

Abstract
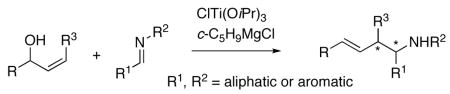
A cross-coupling reaction between an allylic alcohol and an imine is described for stereoselective allylation of aromatic and aliphatic imines. This method provides operationally simple, enantioselective access to functionalized homoallylic amines. Particularly noteworthy is direct use of a functionalized allylic alcohol as an allylating reagent without pre-derivatization, which obviates the use of preformed organometallic reagents or activated imine derivatives.
Addition of organometallic reagents to imines provides a useful method for the stereoselective preparation of amines.1 An enantioselective allylation/crotylation reaction to aldimines is a valuable tool in organic synthesis, as homoallylic amines are useful building blocks in natural product synthesis and medicinal chemistry.2,3 Imines are less electrophilic than carbonyl groups, and addition of organometallic reagents to imines can be complicated by accompanying enolization, reduction, or dimerization.4 This reactivity issue requires a judicious choice of an allylic metal reagent and/or activation of an imine by a suitable Lewis acid. Additionally, there is a paucity of convenient methods for generating functionalized allylic nucleophiles despite impressive advances in this field.5 Direct use of an allylic alcohol as an allylating reagent is particularly attractive, as it obviates pre-derivatization of an allylic alcohol substrate. We report herein regio- and stereoselective cross-coupling between an allylic alcohol and an imine by the action of the Kulinkovich reagent.
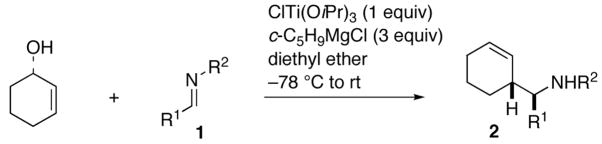
A cross-coupling reaction between an allylic alcohol and a vinylsilane (or a styrene) was recently developed by use of the Kulinkovich reagent, in which directing effects of an allylic alkoxide were exploited via a temporary linker.6,7 An imine was already shown by the Sato group to react with an alkyne–Kulinkovich reagent complex to afford an allylic amine.8 Thus, we reasoned that the use of an aldimine in place of a vinylsilane could provide a new approach to regio- and stereoselectively preparing homoallylic amines. Our study began with the coupling reaction between 2-cyclohexen-1-ol and several imines 1a–h (Table 1). Thus, coupling of 2-cyclohexen-1-ol and 1a under previously reported conditions afforded homoallylic amine 2a in 90% yield in >20:1 diastereoselectivity (entry 1). A broad scope with respect to imines (i.e., different R1 and R2) can be seen from entries 1–8: not only aromatic, but also aliphatic imines are amenable to cross-coupling. The resulting homoallylic amines 2a–h were obtained as virtually single isomers. In the case of imine 1g having an isopropyl branch, a 4:1 mixture of 2g and the by-product (structure not shown) from addition of the cyclopentyl Grignard reagent to the imine was obtained (entry 7). This result could be attributed to steric effects.
Table 1.
Cross-Coupling between 2-Cyclohexen-1-ol and Imines
 | |||||
|---|---|---|---|---|---|
| entry | imine | R1 | R2 | product | yield |
| 1 | 1a | Ph | Ph | 2a | 90% |
| 2 | 1b | p-MeOC6H4 | 2b | 75% | |
| 3 | 1c | Bn | 2c | 78% | |
| 4 | 1d | CH2-o-MeOC6H4 | 2d | 69% | |
| 5 | 1e | n-Bu | 2e | 76% | |
| 6 | 1f | 2-furyl | Bn | 2f | 55% |
| 7 | 1g | i-Pr | Bn | 2g | 40% |
| 8 | 1h | n-C7H15 | Bn | 2h | 60% |
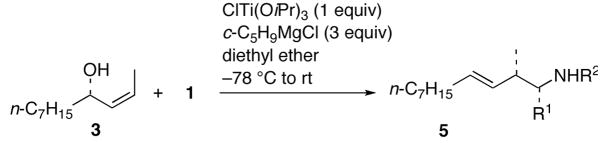
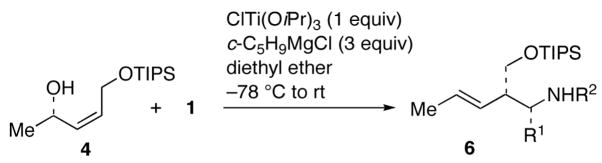
Coupling of acyclic Z-allylic alcohols 3 and 4 with imines 1a–c,e was next examined (Table 2). As was the case with cross-coupling with vinylsilanes or styrenes,6 high levels of diastereocontrol was achieved to provide E-homoallylic amines 5a–c,e and 6a–c,e. Full compatibility with the presence of an allylic ether is clearly seen with allylic alcohol 4 (entries 5–8).
