposed the corresponding PR/Ac-PEP structure (5HVP) by alignment of the pepstatin moieties. Superimposition of the PR/APV structure (1HPV)45 by alignment of the PR chain with 5HVP was used to position the APV inhibitor. Deletion of the pepstatin and PR moieties resulted in a conceptual picture of how APV might orient relative to pepsin (Fig.7) based on the available crystal data. On the basis of this hypothetical structure the following observations are of interest and may explain why APV binds reasonably well to pepsin, albeit more weakly than to PR. Both enzyme/APV complexes contain a single hydrogen bond between O6 of the tetrahydrofuran ring and Asp29 or the equivalent Ser219, and exhibit potential interactions between the central OH of the inhibitor and the crucial Asp catalytic pair. However, in the case of pepsin these latter hydrogen bonds are much less symmetrical than in the case of PR, such that Asp32 appears to be farther away and consequently less favorably located than Asp215. In contrast, both Asp25 residues of PR are symmetrically positioned at virtually the same, ∼3-Å distance from the OH group of APV. Given the crucial significance of these Asp residues for binding of the clinical inhibitors to PR, as demonstrated by our large effects observed on binding when Asp25 is replaced in the mutant PRD25N, we speculate that the less-favorable interaction between APV and the catalytic Asp residues could provide, at least in part, a reasonable explanation for its ∼3 orders of magnitude lower affinity for pepsin relative to PR. ATV, a highly potent inhibitor of PR, exhibited no detectable inhibition of pepsin even at 16 μM concentration. Its PR complex (2O4K)46, when overlaid with pepsin as described above, suggested the possibility of a steric clash between the aza-biphenyl moiety of ATV and phenylalanines 111 and 117, which might account at least in part for its failure to inhibit pepsin.
Fig. 7. Predicted molecular interactions of pepsin with APV at its active site based on structure overlays of HIV-1 PR/APV (1HPV), HIV-1 PR/Ac-PEP (5HVP) and pepsin/pepstatin A (1PSO) crystal complexes.

Essential aspartyl residues, Asp25 and Asp29 of HIV-1 PR are indicated. The catalytic Asp32 and Asp215 residues of pepsin, and Ser219 and Ser36 residues that are in equivalent positions to that of Asp29 and Asp29′ of PR are also indicated. Distances are indicated in angstroms.
An interesting consequence of the active-site homologies between the monomeric, pepsin-like aspartyl proteases of higher organisms and the homodimeric viral proteases is the observation that some HIV PR inhibitors (PIs) may be effective against other pathogenic organisms that depend on aspartyl proteases for viability and/or virulence. Candida albicans, the most frequent cause of opportunistic infections in patients with HIV/AIDS, produces secreted aspartyl proteases (Saps) that are associated with pathogenicity47. Saps from C. albicans and several other Candida species are inhibited by PIs, most notably RTV and SQV32,48. The relative orientations of the catalytic Asp32 and 218, Ser36 and Thr222 in Sap3 of C. albicans (2H6T)49 when overlaid with the corresponding Asp32 and 215, Ser36 and 219 of pepsin, are almost superimposable. It has also been observed that several PIs inhibit the growth of the malaria parasite Plasmodium falciparum and are active in vitro against aspartyl proteases (plasmepsins) of this organism34,50. Plasmepsins I and II initiate the degradation of hemoglobin, an essential nutrient source for the parasite51. As in the case of C. albicans Sap3, a structural overlay of plasmepsin II (1XDH)52 with pepsin showed a close correspondence between the positions of the catalytic Asp34/Asp214 pair of plasmepsin with Asp32/Asp215 of pepsin and of Ser218 of plasmepsin with Ser219 of pepsin; Ser36 of pepsin, which appeared too far away to interact with APV in the overlaid structure shown in Fig. 7, is equivalent to Ala38 in plasmepsin II.
Although relative inhibitory potencies vs. pepsin, Candida Saps and plasmepsins vary for the PIs and enzymes that have been reported, RTV, the second-best inhibitor of pepsin among the PIs tested in the present study, appears to represent a “consensus”. Of the HIV PIs, RTV was the most tightly bound to Saps 1 and 2 (binding constants in the low μM range33. It was also the most inhibitory PI tested against Saps 1-3 of Candida albicans48 and against Saps from several other Candida species (Ki 0.3-2 μM)32. RTV was also highly effective against P. falciparum in culture50 and only 2-5 times less potent than the most-effective PI, SQV, as an inhibitor of plasmepsins II and IV in solution34. Somewhat surprisingly, APV, which was about twice as effective as RTV as an inhibitor of pepsin, was about four times more weakly bound to C. albicans Sap2 than RTV33 and did not inhibit the growth of P. falciparum53. INV and ATV, which did not inhibit pepsin in the present work, exhibited activities of the same order of magnitude as RTV against P. falciparum in culture34. Despite these variations, the overall similarities among the aspartyl proteases from viral, fungal, protozoan and mammalian sources highlight the crucial role of geometry both of the catalytic aspartates and the residues corresponding to Asp29/Ser219 in HIV-1 PR and pepsin, respectively, as determinants of inhibitor binding.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Annie Aniana and Sharon J. Durham for technical assistance. The nine clinical protease inhibitors (Nelfinavir, Ritonavir, Atazanavir sulfate, Saquinavir, Indinavir sulfate, Amprenavir, Lopinavir, Tipranavir and Darunavir) used in this research were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. This work was supported by the Intramural Research Program of the NIDDK, NIH.
