

Abstract
Purpose
To report the identification and characterization of stromal amyloid deposits in patients with keratoconus.
Methods
The excised corneal buttons from two patients diagnosed clinically with keratoconus underwent histochemical analysis with Masson trichrome, Congo red, Alcian blue and periodic acid-Schiff stains as well as immunohistochemical analysis for the TGFBI protein product (TGFBIp), prealbumin, lysozyme, kappa and lambda light chain expression. Following the collection of DNA from both patients, exons 4 and 11–14 of TGFBI were amplified and sequenced to search for mutations previously associated with dystrophic corneal stromal amyloid deposition.
Results
Light microscopic examination of the corneal buttons revealed stromal thinning, epithelial basement membrane abnormalities and focal disruption of Bowman’s layer. Multiple stromal deposits were identified that stained red with Masson trichrome, pink with periodic acid-Schiff, and red with Congo red; the Congo red-stained deposits demonstrated birefringence and dichroism with crossed polarized lenses. Immunohistochemical staining demonstrated reactivity of the stromal deposits with antibodies to TGFBIp, but no reactivity with antibodies against prealbumin, lysozyme, or kappa and lambda light chains. Screening of TGFBI exons 4, 11–14 revealed two previously identified SNPs present in the heterozygous state in both individuals, but no other coding region variants.
Conclusions
Two cases of keratoconus with clinically unsuspected, presumed secondary stromal amyloid deposition are described. Although TGFBIp is identified in the stromal deposits, no previously reported amyloidogenic mutations are identified in TGFBI in either affected individual, indicating a previously undescribed mechanism of stromal amyloid deposition.
Keywords: Keratoconus, amyloid, keratoepithelin, TGFBI
INTRODUCTION
Amyloid is a term given to a variety of proteins that appear as amorphous, eosinophilic, hyaline material on routine hematoxylin-eosin staining. When stained with Congo red and viewed with polarized light, amyloid exhibits a characteristic birefringence and red-green dichroism. Ultrastructurally, amyloid has a fibrillar appearance with the fibers ranging from 70–100Ǻ in diameter.
Amyloid deposition may be classified as primary or secondary, localized or systemic. Lattice and combined granular-lattice corneal dystrophies are autosomal dominant disorders that feature primary localized dystrophic corneal amyloid deposition. Missense mutations in the transforming growth factor beta-induced gene (TGFBI), which codes for a protein product known as TGFBIp, are associated with these corneal dystrophies, and the corneal deposits in these dystrophies contain TGFBIp.1 More than 35 different pathogenic mutations have been identified in TGFBI, located in exons 4, 11, 12, 13 and 14.2, 3 In contrast, Meretoja’s syndrome, multiple myeloma, and familial cutaneous amyloidosis are associated with systemic amyloid deposition and are well-recognized causes of secondary corneal amyloidosis.4–6 Secondary corneal amyloid deposition has also been associated with localized causes such as trachoma, trichiasis, interstitial keratitis, phlyctenular keratitis, and uveitis.7–9 In spite of the significant number of localized and systemic disorders that have been associated with corneal amyloid deposition, amyloid deposits have only been reported a few times in association with keratoconus, a relatively common corneal degeneration and frequent indication for corneal transplantation worldwide.10–13
Screening of TGFBI to exclude the most common dystrophic cause of corneal amyloid deposition was not performed in any of these previous reports. Additionally, neither a determination of the origin of the amyloid deposits nor a characterization of the deposits was performed.13 In this report, we describe two cases of keratoconus associated with clinically unsuspected corneal amyloidosis. Histochemical, immunohistochemical and molecular genetic analysis were performed as well to investigate the nature and origin of the amyloid deposits.
MATERIALS AND METHODS
The researchers followed the tenets of the Declaration of Helsinki in the treatment of the subjects reported herein. Study approval was obtained from the institutional review board at The University of California, Los Angeles (UCLA IRB # 94-07-243-24B).