Table 2.
Cross-Coupling between (Z)-Allylic Alcohols and Imines
 | |||||
|---|---|---|---|---|---|
| entry | imine | R1 | R2 | product | yield |
| 1 | 1a | Ph | Ph | 5a | 78% |
| 2 | 1b | p-MeOC6H4 | 5b | 72% | |
| 3 | 1c | Bn | 5c | 74% | |
| 4 | 1e | n-Bu | 5e | 72% | |
| |||||
| entry | imine | R1 | R2 | product | yield |
|
| |||||
| 5 | 1a | Ph | Ph | 6a | 62% |
| 6 | 1b | p-MeOC6H4 | 6b | 64% | |
| 7 | 1c | Bn | 6c | 68% | |
| 8 | 1e | n-Bu | 6e | 57% | |
Complete chirality transfer was established by the use of enantiopure allylic alcohols 7–11 (Table 3). Coupling of (S)-7 with 1a and 1c proceeded diastereo- and enantioselectively to afford amines 12 and 13 in 60% and 68% yield, respectively (entries 1 and 2). Similarly, 14 and 15 were obtained from 8 and 9, respectively (entries 3 and 4). Comparative evaluation of E-allylic alcohols 10 and 11 was undertaken next for additional stereochemical studies. As expected by analogy to ethylation and cross-coupling with vinylsilanes,6 these E-allylic alcohol substrates produced a mixture of two diastereomers: 10 gave a 1.3:1 separable mixture of 16 and 15 in 71% yield (entry 5), whereas a 1.2:1 mixture of 17 and 14 was obtained from 11 (entry 6). The unequivocal determination of the absolute and relative stereochemistry of the homoallylic amine products 12–17 was possible by these correlation studies, as well as an independent synthesis of 13.9
Table 3.
Enantioselective Synthesis of Homoallylic Amines
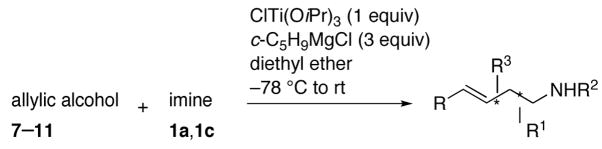 | ||||
|---|---|---|---|---|
| entry | imine | allylic alcohol | product | yield (%) |
| 1 | 1a |
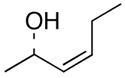 (S)-7 |
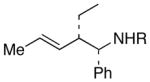 12: R = Ph |
60 |
| 2 | 1c | (S)-7 | 13: R = Bn | 68 |
| 3 | 1c |
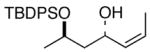 8 |
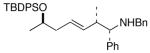 14 |
68 |
| 4 | 1c |
9 |
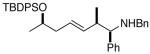 15 |
65 |
| 5 | 1c |
10 |
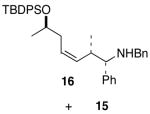
|
71 1.3:1 dr |
| 6 | 1c |
11 |
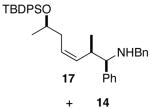
|
69 1.2:1 dr |
The observed stereochemical outcome can be rationalized by formation of a temporary alkoxide tether and subsequent syn addition/syn β-elimination for the 1,3-transpositive cross-coupling reactions of acyclic and cyclic allylic alcohols. High diastereoselectivity displayed by Z-allylic alcohols is in accord with the involvement of conformer A to minimize A(1,3) strain (Scheme 1). The lack of selectivity for E-allylic alcohols and the attendant formation of both E- and Z-alkenes suggest the co-involvement of both conformers B and C.6
Scheme 1.

