

Abstract
Purpose
To investigate in vitro, the cellular network activity of the hippocampus in Q54 mice that display spontaneous seizures because of a gain-of-function mutation of the Scn2a sodium channel gene.
Methods
Extacellular recordings were obtained from CA1 and CA3 pyramidal neurons in hippocampal slices prepared from Q54 transgenic and nontransgenic littermates (WT) under physiologic conditions as well as during periods of orthodromic stimulation of the Schaffer collaterals. Cerebral spinal fluid samples were analyzed and cresyl violet histology of the hippocampus was conducted.
Results
Increased spontaneous extracellular activity was found in both CA1 and CA3 regions of Q54 hippocampal slices. Q54 slices also demonstrated significantly greater spontaneous and afterdischarge activity as well as population spike amplitude and duration following tetanic stimulus in comparison to WT slices. Frequency analysis of tetanically stimulated recordings indicated high-frequency components (100 and 200 Hz) unique to Q45 slices. Analysis of cresyl violet histology supports healthy Q54 slices up to 10 weeks, while Q54 cerebral spinal fluid shows elevated osmolarity.
Conclusion
Evidence for hyperexcitability and increased synaptic efficacy in Q54 mice was found by observing spontaneous activity as well as evoked activity. Response to tetanic stimulation included unique high-frequency oscillations, and resulted in an increased population spike amplitude and duration. Histological assessment shows equivalent neuronal development in both experimental groups. The data support the hypothesis that modified Scn2a channels in Q54 mice result in network hyperexcitability of the hippocampus necessary for the development and maintenance of temporal lobe seizures.
Keywords: Hippocampus, Epilepsy, Sodium Channel, SCN2A, Q54
INTRODUCTION
Epilepsy is the most prevalent chronic neurological disorder, affecting over 3 million Americans with approximately 200,000 new cases reported each year (epilepsyfoundation.org). Although there are multiple epilepsy syndromes, all are characterized and identified by the presence of seizures. Seizures are the result of a chemical imbalance affecting neuronal populations that leads to uncontrolled neuronal firing characterized by large electrical field oscillations in one or more areas of the brain.
Many ion channels are involved in neuronal activity, but sodium channels play a key role in epilepsy. Two well known antiepileptic drugs (AEDs), carbamazepine and phenytoin, act by enhancing the inactivation property of the sodium channel. Enhancing inactivation increases the refractory period of the cell, thus slowing or preventing the occurrence of rapid repetitive firing. In addition, increased brain sodium channel transcript levels are known to occur in the epileptic human brain (Lombardo et al., 1996), further supporting the conclusion that sodium channels are likely to be the primary mechanism for most, if not all forms of epilepsy (Tian et al., 1995).
Several sodium channel mutations have been identified in the development of epileptic seizures. Voltage-gated sodium channels consist of a single pore-forming α-subunit as well as one or more auxiliary β-subunits. There are at least four sodium channel β-subunits (Scn1b-4b) and nine α-subunits (Scn1a-11a), four of which are primarily responsible for encoding sodium currents in the brain: Scn1a, Scn2a, Scn3a, and Scn8a (Catterall et al., 2005). Slight variations in protein-coding sequences can significantly modify these structures and their gating kinetics. Mutations in three genes encoding isoforms of the voltagegated sodium channel subunits (Scn1a, Scn2a, Scn1b) have been identified in human idiopathic epilepsies including generalized epilepsy with febrile seizures plus (GEFS+) (Wallace et al., 1998; Escayg et al., 2000; Escayg et al., 2001; Lerche et al., 2001; Sugawara et al., 2001a; Sugawara et al., 2001b; Wallace et al., 2001; Wallace et al., 2002; Kearney et al., 2006b), severe myoclonic epilepsy of infancy (SMEI) (Claes et al., 2001; Ohmori et al., 2002; Sugawara et al., 2002; Claes et al., 2003; Fujiwara et al., 2003; Ceulemans et al., 2004a; Kamiya et al., 2004; Ohmori et al., 2006; Suls et al., 2006), and benign familial neonatal-infantile seizures (BFNI) (Heron et al., 2002; Berkovic et al., 2004). The substantial incidence of mutations in genes encoding sodium channel subunits that have been associated with the development of epilepsy confirm that alteration in sodium channel genetics is a primary cause of inherited epilepsy syndromes.