Clinical Examination
Clinical examination of the patients included slit lamp biomicroscopy and corneal topography. The diagnosis of keratoconus was based on the presence of characteristic topographic features, such as inferior or central corneal steepening, or an asymmetric bow-tie pattern with skewing of the radial axes, and the presence of one or more of the following characteristic clinical features in one or both eyes: conical corneal deformation, corneal stromal thinning, a Fleischer ring, or Vogt’s striae.
Histopathologic and Immunohistochemical Examination
The corneal buttons excised from the subjects at the time of penetrating keratoplasty were fixed in 10% neutral buffered formaldehyde. After embedding in paraffin, thin sections were stained with hematoxylin and eosin, Alcian blue, periodic acid-Schiff, Congo red and Masson trichrome stains. Immunohistochemistry was performed on sections obtained from the right corneal button from Case 1 using antibodies directed against prealbumin, lysozyme, kappa light chains, lambda light chains and TGFBIp. The antibody for TGFBIp was produced in goats immunized with purified, NSO-derived, recombinant human transforming growth factor beta Induced Gene H3 (rhβIG-H3). Human βIGH-H3 specific IgG was purified by human βIG-H3 affinity chromatography (R&D Systems, Minneapolis, MN). Normal goat IgG (Jackson Laboratories, West Grove, PA) at the same concentration as the specific antibody was used as the negative antibody control. Immunohistochemical staining of slides prepared from the corneal button of an individual with a confirmed TGFBI dystrophy using the same antibody for TGFBIP was used as the positive control.14 TGFBIp antibody binding was detected using a RTU Vectastain-HRP Kit (Vector Laboratories, Burlingame, CA).
DNA Collection and Analysis
Buccal epithelial swabs were collected from the patients using CytoSoft CP-5B brushes (Medical Packaging Corporation, Camarillo, California). Genomic DNA was isolated from the buccal epithelial cells using the QIAamp DNA Mini Kit spin protocol (Qiagen, Valencia, California). Polymerase chain reaction amplification and automated sequencing of exons 4, 11, 12, 13 and 14 of TGFBI were performed using previously described primer sequences and conditions15. Nucleotide sequences were compared with the published TGFBI cDNA sequence (Gen Bank accession no. NM_000358).
RESULTS
Case Histories
Case 1
A 74-year-old man with a 40 year history of bilateral keratoconus was referred to one of the authors (JDH) for combined penetrating keratoplasty and cataract extraction in the right eye. The patient had noticed increasingly poor rigid gas permeable contact lens-corrected vision, which was attributed to corneal scarring. The patient’s past medical history was significant only for hypertension and coronary artery disease, and he denied a family history of keratoconus or other eye disorders.
On presentation, contact lens-corrected visual acuity was 20/80 in the right eye and 20/70 in the left eye. Both corneas demonstrated central conical deformation with apical stromal thinning and scarring, right eye more than left. No other stromal opacities were noted. A 4+ nuclear sclerotic cataract was present in each eye.
A combined penetrating keratoplasty and cataract extraction was performed in the left eye, followed two years later by a combined penetrating keratoplasty and cataract extraction in the right eye.
Four months after surgery in the right eye, both corneal transplants were clear and compact, devoid of focal stromal deposits.
Case 2
A 37-year-old man with a 10 year history of bilateral keratoconus presented to one of the authors (RC) complaining of increasingly poor contact lens-corrected vision for the previous 2 years. The patient's medical history was significant for panhypopituitarism secondary to head trauma, hypertension, asthma and acid reflux, but he denied a family history of keratoconus or other ocular disorders.
Contact lens-corrected visual acuities measured 20/25 in the right eye and 20/100 in the left eye. Significant central conical deformation and apical stromal thinning were noted in each cornea. Additionally, apical scarring was noted in each cornea, more prominent in the left eye than in the right. Topographic imaging of the left cornea revealed irregular central corneal steepening (simulated K readings were 67.91 D @ 125 degrees × 58.70 D).
A penetrating keratoplasty was performed in the left eye and the corneal button was submitted for histopathologic examination. Four months following the corneal transplant, the graft was clear without evidence of stromal deposits.