In conclusion, we have developed convenient cross-coupling reactions between allylic alcohols and imines for stereoselective allylation of aromatic and aliphatic imines. Particularly noteworthy is direct use of a functionalized allylic alcohol as an allylating reagent without pre-derivatization. This convenient method obviates the use of preformed organometallic reagents or activated imine derivatives.10
Supplementary Material
Acknowledgments
We thank NSF (CHE-0615604) and NIH (GM35956) for generous financial support.
Footnotes
Supporting Information Available Experimental details and spectroscopic data for key intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews: Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Enders D, Reinhold U. Tetrahedron: Asymmetry. 1997;8:1895.Alvaro G, Savoia D. Synlett. 2002:651.Friestad GK, Mathies AK. Tetrahedron. 2007;63:2541. doi: 10.1016/j.tet.2007.06.117.
- 2.For reviews: Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Ding H, Friestad GK. Synthesis. 2005:2815.Kargbo RB, Cook GR. Curr Org Chem. 2007;11:1287.Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.
- 3.For selected recent examples: Schaus JV, Jain NF, Panek JS. Tetrahedron. 2000;56:10263.Ferraris D, Young B, Cox C, Dudding T, Drury WJ, Ryzhkov L, Taggi AE, Lectka T. J Am Chem Soc. 2002;124:67. doi: 10.1021/ja016838j.Kobayashi S, Ogawa C, Konishi H, Sugiura M. J Am Chem Soc. 2003;125:6610. doi: 10.1021/ja035061m.Berger R, Rabbat PMA, Leighton JL. J Am Chem Soc. 2003;125:9596. doi: 10.1021/ja035001g.Huber JD, Leighton JL. J Am Chem Soc. 2007;129:14552. doi: 10.1021/ja076035h.Huber JD, Perl NR, Leighton JL. Angew Chem Int Ed. 2008;47:3037. doi: 10.1002/anie.200705621.Fernandes RA, Stimac A, Yamamoto Y. J Am Chem Soc. 2003;125:14133. doi: 10.1021/ja037272x.Shimizu M, Kimura M, Watanabe T, Tamaru Y. Org Lett. 2005;7:637. doi: 10.1021/ol047609f.Ramachandran PV, Burghardt TE, Bland-Berry L. J Org Chem. 2005;70:7911. doi: 10.1021/jo0508200.Tan KL, Jacobsen EN. Angew Chem Int Ed. 2007;46:1315. doi: 10.1002/anie.200603354.Wada R, Shibuguchi T, Makino S, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2006;128:7687. doi: 10.1021/ja061510h.Kargbo R, Takahashi Y, Bhor S, Cook GR, Lloyd-Jones GC, Shepperson IR. J Am Chem Soc. 2007;129:3846. doi: 10.1021/ja070742t.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v.Fujita M, Nagano T, Schneider U, Hamada T, Ogawa C, Kobayashi S. J Am Chem Soc. 2008;130:2914. doi: 10.1021/ja710627x.
- 4.Cf. Stock G, Dowd SRJ. Am Chem Soc. 1963;85:2178.Bloch R. Chem Rev. 1998;98:1407. doi: 10.1021/cr940474e.
- 5.For recent developments in transition metal-catalyzed reductive coupling of imines and carbonyl compounds see: Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Montgomery J. Angew Chem Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634. and references therein.
- 6.Lysenko IL, Kim K, Lee HG, Cha JK. J Am Chem Soc. 2008;130:15997. doi: 10.1021/ja806440m. [DOI] [PubMed] [Google Scholar]
- 7.Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870. doi: 10.1021/ja8074242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Harada K, Sato F. Tetrahedron Lett. 1995;36:5913.. See also: Gao Y, Sato F. J Org Chem. 1995;60:8136.
- 9.(a) Amine 13 was independently prepared by the Claisen-Ireland rearrangement of the phenyl acetate of (S)-7, followed by the Curtius rearrangement, hydrolysis, and reductive amination with benzaldehyde. (b) The relative stereochemistry was also established by measurement of the key vicinal coupling constant of a tetrahydropyridine derived from 5c via allylation and ring-closing metathesis. (c) See Supporting Information for these stereochemical studies.
- 10.During the course of manuscript preparation, a related study was reported by Professor Micalizio: Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648. doi: 10.1002/anie.200900236.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.