

Abstract
Juvenile Neuronal Ceroid Lipofuscinosis (JNCL) results from a deficiency of CLN3, a protein recently identified within detergent-resistant membranes (DRMs). To study the function of CLN3 within these domains we isolated DRMs from control and JNCL-brain and noted that JNCL-derived DRMs are less buoyant than control. Analysis of DRM phospholipids derived from JNCL-brain revealed a reduction of bis(monoacylglycerol)phosphate. Metabolic labeling of JNCL-fibroblasts demonstrated a reduction in the synthesis of bis(monoacylglycerol)phosphate which was restored following complementation with wild-type-CLN3, substantiating our initial observation in brain. Metabolic labeling of cell lines overexpressing wild-type-CLN3 resulted in increased bis(monoacylglycerol)phosphate synthesis, while overexpression of mutant CLN3-L170P decreased bis(monoacylglycerol)phosphate synthesis. These data illustrate a new finding, a strong correlation between CLN3 protein expression and synthesis of bis(monoacylglycerol)phosphate.
Keywords: Neuronal Ceroid Lipofuscinosis, Batten disease, CLN3, bis(monoacylglycerol)phosphate, lysobisphosphatidic acid, detergent-resistant microdomains
1. Introduction
Deficiency of CLN3 protein is the underlying cause of Juvenile Neuronal Ceroid Lipofuscinosis (JNCL). This lysosomal storage disease typically manifests between the ages of 4 and 10 and is characterized by progressive vision loss, seizures and dementia. JNCL belongs to a group of disorders known as the Neuronal Ceroid Lipofuscinoses (NCLs) that share a common pathology of lysosomal accumulation of autofluorescent lipopigment and collectively are one of the most common causes of inherited childhood neurodegeneration. The genes associated with NCL encode a diverse group of both soluble and membrane bound proteins that reside in multiple cellular compartments. Single deficiencies in any of four soluble proteins, palmitoyl-protein thioesterase-1 [1], tripeptidyl peptidase-1 [2], cathepsin D [3] or cathepsin F [4], or of six membrane bound proteins, CLN3 [5], CLN5 [6], CLN6 [7], CLN8 [8], CLC-3 [9; 10] or CLC-7 [11], have been associated with the development of NCL in either human or animal models.
CLN3 protein is highly hydrophobic and its expression has been detected in multiple human tissues including brain, pancreatic islets, peripheral nerve, spleen and testis [12]. Topology prediction studies suggest that the CLN3 protein contains 5–6 transmembrane spanning domains [13], and detergent partitioning experiments revealed that it spontaneously integrates with cellular membranes [14]. Western blot analysis of lipid rafts, or detergent-resistant microdomains (DRMs), isolated from bovine brain demonstrated that CLN3 protein exists within these hydrophobic domains [15]. To further examine the role of CLN3 protein within DRMs we isolated DRMs from normal and JNCL-brain and noted that DRMs derived from JNCL-brain were less buoyant than control. As the buoyancy of DRMs is directly related to their lipid composition, we analyzed the phospholipid composition of DRMs and total lipids isolated from CLN3-deficient human brain. In addition, we studied the ability of CLN3-deficient fibroblasts and LA-N-5 cell lines overexpressing wild-type or mutant CLN3-L170P protein to synthesize phospholipids. Our data demonstrate that expression of CLN3 protein is directly correlated with the synthesis of the phospholipid bis(monoacylglycerol)phosphate.
2. Materials and methodsa
2.1. Chemicals and reagents
[3H]-Palmitic acid (43 Ci/mmol) was purchased from New England Nuclear (Boston, MA). The protein assay kit was from Bio-Rad (Hercules, CA). Human autopsy brains HSB 635 (control, 15 year old male, frontal cortex) and HSB 3187 (CLN3-deficient, 16 year old male, frontal cortex) were obtained from Human Brain and Spinal Fluid Resource Center, Los Angeles California. CLN3-deficient fibroblasts were a kind gift of Dr. K. E. Wisniewski, Staten Island, New York. The neuron-like LA-N-5 cell line was obtained from Dr. J. Kanfer, University of Manitoba, Canada [16].