Histopathologic and Immunohistochemical Findings
Microscopic examination of the stained corneal sections from both cases revealed irregular epithelial surfaces with areas of epithelial thickening. Thickened and duplicated basement membrane and areas of irregular thinning and focal disruptions in Bowman's layer were noted, consistent with keratoconus. The stroma demonstrated scar formation and central thinning. Central amorphous, eosinophilic deposits, extending from the anterior stroma to the posterior two-thirds of the stroma, were noted in each of the corneal buttons submitted from cases 1 and 2. The deposits did not stain with Alcian blue, but appeared pink with periodic acid-Schiff, and red with Masson trichrome and Congo red stains. The Congo red deposits demonstrated green birefringence and dichroism with crossed polarizing lenses. Descemet's membrane and endothelium appeared unremarkable in each of the corneal buttons (Figure 1 and Figure 2).
Figure 1.

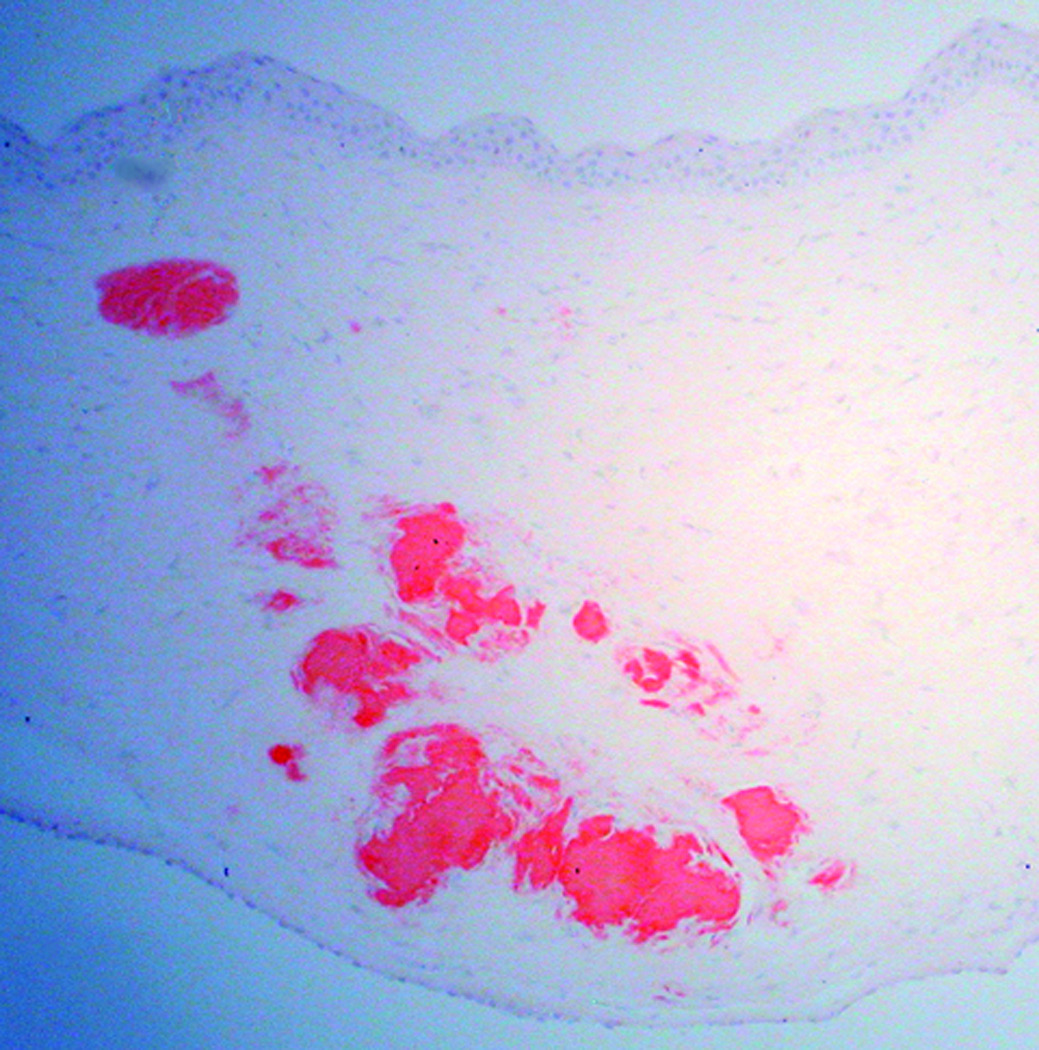
Congo red-stained sections of the right cornea from a 74-year-old man (Case 1 - Figure 1a) and the left cornea from a 37-year-old man (Case 2 - Figure 1b) who both underwent corneal transplantation for keratoconus. Red deposits that extend from the anterior to the deep stroma are noted in each section (original magnification, 200x).