One Scn2a mutation, GAL879-881QQQ, alters the structure of the S4–S5 linker of alpha-subunit domain two and results in delayed channel inactivation and increased persistent current when expressed in Xenopus oocytes (Smith and Goldin, 1997; Kearney et al., 2001). Introduction of this missense mutation into mice resulted in the development of the Q54 model of temporal lobe epilepsy (Smith and Goldin, 1997; Kearney et al., 2001). Seizure behaviors were observed in Q54 transgenic mice beginning with focal seizures that seem to originate in the hippocampus between 2 and 3 months of age. Frequency and duration of seizure episodes increased with age, with generalized seizures observed as early as 3 months of age. Whole-cell patch clamp recordings of CA1 pyramidal cells detected a significant increase in persistent current for sodium channels in Q54 transgenic mice at the age of 1 month. Finally, premature death commonly occurred at between 5 and 6 months of age (Smith and Goldin, 1997; Kearney et al., 2001) Additional work with the Q54 model has revealed that genetic background can significantly influence the severity of phenotypic expression (Bergren et al., 2005; Kearney et al., 2006a). Nevertheless, the mechanisms behind the development from genotype to phenotype in this unique model of temporal lobe epilepsy remain unclear and further investigation is required.
Brain slice recording is a well-established method for in vitro study of intracellular and extracellular CNS activity. In particular, murine hippocampal slices are economical and well-understood models that have yielded a wealth of information in epilepsy studies (Fisher, 1989; Durand, 1993). Unfortunately, seizures are primarily induced in this preparation via pharmacological or stimulatory application and the resultant seizure activity is often termed epileptiform, rather than epileptic, in order to distinguish it from its naturally occurring counterpart. Alternatively, human epileptic tissue slices are difficult to obtain, often damaged by surgical removal techniques, and limited by inadequate control tissue (Avoli et al., 2005). New developments in murine genetics have led to a wealth of disease models based on human mutations and matching their phenotypes, notably the Q54 model of temporal lobe epilepsy. Although patch-clamp and EEG studies have led to valuable insights into the possible brain locations and channel involvement during phenotypic seizures, our understanding of the network activity involved in seizure generation and propagation is incomplete. In this study, we investigated extracellular properties of a genetically epileptic, Q54 hippocampal slice model in order to uncover the mechanisms behind phenotypic development in genetic models of epilepsy.
METHODS
Animals
Experiments were performed on the Q54 strain of transgenic mice expressing the GAL879-881QQQ mutation of Scn2a (Kearney et al., 2001). Q54 transgenic mice were originally generated by microinjection of (C57Bl/6J × SJL/J)F2 oocytes, and a congenic line C57Bl/6J × Q54 (B6Q54) was established though ten successive generations of backcrossing to recipient strain, C57Bl/6J (B6), as previously described (Kearney et al., 2001; Bergren et al., 2005). Founder B6Q54 mice were obtained from the laboratory of M.H. Meisler (Department of Human Genetics, University of Michigan) and a colony was established at Case Western Reserve University. B6Q54 founder males were bred to C57BL/6J (B6) (Stock 000664) and SJL/J (SJL) (Stock 000686) female mice obtained from Jackson Laboratory (Bar Harbor, ME, U.S.A.). These standard inbred mouse strains were selected because they are both well-established, readily available, and similarly invulnerable to kainic acid-induced neurotoxicity (Schauwecker, 2002). B6Q54 founder males were backcrossed to B6 female mice to maintain the inbred B6Q54 line, and outcrossed to female SJL/J mice to create experimental mice, (B6Q54 × SJL)F1 (SJLQ54). Approximately 50% of all B6Q54 offspring were heterozygous for the Scn2a missense mutation (tg/+), with the remainder homozygous wild type (WT) (+/+). For our experiments, transgenic SJLQ54 (tg/+) mice were used for experimental data and age-matched sibling SJLQ54 (+/+) mice as WT controls. Both male and female mice were used in experiments, but their age was limited to a window of 3–10 weeks, prior to the onset of generalized seizures. All animals were housed in microisolator cages maintained in a veterinarianmonitored animal care facility.