2.2. CLN3 expression plasmid construction and generation of stable cell lines
The complete wild-type-CLN3 cDNA was amplified by PCR using oligonucleotide primers 5′-ATGGGAGGCTGTGCA-3′ and 5′-TCAGGAGAGCTGGCAGAGGAA-3′ designed to allow for directional cloning into the p3XFLAG-CMV-14 vector multiple cloning site (Sigma) to produce a C-terminal FLAG CLN3. Likewise, complete WT CLN3 cDNA was amplified by PCR using oligonucleotide primers 5′-TTGAATTCGGCCACCATGGGAGGCTGTGCA-3′ and 5′-TAAGGATCCTCAGGAGAGCTGGCAGAGG AA-3′ designed to allow for directional cloning into the p3XFLAG-CMV-9 vector multiple cloning site (Sigma) to produce a N-terminal FLAG CLN3. LA-N-5 cells were electroporated with 5μg of either p3XFLAG-CMV-14-CLN3 or p3XFLAG-CMV-9-CLN3 expression vectors. Stable clones were selected in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with neomycin (500 μg/mL), and individual clones isolated by limiting dilution. Clones were screened for CLN3 protein expression by western blot analysis using anti-FLAG antibody (Sigma) according to manufacturers specifications.
The CLN3-L170P point mutant was generated using the megaprimer method [17]. Briefly, WT CLN3 cDNA was amplified by PCR using oligonucleotide primers 5′-ATGGGAGGCTGTGCA-3′ and 5′-AGGCAGTGAGGGAGGGGAAGG-3′ to generate a megaprimer containing the point mutation. This megaprimer was then gel purified and utilized in a subsequent PCR reaction with 5′-TCAGGAGAGCTGGCAGAGGAA-3′ to amplify the entire CLN3-L170P cDNA. The PCR product was cloned into pGEM-T (Invitrogen) and subsequently cloned into the p3XFLAG-CMV-14 expression vector. Stable cell lines were generated as described above.
All plasmid constructs were assessed for correct orientation by restriction digestion analysis, and all PCR-amplified DNA sequences were verified by DNA sequencing.
2.3. Cell culture and transient transfection
Fibroblasts and LA-N-5 cells were grown in monolayers on 100-mm tissue culture dishes in DMEM supplemented with 10% fetal bovine serum and 1% gentamicin. LA-N-5 cells were originally derived from a human adrenal gland tumor and resemble neurons [16]. The LA-N-5 cell lines expressing FLAG-tagged CLN3 and CLN3-L170P were grown in DMEM supplemented with 10% fetal bovine serum, 1% gentamicin and 500 μg/mL neomycin. All cells were maintained in a humidified incubator at 37°C and 10% CO2.
CLN3-deficient patient derived fibroblasts were transiently transfected with p3XFLAG-CMV-14 vector or the p3XFLAG-CMV-14 vector containing wild-type (WT) CLN3 cDNA. Surviving cells were allowed to adhere for 6 hours at 37°C, at which time media was removed, cells were gently washed with PBS, and used in experiments.
2.4. Sucrose gradient fractionation
Detergent-resistant microdomains, were isolated by their insolubility in Triton-X 100 at 4°C as previously described [18; 19]. Briefly, cells and brain tissue were lysed in 1.5 mL of 25mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 6.5, 150 mM NaCl, 1% Triton-X 100, supplemented with protease inhibitor cocktail (leupeptin, phenylmethylsulfonyl fluoride and aprotinin) (Pierce) and incubated on ice for 1 hour. The lysates were homogenized by 10–20 strokes in a loose-fit Dounce homogenizer. Homogenates were mixed with 1.5 mL of 80% sucrose in MBS (25mM MES, pH 6.5, 150 mM NaCl) and overlaid with 3 mL of 30% sucrose in MBS followed by 3 mL of 5% sucrose in MBS. Homogenates were centrifuged for 18 hours at 31,000 rpm in an SW41 swinging-bucket rotor and 1 mL fractions were collected from the top down for Western blot analysis. The DRM fraction was typically found in fractions 3 and 4.