Figure 2.

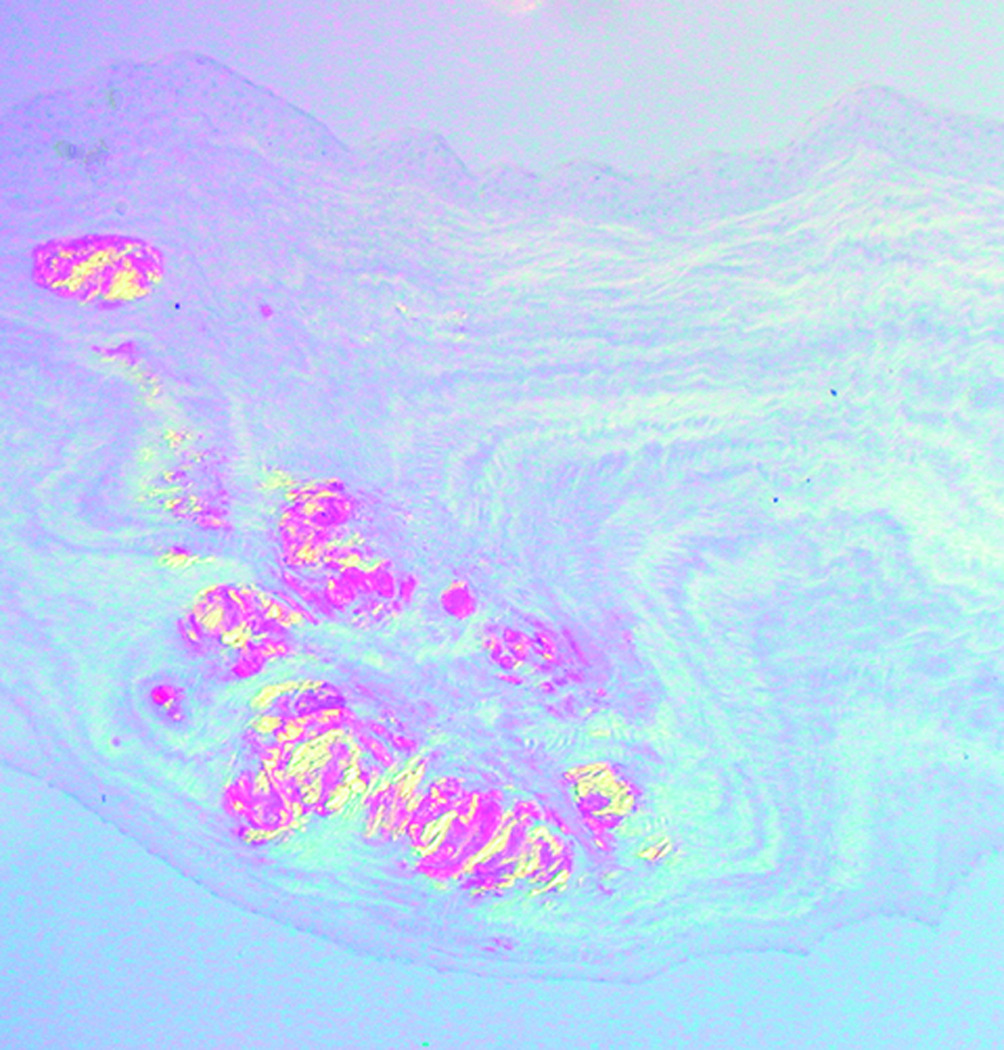
Congo red-stained sections from Case 1 (Figure 2a) and Case 2 (Figure 2b) demonstrate birefringence with crossed polars (original magnification, 200x).