Offspring were weaned and tagged at 3 weeks of age, and a small tail biopsy was taken. Genomic DNA was isolated from tail biopsies via proteinase K digestion, phenol/cholorform extraction and ethanol precipitation. Mice were genotyped by polymerase chain reaction (PCR) and electrophoresis of extracted tail DNA. PCR mix for each tail sample composed of (in ul): 2.5 10 × PCR buffer (without MgCl2), 1.7 MgCl2, 5 1 mM dNTPs, 1.5 Primer A1 (5_ GAT GCT CTT CTC CAC AAT GCT AAC C 3_), 1.5 Primer A2 (5_ GGG GAA ATC TTA ACA CCA GTC ACA C3_), 1.0 Primer N1 (5_ ATC CTT CCT TGG CTG CTT CAG ACT TG 3_), 1.0 Primer N2 (5_ CTC TTC TGC AAT GCG CTG TTC GAT AG 3_), 8.6 DII H2O, 0.2 Taq (5U/µl), and 2 tail DNA for a total volume of 25 µl. Two primer sets were used; N1-2 primers (product length = 960 bp) provided mutation detection while A1–2 primers (product length = 146 bp) served as PCR control. DNA amplification was conducted by PCR on a Mastercycler (Eppendorf AG, Hamburg, Germany). Samples were denatured for 2 min n at 94°C, followed by 33 cycles of 30 s at 94°C, 30 s at 65°C, 75 s at 72°C. Once PCR cycles were completed, 5 µl bromophenol blue marker was added to each sample and the mixture was electrophoresed on a 1% agarose gel (120V), visualized by ethidium bromide fluorescence, and measured against a 1 Kb DNA ladder.
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
Histology and cerebral spinal fluid extraction
The brain was removed from Q54 and WT mice at 4, 6, 8, and 10 weeks using the method described above. Following hemisphere sectioning, each half of the brain was immediately fixed in 10% formalin and later embedded in paraffin. Six sagittal hippocampal slices (20 µm thick) were cut from each animal. Cresyl violet stain was applied to mark Nissel bodies. Final sections were viewed with a light microscope and captured with digital imaging software. Quantitative histological measurements of the CA1 and CA3 pyramidal cell layers were completed with Java image processing software, ImageJ (NIH, Bethesda, MD, U.S.A.). Both cell layer measurements were taken a minimum of three times per slice, combined and averaged measurements were rounded to the nearest 10 µm. Murine cerebral spinal fluid (CSF) samples were isolated through a microdissection technique with limited blood contamination (<5%) as described in DeMattos et al. (DeMattos et al., 2002). 50 µl CSF samples obtained from Q54 and WT siblings were analyzed for [Na+], [K+], [Cl−], and osmolarity using a Dimension RXL chemical analyzer (Dade Behring, Deerfield, IL, U.S.A.). In addition, 100 µl “normal” artificial cerebral spinal fluid (nACSF) samples were tested for comparison with electrolyte composition of experimental perfusion solution.
Hippocampal slice preparation and perfusion
Three to ten week old Q54 and age-matched WT mice were decapitated under ethyl ether anesthesia. The brain was then rapidly removed and sectioned twice, removing the cerebellum and separating the two hemispheres by cutting midsagittally. Each hemisphere was glued (sagital face down) to a specimen disc with cyanoacrylate and placed into the cutting chamber of a vibratome (VT1000S, Leica, Nusslock, Germany) filled with ice-cold (3–4°C), sucroserich artificial cerebrospinal fluid (sACSF), containing (in mM): 220 sucrose, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 dextrose, saturated with 95% O2/5%CO2 gases (pH, 7.4). Sagittal hippocampal slices (350–400 µm thick) were cut and transferred to an incubation chamber filled with nACSF containing (in mM): 124 NaCl, 3.75 KCl, 1.25 KH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, 10 dextrose, saturated with 95% O2/5% CO2 gases (pH, 7.4) at room temperature (25°C) for at least 1h. Postincubation period, brain slices were transferred to an interface-recording chamber (Harvard Apparatus, Holliston, MA). Stimulation and recording of slices were conducted at 34°C ± in 2 nACSF, unless otherwise noted. Slices were discarded 6–8 h postincubation.