2.5. Western blot analysis
Sucrose gradient fractions were separated on 12% SDS-PAGE gels, transferred to Immobilon-P membranes (Milli-Q) and blocked as directed by the antibody manufacturer. The Flotillin-2/ESA monoclonal antibody (BD Biosciences) was detected with horseradish peroxidase coupled anti-mouse secondary antibody and protein bands were detected by chemiluminescence.
To determine the relative expression of CLN3-FLAG protein in LA-N-5 cell lines whole cell lysates containing 20 μg of total protein were separated on 12% SDS-PAGE gels. Following electrophoresis, proteins were transferred to Immobilon-P membranes (Milli-Q) and blocked as directed by the antibody manufacturer. M2-antibody (Sigma) was detected using horseradish peroxidase-coupled anti-mouse secondary antibody protein bands were detected by chemiluminescence.
2.6. Lipid synthesis, extraction and HPTLC
Cells were cultured for 18 hours in media containing 1 mCi/mL [3H]-palmitate. Under these conditions, sphingolipids are labeled and palmitate is converted into acetate and recycled such that de novo-synthesized cholesterol is also labeled. Lipids were extracted as previously described [20]. Briefly, for total lipid analysis, cells were washed three times with phosphate buffered saline, pH 7.4 and collected, centrifuged, and the cell pellet resuspended in Milli-Q water. Following sonication, lipids were extracted according to Folch [20]. For analysis of DRM lipids, sucrose fraction four was dialyzed overnight against 5L water at 4°C to remove sucrose. A Folch extraction was then performed using the total volume after dialysis.
Lipids were analyzed by 2D-high performance thin layer chromatography (HPTLC) using 10 × 10-cm LHP-K TLC plates (Whatman, Inc.). Lipids from cell extracts were separated in the first dimension using chloroform: methanol: acetic acid: water (75:25:8.8:4.5 v/v) and second dimension using chloroform: methanol: ammonium hydroxide: water (92:36:2.8:3.1 v/v). TLC plates were sprayed with EN3HANCE (Perkin-Elmer) to facilitate autoradiography and bands were excised for radioactivity determination by liquid scintillation counting.
Lipids from brain extracts were separated two times in the first dimension using chloroform: methanol: ammonium hydroxide (65:20:4 v/v) and second dimension using chloroform: acetone: methanol: acetic acid: water (50:20:10:10:5 v/v). Brain lipids were visualized by charring; HPTLC plates were sprayed with a solution of 10% CuSO4, 8% H3PO4 and heated to 180°C.
Individual lipids were unequivocally identified by the migration of authentic lipid standards in two independent solvent systems.
2.7. Lipid phosphorus assay
Lipid phosphorus assay was done as previously described [21].
3. Results
3.1. CLN3-deficient DRMs are less buoyant than control
Following centrifugation of sucrose gradients during the isolation of DRMs we visually observed that DRMs derived from JNCL-brain appeared to be more diffuse than control. To verify this observation, we assessed the localization of Flotillin-2 ESA, a known DRM protein marker. Western blot analysis of brain sucrose gradient fractions revealed that, unlike control, CLN3-deficient brain exhibited a downward shift of Flotillin-2 localization within the sucrose gradient, indicating that CLN3-deficient brain (HSB 3187) DRMs are less buoyant than those derived from control brain (HSB 635) (Fig. 1).
Figure 1. DRMs isolated from CLN3-deficient brain are less buoyant than control.

Equivalent weights of control and CLN3-deficient human autopsy brains were homogenized, protein standardized, and fractionated on a sucrose gradient. A portion of each sucrose gradient fraction was probed for Flotillin-2 by Western blot. Results are representative of at least three independent experiments.
3.2. BMP content of CLN3-deficient human brain is reduced
To investigate whether an alteration in relative lipid composition might explain the observed decrease in the buoyancy of CLN3-deficient brain DRMs, we isolated DRMs and total lipids from normal and CLN3-deficient brain. Equivalent amounts of lipids, as determined by lipid phosphorus assay, were separated by 2D-HPTLC. Comparison of phospholipids extracted from DRMs or total brain revealed a substantial reduction in the total amount of BMP exists in JNCL-brain (Fig. 2A and 2B).