The abbreviations used are
- HIV-1
human immunodeficiency virus type 1
- PR
mature HIV-1 protease
- PI
protease inhibitor
- RPB
reduced peptide bond inhibitor
- APV
Amprenavir
- ATV
Atazanavir
- DRV
Darunavir
- LPV
Lopinavir
- INV
Indinavir (as the sulfate)
- NFV
Nelfinavir
- RTV
Ritonavir
- SQV
Saquinavir
- TPV
Tipranavir
- Ac-PEP
acetyl pepstatin
Footnotes
This article contains supplemental Figs. S1 to S3
REFERENCES
- 1.Tang J, Wong RN. Evolution in the structure and function of aspartic proteases. J Cell Biochem. 1987;33:53–63. doi: 10.1002/jcb.240330106. [DOI] [PubMed] [Google Scholar]
- 2.Khan AR, James MN. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin XL, Lin YZ, Koelsch G, Gustchina A, Wlodawer A, Tang J. Enzymic activities of two-chain pepsinogen, two-chain pepsin, and the amino-terminal lobe of pepsinogen. J Biol Chem. 1992;267:17257–17263. [PubMed] [Google Scholar]
- 4.Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature. 1987;329:351–354. doi: 10.1038/329351a0. [DOI] [PubMed] [Google Scholar]
- 5.Blundell TL, Johnson MS. Catching a common fold. Protein Sci. 1993;2:877–883. doi: 10.1002/pro.5560020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin XL, Lin YZ, Tang J. Relationships of human immunodeficiency virus protease with eukaryotic aspartic proteases. Methods Enzymol. 1994;241:195–224. doi: 10.1016/0076-6879(94)41066-6. [DOI] [PubMed] [Google Scholar]
- 7.Oroszlan S, Luftig RB. Retroviral proteinases. Curr Top Microbiol Immunol. 1990;157:153–185. doi: 10.1007/978-3-642-75218-6_6. [DOI] [PubMed] [Google Scholar]
- 8.Louis JM, Weber IT, Tozser J, Clore GM, Gronenborn AM. HIV-1 protease: maturation, enzyme specificity, and drug resistance. Adv Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- 9.Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 Protease: Structure, Dynamics, and Inhibition. Adv Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- 10.Louis JM, Nashed NT, Parris KD, Kimmel AR, Jerina DM. Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc Natl Acad Sci U S A. 1994;91:7970–7974. doi: 10.1073/pnas.91.17.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 12.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louis JM, Smith CA, Wondrak EM, Mora PT, Oroszlan S. Substitution mutations of the highly conserved arginine 87 of HIV-1 protease result in loss of proteolytic activity. Biochem Biophys Res Commun. 1989;164:30–38. doi: 10.1016/0006-291x(89)91678-1. [DOI] [PubMed] [Google Scholar]
- 15.Weber IT. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J Biol Chem. 1990;265:10492–10496. [PubMed] [Google Scholar]
- 16.Ishima R, Ghirlando R, Tozser J, Gronenborn AM, Torchia DA, Louis JM. Folded monomer of HIV-1 protease. J Biol Chem. 2001;276:49110–49116. doi: 10.1074/jbc.M108136200. [DOI] [PubMed] [Google Scholar]
- 17.Sayer JM, Liu F, Ishima R, Weber IT, Louis JM. Effect of the active-site D25N mutation on the structure, stability and ligand binding of the mature HIV-1 protease. J Biol Chem. 2008 doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahalingam B, Boross P, Wang YF, Louis JM, Fischer CC, Tozser J, Harrison RW, Weber IT. Combining mutations in HIV-1 protease to understand mechanisms of resistance. Proteins. 2002;48:107–116. doi: 10.1002/prot.10140. [DOI] [PubMed] [Google Scholar]
- 19.Korting HC, Schaller M, Eder G, Hamm G, Bohmer U, Hube B. Effects of the human immunodeficiency virus (HIV) proteinase inhibitors saquinavir and indinavir on in vitro activities of secreted aspartyl proteinases of Candida albicans isolates from HIV-infected patients. Antimicrob Agents Chemother. 1999;43:2038–2042. doi: 10.1128/aac.43.8.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atzori C, Angeli E, Mainini A, Agostoni F, Micheli V, Cargnel A. In vitro activity of human immunodeficiency virus protease inhibitors against Pneumocystis carinii. J Infect Dis. 2000;181:1629–1634. doi: 10.1086/315437. [DOI] [PubMed] [Google Scholar]