Electrophysiology and data analysis
Field potentials were recorded with a low-impedance glass micropipette (2–6 MΩ) filled with 150 mM NaCl. CA1 pyramidal cell population spikes (PSs) were evoked using a cathodic stimulus pulse (100 µsec, 10–350 µA, 0.05–0.1 Hz) delivered to the Schaffer Collaterals (orthodromic) by a tungsten electrode. All slices evoked a PS ≥1.0 mV to ensure consistent slice viability between experiments.
Paired-pulse and tetanic stimulation were applied to Q54 and WT brain slices to evaluate local circuitry. Paired stimuli (100 75 – 250 µA, 10 – 100 ms delay) were applied to Schaeffer collaterals at a frequency of 0.05 Hz. Delay between stimuli was varied from 100 to 10 ms at 10 ms intervals, while amplitude of stimulation was constant and determined during initial health check from the stimulus required to activate 75% of the maximum evoked PS in CA1 pyramidal neurons. Furthermore, a tetanization protocol was adapted from previous studies where sustained epileptiform discharges, or primary after discharges (ADs), were evoked in CA1 pyramidal neurons (Rafiq et al., 1995; Jahromi et al., 2002). Ten, two second stimulus trains (100 Hz, 100 µs, 75 – 250 µA) were applied to the Schaeffer collaterals of each slice at 10 min intervals. Stimulus amplitude was determined during initial health check as above.
Epileptiform activity was determined and quantified by evaluation of burst frequency (Hz), interburst frequency (Hz), and burst duration (ms). Burst frequency was measured by counting the number of interictal-like events per second during set time duration. Interburst frequency was measured by counting the number of recorded depolarization spikes per event per second over a set time duration (minimum peak-to-peak threshold was 0.25– 0.5 mV). Burst duration was determined by measuring the average time between burst onset and burst termination in ten consecutive bursts. All signals were amplified with an Axoprobe-1A microelectrode amplifier (Axon Instruments, Inc., Union City, CA, U.S.A.), further amplified and filtered by an FLA-01 eight pole Bessel filter/amplifier (Cygnus Technology, Delaware Water Gap, PA, U.S.A.), and recorded on a digital tape recorder DT-200 (Microdata Instruments, S. Plainfield, NJ, U.S.A.). Off-line data analyses were performed using Matlab Software (The Math-Works, Inc., Natick, MA, U.S.A.).
Measurements throughout this text are expressed as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM), with n indicating the number of animals, or slices, or samples of CSF per test group in each study. Results obtained were evaluated using statistical analysis methods including analysis of variance (ANOVA) and Student’s t-test, with a significance level, p < 0.05.
RESULTS
Spontaneous activity in Q54 slices
Initially, extracellular activity was recorded in Q54 (tg/+) and WT (+/+) slices to examine cellular activity under “normal” physiologic conditions. For several hours, spontaneous extracellular signals were recorded from the CA1 or CA3 pyramidal cell layer of hippocampal slices perfused with nACSF (saturated with 95% O2/5% CO2 gases, pH 7.4) at 34°C. As expected, spontaneous activity was not seen in either CA1 or CA3 pyramidal cell layer recordings from WT slices (n = 8). An example of a CA1 recording taken from WT slices is shown in Fig. 1A, (top). Approximately one-third of the field potential recordings obtained from the CA1 pyramidal cell layer of slices from Q54 mice (n = 12) showed abnormal spontaneous activity (Fig. 1B, middle). This spontaneous activity is similar to that of inter-ictal activity, with isolated or small groups of primarily unipolar field potential spiking and a mean burst frequency range of 0–15 Hz. In separate recordings, just over one-third of the field potential recordings from the CA3 pyramidal cell layer of slices from Q54 mice (n = 8) also showed similar abnormal spontaneous activity to that found in CA1 (data not shown). Moreover, a power analysis of field activity from all recordings revealed a significant increase in activity in both CA1 (+380%, p < 0.01) and CA3 (+120%, p < 0.01) from Q54 slices when compared to activity in WT slices (CA1, +9%, CA3, +6%). Power analysis was determined from an average of 15 power calculations, conducted over twenty second windows of recorded data taken between 55 and 65 min into each recording. Although no seizure-like-event (SLE) was observed in any slice, the increase of the network excitability was investigated by first testing the response to orthodromic stimulation.
Figure 1. Spontaneous Activity in Q54 hippocampal slices.