Figure 2. BMP is reduced in detergent resistant microdomains and total lipids isolated from JNCL-brain.
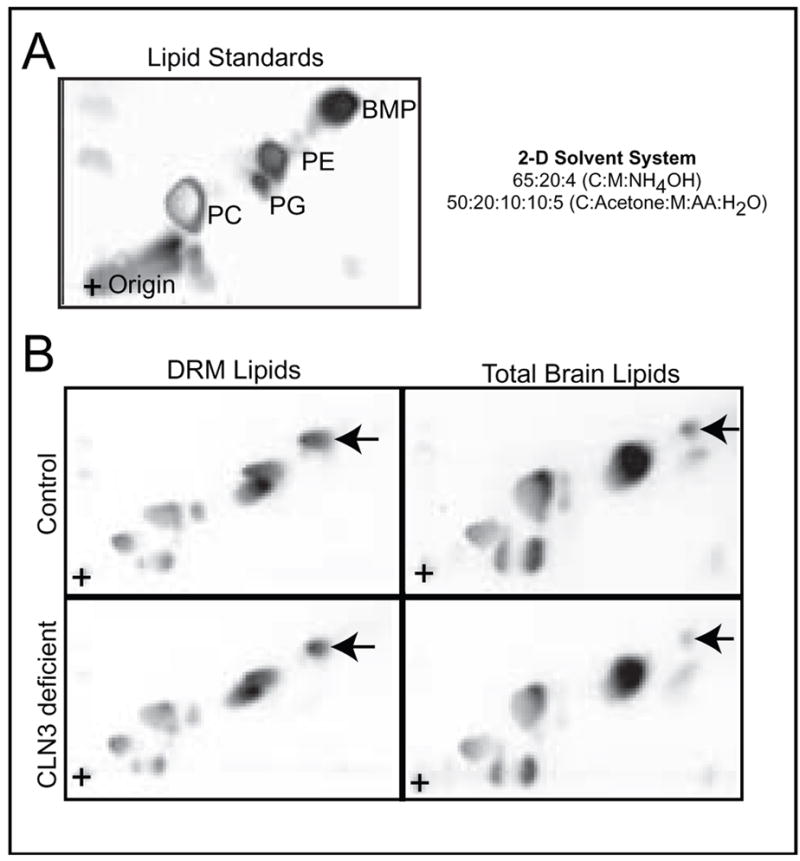
(A) Lipid standards. (B) DRM lipids (Left) and total lipids (Right) were isolated from control and CLN3-deficient brain. Equivalent amount of brain lipids were separated by 2D-HPTLC and visualized by charring. BMP is indicated by the arrow. Results are representative of at least three independent experiments.
3.3. BMP synthesis is reduced in CLN3-deficient patient derived fibroblasts
Since autopsy-derived patient brain tissue is no longer metabolically active we could only examine the steady-state levels of lipids. To assess the relative ability of normal and CLN3-deficient fibroblasts to synthesize BMP, equal numbers of cells were metabolically labeled with [3H]-palmitate, which is incorporated into newly synthesized lipids. After harvesting, DRMs were isolated and phospholipids were extracted and analyzed by 2D-HPTLC followed by autoradiography. Comparison of normal and CLN3-deficient fibroblasts revealed that de novo synthesis of [3H]-labeled BMP was decreased in CLN3-deficient cell lines (Fig. 3A and 3B), corroborating our steady state measurement in brain.
Figure 3. BMP synthesis is reduced in detergent resistant microdomains isolated from JNCL patient derived fibroblasts.