Immunohistochemical staining of the right corneal button from Case 1 did not reveal significant reactivity with antibodies for prealbumin, lysozyme, kappa and lambda light chains. However, the Congo red-stained stromal deposits demonstrated positive reactivity with an antibody to TGFBIp (Figure 3). Immunohistochemical staining of the corneal button from Case 2 was not performed.
Figure 3.

Section of cornea from Case 1 stained with antibody directed against TGFBIp reveals a positive reaction of the stromal deposits that demonstrated staining with Congo red and birefringence with the use of polarizing filters.
Genetic Screening
Screening of TGFBI exons 4 and 11–14 in each patient revealed only two previously reported synonymous substitutions, both in the heterozygous state: c.1463C>T (p.Leu472Leu; rs1133170) and c.1667T>C (p.Phe540Phe; rs4669).
DISCUSSION
We identified stromal amyloid deposits associated with TGFBIp in the corneal buttons of two patients with keratoconus and excluded previously reported amyloidogenic and novel coding region mutations in TGFBI. This represents only the fourth report of corneal amyloidosis associated with keratoconus, and is the first to characterize the nature of the amyloid protein as containing TGFBIp and to exclude a TFGBI mutation (Table). McPherson and associates identified corneal amyloidosis as an incidental finding in 3.5% of 210 ocular pathology specimens (75 whole eyes and 135 corneal buttons),11 one of which was a corneal button collected from a patient with keratoconus. A subsequent examination of 17 corneal buttons from patients with keratoconus revealed amyloid deposits in 3 (17.6%).10 The amyloid deposits were small in these patients and were scattered in various depths of the cornea, from just below Bowman's layer to just above Descemet's membrane. It was not noted in these reports whether corneal scarring was present, as was observed in the two corneal specimens that we report. Stern and colleagues reported one case of amyloidosis associated with keratoconus, in which a central gray-white opacity was noted in the anterior corneal stroma on clinical examination, and panstromal amyloid deposition was found on histopathologic analysis.13 The authors suggested that long term use of hard contact lenses with significant central corneal touch could have contributed to the development of the central scarring and presumed amyloid deposition. Genetic analysis was not performed in any of these previously reported cases.
Table 1.
Reported cases of keratoconus associated with corneal amyloidosis
| Study | Number of patients | Location of stromal amyloid | PAS | Masson Trichrome | Congo red | Tests for secondary amyloid |
|---|---|---|---|---|---|---|
| McPherson et al.11 | 1 | Superficial | Not done | Not done | + | Not done |
| McPherson et al.10 | 3 | Superficial and mid stromal | Done, but PAS staining of amyloid deposits not described | Not done | + | Not done |
| Stern et al.13 | 1 | Superficial, mid and deep stromal | Not done | Not done | + | Not done |
| Present Study | 2 | Superficial, mid and deep stromal | + | + | + | Prealbumin (−), lysozyme (−), kappa and lambda light chains (−), TGFBIp (+) |
Various theories have been put forth to explain the mechanism of development of secondary localized corneal amyloidosis. In cases involving repeated local trauma to the corneal surface, such as in the setting of trichiasis, lactoferrin has been proposed as playing a role in corneal amyloid formation.15, 16 Suesskind and colleagues demonstrated corneal amyloid deposits in cases of Fuchs endothelial dystrophy that stained with antibodies to TGFBIp.9 The authors explained this observation by proposing that proteolysis, in association with certain environmental factors, may alter the structure of TGFBIp in such a way that it forms fibrils capable of aggregating into amyloid deposits. In this report, we also demonstrate the presence of TGFBIp in stromal amyloid deposits in a patient with a non-TGFBI corneal dystrophy, leading to the conclusion that TGFBIp aggregation in the corneal stroma may be found in both dystrophic and non-dystrophic corneal disorders.