- 21.Ishima R, Torchia DA, Louis JM. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor. J Biol Chem. 2007;282:17190–17199. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- 22.Katoh E, Louis JM, Yamazaki T, Gronenborn AM, Torchia DA, Ishima R. A solution NMR study of the binding kinetics and the internal dynamics of an HIV-1 protease-substrate complex. Protein Sci. 2003;12:1376–1385. doi: 10.1110/ps.0300703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis JM, Ishima R, Nesheiwat I, Pannell LK, Lynch SM, Torchia DA, Gronenborn AM. Revisiting monomeric HIV-1 protease. Characterization and redesign for improved properties. J Biol Chem. 2003;278:6085–6092. doi: 10.1074/jbc.M209726200. [DOI] [PubMed] [Google Scholar]
- 24.Wondrak EM, Louis JM. Influence of flanking sequences on the dimer stability of human immunodeficiency virus type 1 protease. Biochemistry. 1996;35:12957–12962. doi: 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- 25.Todd MJ, Semo N, Freire E. The structural stability of the HIV-1 protease. J Mol Biol. 1998;283:475–488. doi: 10.1006/jmbi.1998.2090. [DOI] [PubMed] [Google Scholar]
- 26.Yanchunas J, Jr., Langley DR, Tao L, Rose RE, Friborg J, Colonno RJ, Doyle ML. Molecular basis for increased susceptibility of isolates with atazanavir resistance-conferring substitution I50L to other protease inhibitors. Antimicrob Agents Chemother. 2005;49:3825–3832. doi: 10.1128/AAC.49.9.3825-3832.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandts JF, Lin LN. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry. 1990;29:6927–6940. doi: 10.1021/bi00481a024. [DOI] [PubMed] [Google Scholar]
- 28.Ohtaka H, Velazquez-Campoy A, Xie D, Freire E. Overcoming drug resistance in HIV-1 chemotherapy: the binding thermodynamics of Amprenavir and TMC-126 to wild-type and drug-resistant mutants of the HIV-1 protease. Protein Sci. 2002;11:1908–1916. doi: 10.1110/ps.0206402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luque I, Todd MJ, Gomez J, Semo N, Freire E. Molecular basis of resistance to HIV-1 protease inhibition: a plausible hypothesis. Biochemistry. 1998;37:5791–5797. doi: 10.1021/bi9802521. [DOI] [PubMed] [Google Scholar]
- 30.Todd MJ, Luque I, Velazquez-Campoy A, Freire E. Thermodynamic basis of resistance to HIV-1 protease inhibition: calorimetric analysis of the V82F/I84V active site resistant mutant. Biochemistry. 2000;39:11876–11883. doi: 10.1021/bi001013s. [DOI] [PubMed] [Google Scholar]
- 31.Dunn BM, Kammermann B, McCurry KR. The synthesis, purification, and evaluation of a chromophoric substrate for pepsin and other aspartyl proteases: design of a substrate based on subsite preferences. Anal Biochem. 1984;138:68–73. doi: 10.1016/0003-2697(84)90770-x. [DOI] [PubMed] [Google Scholar]
- 32.Pichova I, Pavlickova L, Dostal J, Dolejsi E, Hruskova-Heidingsfeldova O, Weber J, Ruml T, Soucek M. Secreted aspartic proteases of Candida albicans, Candida tropicalis, Candida parapsilosis and Candida lusitaniae. Inhibition with peptidomimetic inhibitors. Eur J Biochem. 2001;268:2669–2677. doi: 10.1046/j.1432-1327.2001.02152.x. [DOI] [PubMed] [Google Scholar]
- 33.Backman D, Monod M, Danielson UH. Biosensor-based screening and characterization of HIV-1 inhibitor interactions with Sap 1, Sap 2, and Sap 3 from Candida albicans. J Biomol Screen. 2006;11:165–175. doi: 10.1177/1087057105284270. [DOI] [PubMed] [Google Scholar]
- 34.Andrews KT, Fairlie DP, Madala PK, Ray J, Wyatt DM, Hilton PM, Melville LA, Beattie L, Gardiner DL, Reid RC, Stoermer MJ, Skinner-Adams T, Berry C, McCarthy JS. Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria. Antimicrob Agents Chemother. 2006;50:639–648. doi: 10.1128/AAC.50.2.639-648.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA, Green BE, Fino L, Park CH, Kong XP. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci U S A. 1995;92:2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A, et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248:358–361. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- 37.Dee D, Pencer J, Nieh MP, Krueger S, Katsaras J, Yada RY. Comparison of solution structures and stabilities of native, partially unfolded and partially refolded pepsin. Biochemistry. 2006;45:13982–13992. doi: 10.1021/bi061270i. [DOI] [PubMed] [Google Scholar]