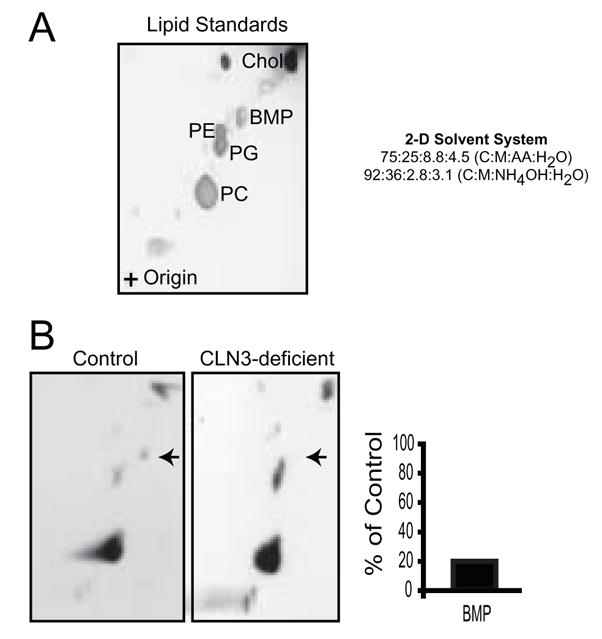
(A) Lipid standards. (B) Control and CLN3-deficient human fibroblast cell lines were incubated with [3H]-palmitate for 18 hours. Equivalent number of cells were harvested and DRMs were isolated. DRM lipids were extracted, separated by 2D-HPTLC, and visualized by autoradiography. BMP is indicated by the arrow. Bands were excised from the TLC plates and [3H]-palmitate incorporation was measured for each of the lipid species. Results are representative of at least three independent experiments in three independent patient cell lines.
3.4. BMP synthesis in DRMs and total lipid extract is directly correlated with CLN3 protein expression
To further evaluate the role of CLN3 protein expression in the synthesis of BMP, we generated LA-N-5 neuroblastoma cell lines that overexpressed either wild-type-CLN3 or its non-functional mutant CLN3-L170P [22]. Western blot analysis of LA-N-5 whole cell lysates demonstrated expression of wild-type-CLN3-FLAG or mutant CLN3-L170P FLAG protein only in extracts derived from transfected cell lines (Fig. 4A). To ascertain the relative abilities of these cell lines to synthesize phospholipids we measured the incorporation of [3H]-palmitate into lipids isolated from DRMs and total lipid extract. Analysis of [3H]-palmitate incorporation into newly synthesized lipids within both DRMs (Fig. 4B) and total lipid extracts (Fig. 4C) revealed a pronounced increase in BMP synthesis in cell lines overexpressing either N or C terminal FLAG CLN3. Conversely, overexpression of the non-functional mutant CLN3-L170P resulted in a reduction in BMP derived from both DRMs (Fig. 4B) and total lipid extracts (Fig. 4C).
Figure 4. Overexpression of CLN3 increases BMP content of DRMs and total lipids and restores BMP synthesis in patient derived cell lines.

(A) Total cell extracts from LA-N-5 cell lines were probed by Western blot using an anti-FLAG antibody Lane 1: LA-N-5 WT; Lane 2: vector only; Lane 3: C-terminal FLAG CLN3; Lane 4: C-terminal FLAG L170P CLN3; Lane 5: N-terminal FLAG CLN3. Results are representative of at least three independent experiments. (B) LA-N-5 cell lines: parental cell line (WT), vector transfected (Vector), transfected with C-terminal FLAG CLN3 cDNA containing missense mutation L170P (C-CLN3 L170P), N-terminal FLAG-CLN3 tranfected (N-CLN3), or C-terminal FLAG-CLN3 transfected (C-CLN3) were incubated with [3H]-palmitate for 18 hours. Cells were harvested, protein standardized, fractionated on a sucrose gradient, lipids were extracted from the DRM fraction, and separated by 2D-HPTLC. Bands were excised from TLC plates and [3H]-palmitate incorporation was measured for each lipid. Results are representative of at least three independent experiments. (C) LA-N-5 cell lines: parental cell line (WT), vector transfected (Vector), transfected with C-terminal FLAG CLN3 cDNA containing missense mutation L170P (C-CLN3 L170P), N-terminal FLAG-CLN3 tranfected (N-CLN3), or C-terminal FLAG-CLN3 transfected (C-CLN3) were incubated with [3H]-palmitate for 18 hours. Cells were harvested, protein standardized, and total lipids were extracted and separated by 2D-HPTLC. Bands were excised from TLC plates and [3H]-palmitate incorporation was measured for each lipid. Results are representative of at least three independent experiments. (D) Patient derived CLN3-deficient fibroblasts were transfected with either an empty C-terminal FLAG vector or the same vector containing WT CLN3 cDNA. Cells were labeled for 24 hours with [3H]-palmitate and harvested 30 hours post-transfection. Total lipids were isolated, separated by 2D-HPTLC, visualized by autoradiography, and quantitated by scintillation counting. Data shown is displayed as % of vector transfected. Results are from a single experiment that are representative at least three independent experiments.