While other investigators have looked for a possible relationship between TGFBI, TGFBIp and keratoconus, no definitive association has been established. As TGFBIp has been proposed to play a role in corneal development, wound healing and lamellar adhesion, investigators have theorized that mutations in TGFBI leading to decreased expression of TGFBIp in the corneal stroma could result in decreased mechanical stability of the cornea and contribute to the development of keratoconus.17 Although levels of TGFBIp have been shown to be decreased in keratoconic corneas,17 no mutations have been identified in TGFBI in affected individuals.18 Paradoxically, other investigators have demonstrated elevated levels of TGFBIp in areas of corneal scarring, likely related to the stimulation of TGFBIp production by TGFB1.19, 20
The current study raises important questions about the mechanism of amyloid formation in the cornea. While it is clear that certain missense mutations in TGFBI lead to the development of a mutant TGFBIp in the form of amyloid fibrils, the current study demonstrates that such mutations are not mandatory. A number of other factors may be involved. The presence of amyloidogenic consensus sequences renders numerous proteins and peptides vulnerable to amyloid fibril formation.21 TGFBIp contains amyloidogenic domains and it is plausible that fibril formation could be initiated by alteration in TGFBIp expression, local pH, molecular chaperones, proteolysis, or even in the unfolded protein response-proteosomal pathway for removal of misfolded proteins.22 Corneal injury, such as during keratorefractive surgery, may lead to a significant acceleration of amyloid formation through TGFB1-mediated upregulation of TGFBIp production by the injured keratocytes.23–25 In the cases we report, local corneal injury that led to stromal scarring evident on clinical and histopathologic examination of the corneas may have also produced an increased expression of TGFB1, with subsequent increased expression of TGFBIp in the corneal stroma. While the increased expression of the wild-type TGFBIp would not be expected to result in the formation of stromal amyloid deposits that are characteristic of mutant TGFBIp, it is possible that local factors in keratoconic corneas predispose to the development of the observed amyloid deposits in the absence of coding region mutations in TGFBI.
Acknowledgments
Sources of support: National Eye Institute K08 EY016079 (AJA)
REFERENCES
- 1.Stix B, Leber M, Bingemer P, et al. Hereditary lattice corneal dystrophy is associated with corneal amyloid deposits enclosing C-terminal fragments of keratoepithelin. Invest. Ophthalmol. Vis. Sci. 2005;46:1133–1139. doi: 10.1167/iovs.04-1319. [DOI] [PubMed] [Google Scholar]
- 2.Aldave AJ, Sonmez B. Elucidating the molecular genetic basis of the corneal dystrophies: are we there yet? Arch Ophthalmol. 2007;125:177–186. doi: 10.1001/archopht.125.2.177. [DOI] [PubMed] [Google Scholar]
- 3.Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mutat. 2006;27:615–625. doi: 10.1002/humu.20334. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein D, Schteingart M, Birnbaum A, Tessler H. Bilateral eyelid ecchymoses and corneal crystals: An unusual presentation of multiple myeloma. Cornea. 2005;24:757–758. doi: 10.1097/01.ico.0000154381.13237.a3. [DOI] [PubMed] [Google Scholar]
- 5.Kivela T, Tarkkanen A, Frangione B, et al. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja's syndrome. Invest. Ophthalmol. Vis. Sci. 1994;35:3759–3769. [PubMed] [Google Scholar]
- 6.Partington M, Marriott P, Prentice R, et al. Familial cutaneous amyloidosis with systemic manifestations in males. Am J Med Genet. 1981;10:65–75. doi: 10.1002/ajmg.1320100109. [DOI] [PubMed] [Google Scholar]
- 7.Hayasaka S, Setogawa T, Ohmura M. Secondary localized amyloidosis of the cornea caused by trichiasis. Ophthalmologica. 1987;194:77–81. doi: 10.1159/000309739. [DOI] [PubMed] [Google Scholar]