- 38.Markgren PO, Schaal W, Hamalainen M, Karlen A, Hallberg A, Samuelsson B, Danielson UH. Relationships between structure and interaction kinetics for HIV-1 protease inhibitors. J Med Chem. 2002;45:5430–5439. doi: 10.1021/jm0208370. [DOI] [PubMed] [Google Scholar]
- 39.Brower ET, Bacha UM, Kawasaki Y, Freire E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem Biol Drug Des. 2008;71:298–305. doi: 10.1111/j.1747-0285.2008.00647.x. [DOI] [PubMed] [Google Scholar]
- 40.Ohtaka H, Schon A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42:13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- 41.Lam PY, Ru Y, Jadhav PK, Aldrich PE, DeLucca GV, Eyermann CJ, Chang CH, Emmett G, Holler ER, Daneker WF, Li L, Confalone PN, McHugh RJ, Han Q, Li R, Markwalder JA, Seitz SP, Sharpe TR, Bacheler LT, Rayner MM, Klabe RM, Shum L, Winslow DL, Kornhauser DM, Hodge CN. Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2′ structure-activity relationship, and molecular recognition of cyclic ureas. J Med Chem. 1996;39:3514–3525. doi: 10.1021/jm9602571. [DOI] [PubMed] [Google Scholar]
- 42.Workman RJ, Burkitt DW. Pepsin inhibition by a high specific activity radioiodinated derivative of pepstatin. Arch Biochem Biophys. 1979;194:157–164. doi: 10.1016/0003-9861(79)90605-2. [DOI] [PubMed] [Google Scholar]
- 43.Fitzgerald PM, McKeever BM, VanMiddlesworth JF, Springer JP, Heimbach JC, Leu CT, Herber WK, Dixon RA, Darke PL. Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0-A resolution. J Biol Chem. 1990;265:14209–14219. [PubMed] [Google Scholar]
- 44.Fujinaga M, Chernaia MM, Tarasova NI, Mosimann SC, James MN. Crystal structure of human pepsin and its complex with pepstatin. Protein Sci. 1995;4:960–972. doi: 10.1002/pro.5560040516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim EE, Baker CT, Dwyer MD, Murcho MA, Rao BG, Tung RD, Navia MA. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117:1181–1182. [Google Scholar]
- 46.Muzammil S, Armstrong AA, Kang LW, Jakalian A, Bonneau PR, Schmelmer V, Amzel LM, Freire E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J Virol. 2007;81:5144–5154. doi: 10.1128/JVI.02706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bein M, Schaller M, Korting HC. The secreted aspartic proteinases as a new target in the therapy of candidiasis. Curr Drug Targets. 2002;3:351–357. doi: 10.2174/1389450023347542. [DOI] [PubMed] [Google Scholar]
- 48.Borg-von Zepelin M, Meyer I, Thomssen R, Wurzner R, Sanglard D, Telenti A, Monod M. HIV-Protease inhibitors reduce cell adherence of Candida albicans strains by inhibition of yeast secreted aspartic proteases. J Invest Dermatol. 1999;113:747–751. doi: 10.1046/j.1523-1747.1999.00747.x. [DOI] [PubMed] [Google Scholar]
- 49.Borelli C, Ruge E, Schaller M, Monod M, Korting HC, Huber R, Maskos K. The crystal structure of the secreted aspartic proteinase 3 from Candida albicans and its complex with pepstatin A. Proteins. 2007;68:738–748. doi: 10.1002/prot.21425. [DOI] [PubMed] [Google Scholar]
- 50.Parikh S, Gut J, Istvan E, Goldberg DE, Havlir DV, Rosenthal PJ. Antimalarial activity of human immunodeficiency virus type 1 protease inhibitors. Antimicrob Agents Chemother. 2005;49:2983–2985. doi: 10.1128/AAC.49.7.2983-2985.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci U S A. 2002;99:990–995. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Binkert C, Frigerio M, Jones A, Meyer S, Pesenti C, Prade L, Viani F, Zanda M. Replacement of isobutyl by trifluoromethyl in pepstatin A selectively affects inhibition of aspartic proteinases. Chembiochem. 2006;7:181–186. doi: 10.1002/cbic.200500180. [DOI] [PubMed] [Google Scholar]
- 53.Skinner-Adams TS, McCarthy JS, Gardiner DL, Hilton PM, Andrews KT. Antiretrovirals as antimalarial agents. J Infect Dis. 2004;190:1998–2000. doi: 10.1086/425584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.