3.5. CLN3-deficient patient fibroblasts are rescued by transcomplementation with wild-type-CLN3
In order to determine if the defective BMP synthesis observed in CLN3-deficient fibroblasts was a direct result of CLN3 deficiency, we asked whether BMP synthesis could be restored by transient expression of wild-type-CLN3. Western blot analysis of transiently transfected patient derived cell lines revealed that CLN3-FLAG protein expression peaked at 24 hours after transfection (data not shown). Transfected patient fibroblasts were then assayed for their ability to synthesize BMP by labeling cells with [3H]-palmitate for 24 hours beginning at 6 hours post transfection. Total lipids were separated by 2D-HPTLC and individual lipids identified and quantified. BMP synthesis was restored to 260% of control cells, while synthesis of other lipids remained the same (Fig. 4D)
4. Discussion
DRM preparations selectively enrich for cholesterol-rich membranes comprised of plasma membrane DRMs and late endosomal DRMs [23]. In this study we show that deficiency of CLN3 protein results in less buoyant DRMs (Fig. 1) and that the change in buoyancy corresponds to altered lipid composition (Fig. 2B). Analysis of total lipids revealed that total BMP level is reduced in JNCL-brain (Fig. 2B). To determine if this observation was a result of CLN3 deficiency we incubated both patient-derived fibroblasts and overexpressing neuroblastoma cell lines with 3[H]-palmitic acid to label newly synthesized phospholipids. Metabolic labeling studies demonstrated that overexpression of wild-type-CLN3 protein resulted in increased synthesis of BMP (Fig. 4A, B, C), whereas CLN3-deficient patient fibroblasts (Fig. 3B) or overexpression of the missence mutant CLN3-L170P (Fig. 4A, B, C), reduced synthesis of BMP, corroborating our steady-state measurements in brain. Further, complementation of JNCL-fibroblasts with C-terminal FLAG CLN3 was sufficient to restore their ability to synthesize BMP (Fig. 4D).
Numerous functions for CLN3 protein have been proposed, including roles in vesicle acidification [24; 25], amino acid transport [26; 27], and more recently, palmitoyl-protein delta-9 desaturase activity [28]. Our work shows, for the first time, a new function of the CLN3 protein: a role in BMP biosynthesis. As is the case for most of the previously proposed roles for CLN3 protein, our data do not establish whether the role of CLN3 protein in BMP synthesis is a primary function or a secondarily mediated effect and further study is needed to address this issue. Nevertheless, in view of the described roles of BMP in cellular function it is readily apparent that significant reductions of BMP within detergent-resistant microdomains could alter the biophysical properties of those membranes and impact membrane dynamics and flow, as well as the function of enzymes that require BMP for optimal functioning [29; 30; 31; 32; 33; 34; 35]. Reduced BMP, therefore, could result in lysosomal pathology.