- 8.Lin PY, Kao SC, Hsueh KF, et al. Localized amyloidosis of the cornea secondary to trichiasis: clinical course and pathogenesis. Cornea. 2003;22:491–494. doi: 10.1097/00003226-200307000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Suesskind D, Auw-Haedrich C, Schorderet D, et al. Keratoepithelin in secondary corneal amyloidosis Graefe's. Arch Clin Exp Ophthalmol. 2006;244:725–731. doi: 10.1007/s00417-005-0153-x. [DOI] [PubMed] [Google Scholar]
- 10.McPherson SJ, Kiffney GJ. Some histologic findings in keratoconus. Arch Ophthalmol. 1968:79. doi: 10.1001/archopht.1968.03850040671004. [DOI] [PubMed] [Google Scholar]
- 11.McPherson SJ, Kiffney GJ, Freed C. Corneal amyloidosis. Am J Ophthalmol. 1966;62:1025–1033. doi: 10.1016/0002-9394(66)92549-9. [DOI] [PubMed] [Google Scholar]
- 12.Rabinowitz YS. Keratoconus. Surv Ophthalmol. 1998;42:297–319. doi: 10.1016/s0039-6257(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 13.Stern G, Knapp A, Hood I. Corneal amyloidosis associated with keratoconus. Ophthalmology. 1988;95:52–55. doi: 10.1016/s0161-6420(88)33225-2. [DOI] [PubMed] [Google Scholar]
- 14.Aldave AJ, Yellore VS, Sonmez B, et al. A novel variant of combined granular-lattice corneal dystrophy associated with the Met619Lys mutation in the TGFBI gene. Arch Ophthalmol. 2008;126:371–377. doi: 10.1001/archopht.126.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldave AJ, Principe AH, Lin DY, et al. Lattice dystrophy-like localized amyloidosis of the cornea secondary to trichiasis. Cornea. 2005;24:112–115. doi: 10.1097/01.ico.0000134194.71981.ab. [DOI] [PubMed] [Google Scholar]
- 16.Araki-Sasaki K, Ando Y, Nakamura M, et al. Lactoferrin Glu561Asp facilitates secondary amyloidosis in the cornea. Br J Ophthalmol. 2005;89:684–688. doi: 10.1136/bjo.2004.056804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takacs L, Csutak A, Balazs E, et al. Expression of betaig-h3 is lower than normal in keratoconus corneas but increases with scarring. Cornea. 1999;18:599–605. [PubMed] [Google Scholar]
- 18.Udar N, Kenney MC, Chalukya M, et al. Keratoconus--no association with the transforming growth factor beta-induced gene in a cohort of American patients. Cornea. 2004;23:13–17. doi: 10.1097/00003226-200401000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Wang M, Munier F, Araki-Saski K, Schorderet D. TGFBI gene transcript is transforming growth factor-beta1-responsive and cell density-dependent in a human corneal epithelial cell line. Ophthalmic Genet. 2002;23:237–245. doi: 10.1076/opge.23.4.237.13884. [DOI] [PubMed] [Google Scholar]
- 20.Takacs L, Csutak A, Balazs E, Berta A. Immunohistochemical detection of betaIG-H3 in scarring human corneas. Graefe's Arch Clin Exp Ophthalmol. 1999;237:529–534. doi: 10.1007/s004170050275. [DOI] [PubMed] [Google Scholar]
- 21.Turnell WG, Finch JT. Binding of the dye congo red to the amyloid protein pig insulin reveals a novel homology amongst amyloid-forming peptide sequences. Journal of Molecular Biology. 1992;227:1205–1223. doi: 10.1016/0022-2836(92)90532-o. [DOI] [PubMed] [Google Scholar]
- 22.Yuan C, Berscheit HL, Huang AJW. Identification of an amyloidogenic region on keratoepithelin via synthetic peptides. FEBS Letters. 2007;581:241–247. doi: 10.1016/j.febslet.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 23.Banning C, Larson P, Randleman J. Outcome of LASIK in fleck corneal dystrophy. Cornea. 2006;25:1262–1264. doi: 10.1097/01.ico.0000230345.64607.8a. [DOI] [PubMed] [Google Scholar]
- 24.Jun RM, Tchah H, Kim T-i, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111:463–468. doi: 10.1016/j.ophtha.2003.06.026. [DOI] [PubMed] [Google Scholar]
- 25.Aldave AJ, Sonmez B, Forstot SL, et al. A clinical and histopathologic examination of accelerated TGFBIp deposition after LASIK in combined granular-lattice corneal dystrophy. Am J Ophthalmol. 2007;143:416–419. doi: 10.1016/j.ajo.2006.11.056. [DOI] [PubMed] [Google Scholar]