Acknowledgments
The authors thank Dr. Marvin R. Natowicz for his critical discussion and thoughtful review of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waliany S, Das AK, Gaben A, Wisniewski KE, Hofmann SL. Identification of three novel mutations of the palmitoyl-protein thioesterase-1 (PPT1) gene in children with neuronal ceroid-lipofuscinosis. Hum Mutat. 2000;15:206–7. doi: 10.1002/(SICI)1098-1004(200002)15:2<206::AID-HUMU14>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.Sohar I, Sleat DE, Jadot M, Lobel P. Biochemical characterization of a lysosomal protease deficient in classical late infantile neuronal ceroid lipofuscinosis (LINCL) and development of an enzyme-based assay for diagnosis and exclusion of LINCL in human specimens and animal models. J Neurochem. 1999;73:700–11. doi: 10.1046/j.1471-4159.1999.0730700.x. [DOI] [PubMed] [Google Scholar]
- 3.Tyynela J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M, Haltia M, Lobel P. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. Embo J. 2000;19:2786–92. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang CH, Lee JW, Galvez MG, Robillard L, Mole SE, Chapman HA. Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease. Mol Cell Biol. 2006;26:2309–16. doi: 10.1128/MCB.26.6.2309-2316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell. 1995;82:949–57. doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 6.Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. 1998;19:286–8. doi: 10.1038/975. [DOI] [PubMed] [Google Scholar]
- 7.Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. 2002;70:324–35. doi: 10.1086/338190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, Sharp J, Wheeler R, Kusumi K, Mole S, Liu W, Soares MB, Bonaldo MF, Hirvasniemi A, de la Chapelle A, Gilliam TC, Lehesjoki AE. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. 1999;23:233–6. doi: 10.1038/13868. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Wang T, Zhao Z, Weinman SA. The ClC-3 chloride channel promotes acidification of lysosomes in CHO-K1 and Huh-7 cells. Am J Physiol Cell Physiol. 2002;282:C1483–91. doi: 10.1152/ajpcell.00504.2001. [DOI] [PubMed] [Google Scholar]
- 10.Yoshikawa M, Uchida S, Ezaki J, Rai T, Hayama A, Kobayashi K, Kida Y, Noda M, Koike M, Uchiyama Y, Marumo F, Kominami E, Sasaki S. CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes Cells. 2002;7:597–605. doi: 10.1046/j.1365-2443.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- 11.Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poet M, Steinfeld R, Schweizer M, Kornak U, Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. Embo J. 2005;24:1079–91. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margraf LR, Boriack RL, Routheut AA, Cuppen I, Alhilali L, Bennett CJ, Bennett MJ. Tissue expression and subcellular localization of CLN3, the Batten disease protein. Mol Genet Metab. 1999;66:283–9. doi: 10.1006/mgme.1999.2830. [DOI] [PubMed] [Google Scholar]
- 13.Mao Q, Foster BJ, Xia H, Davidson BL. Membrane topology of CLN3, the protein underlying Batten disease. FEBS Lett. 2003;541:40–6. doi: 10.1016/s0014-5793(03)00284-9. [DOI] [PubMed] [Google Scholar]
- 14.Kaczmarski W, Wisniewski KE, Golabek A, Kaczmarski A, Kida E, Michalewski M. Studies of membrane association of CLN3 protein. Mol Genet Metab. 1999;66:261–4. doi: 10.1006/mgme.1999.2833. [DOI] [PubMed] [Google Scholar]
- 15.Rakheja D, Narayan SB, Pastor JV, Bennett MJ. CLN3P, the Batten disease protein, localizes to membrane lipid rafts (detergent-resistant membranes) Biochem Biophys Res Commun. 2004;317:988–91. doi: 10.1016/j.bbrc.2004.03.146. [DOI] [PubMed] [Google Scholar]
- 16.Spinelli W, Sonnenfeld KH, Ishii DN. Effects of phorbol ester tumor promoters and nerve growth factor on neurite outgrowth in cultured human neuroblastoma cells. Cancer Res. 1982;42:5067–73. [PubMed] [Google Scholar]
- 17.Ling MM, Robinson BH. Approaches to DNA mutagenesis: an overview. Anal Biochem. 1997;254:157–78. doi: 10.1006/abio.1997.2428. [DOI] [PubMed] [Google Scholar]
- 18.Kilkus J, Goswami R, Testai FD, Dawson G. Ceramide in rafts (detergent-insoluble fraction) mediates cell death in neurotumor cell lines. J Neurosci Res. 2003;72:65–75. doi: 10.1002/jnr.10549. [DOI] [PubMed] [Google Scholar]
- 19.Testai FD, Landek MA, Dawson G. Regulation of sphingomyelinases in cells of the oligodendrocyte lineage. J Neurosci Res. 2004;75:66–74. doi: 10.1002/jnr.10816. [DOI] [PubMed] [Google Scholar]
- 20.Wiesner DA, Dawson G. Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J Neurochem. 1996;66:1418–25. doi: 10.1046/j.1471-4159.1996.66041418.x. [DOI] [PubMed] [Google Scholar]
- 21.Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–6. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 22.Haskell RE, Carr CJ, Pearce DA, Bennett MJ, Davidson BL. Batten disease: evaluation of CLN3 mutations on protein localization and function. Hum Mol Genet. 2000;9:735–44. doi: 10.1093/hmg/9.5.735. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Kobayashi T, Gruenberg J. Separation and characterization of late endosomal membrane domains. J Biol Chem. 2002;277:32157–64. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 24.Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, Wisniewski KE. CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Mol Genet Metab. 2000;70:203–13. doi: 10.1006/mgme.2000.3006. [DOI] [PubMed] [Google Scholar]
- 25.Holopainen JM, Saarikoski J, Kinnunen PK, Jarvela I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs) Eur J Biochem. 2001;268:5851–6. doi: 10.1046/j.0014-2956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y, Ramirez-Montealegre D, Pearce DA. A role in vacuolar arginine transport for yeast Btn1p and for human CLN3, the protein defective in Batten disease. Proc Natl Acad Sci U S A. 2003;100:15458–62. doi: 10.1073/pnas.2136651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramirez-Montealegre D, Pearce DA. Defective lysosomal arginine transport in juvenile Batten disease. Hum Mol Genet. 2005;14:3759–73. doi: 10.1093/hmg/ddi406. [DOI] [PubMed] [Google Scholar]
- 28.Narayan SB, Rakheja D, Tan L, Pastor JV, Bennett MJ. CLN3P, the Batten’s disease protein, is a novel palmitoyl-protein Delta-9 desaturase. Ann Neurol. 2006;60:570–7. doi: 10.1002/ana.20975. [DOI] [PubMed] [Google Scholar]
- 29.Hepbildikler ST, Sandhoff R, Kolzer M, Proia RL, Sandhoff K. Physiological substrates for human lysosomal beta -hexosaminidase S. J Biol Chem. 2002;277:2562–72. doi: 10.1074/jbc.M105457200. [DOI] [PubMed] [Google Scholar]
- 30.Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 31.Linke T, Wilkening G, Lansmann S, Moczall H, Bartelsen O, Weisgerber J, Sandhoff K. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol Chem. 2001;382:283–90. doi: 10.1515/BC.2001.035. [DOI] [PubMed] [Google Scholar]
- 32.Linke T, Wilkening G, Sadeghlar F, Mozcall H, Bernardo K, Schuchman E, Sandhoff K. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J Biol Chem. 2001;276:5760–8. doi: 10.1074/jbc.M006846200. [DOI] [PubMed] [Google Scholar]
- 33.Werth N, Schuette CG, Wilkening G, Lemm T, Sandhoff K. Degradation of membrane-bound ganglioside GM2 by beta -hexosaminidase A. Stimulation by GM2 activator protein and lysosomal lipids. J Biol Chem. 2001;276:12685–90. doi: 10.1074/jbc.M007970200. [DOI] [PubMed] [Google Scholar]
- 34.Wilkening G, Linke T, Sandhoff K. Lysosomal degradation on vesicular membrane surfaces. Enhanced glucosylceramide degradation by lysosomal anionic lipids and activators. J Biol Chem. 1998;273:30271–8. doi: 10.1074/jbc.273.46.30271. [DOI] [PubMed] [Google Scholar]
- 35.Wilkening G, Linke T, Uhlhorn-Dierks G, Sandhoff K. Degradation of membrane-bound ganglioside GM1. Stimulation by bis(monoacylglycero)phosphate and the activator proteins SAP-B and GM2-AP. J Biol Chem. 2000;275:35814–9. doi: 10.1074/jbc.M006568200. [DOI] [PubMed] [Google Scholar]