Extracellular recordings from CA1 or CA3 pyramidal cell layers in the hippocampus show spontaneous activity in some CA1 (33%, n = 12) and CA3 (37.5%, n = 8) pyramidal cell layer of Q54 slices, but was not found in CA1 (n = 8) or CA3 (n = 6) pyramidal cell layer of WT slices from Q54 and WT mice aged 8–10 weeks. A) extracellular trace from CA1 of WT slice in nACSF; B) extracellular trace from CA1 of Q54 slice in nACSF showing spontaneous activity (scale = 0.5mV, 10s); C) expanded segment (indicated by bar) of extracellular Q54 trace shown in Fig. 1B. (scale = 0.5mV, 0.2s). D) Power analysis of recorded signals supports a significant increase in spontaneous activity in Q54 mice. Error bars represent SD.
Evoked PS amplitude response curve in Q54 slices
CA1 pyramidal cell PSs were evoked by a range of orthodromic stimuli (20–300 µA, 100 µs, 0.2 Hz) applied to Schaeffer collaterals in both Q54 (n = 5) and WT (n = 7) hippocampal slices. A linear regression fit to the linear portion of each input-output curve (60–160 µA) results in a slope of 0.03 mV/µA for both Q54 and WT response curves. Although both groups maintain nearly identical response curves during initial stimulus magnitudes, Q54 evoked PS amplitudes significantly deviate at stimuli ≥ 200 µA, reaching a maximum of 4.03 ± 0.02 at 280 µA (Fig. 2). Conversely, WT amplitudes continue to increase in amplitude with increasing stimulus magnitude, and do not reach maximum of 6.08 ± 0.61 until 320 µA. Notably, Q54 mean PS amplitudes were found to be significantly different (p < 0.05) from WT amplitudes at stimulus magnitudes ≥ 200 µA (see Fig. 2).
Figure 2. Evoked CA1 population spike (PS) response curve.

Q54 hippocampal slice (n = 5) exhibit significantly smaller CA1 PS amplitudes when evoked by Schaffer collateral stimuli ≥ 200 µA in comparison with WT slices (n = 7) from mice aged 4–10 weeks (* indicates p<0.05); A) characteristic CA1 PS in WT slice at 320 µA; (scale is 1mV, 5ms); B) characteristic CA1 PS in Q54 slice at 320 µA; (scale is the same as in Fig. 2A) (▼ indicates stimulus artifact; error bars represent SD).
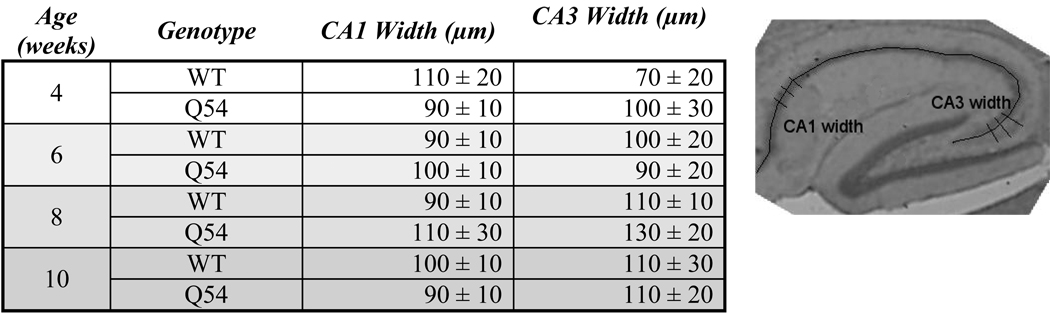
Cresyl violet histology of Q54 slices
The reduced PS amplitude in Q54 mice suggested the possibility of cellular damage, therefore a histological analysis of cresyl violet stained hippocampal slices from both Q54 and WT mice was carried out. Sagittal sections (20 µm, n = 6) were cut at four consecutive two week intervals for both Q54 (n = 3) and WT (n = 3) mice at each age group ranging from four to ten weeks of age. Visual inspection of cresyl violet stained sections did not indicate any significant differences between Q54 and WT at any age level. An example of ten week old Q54 and WT sections are shown in Fig. 3. Normal neuronal development was further supported by quantitative analysis of pyramidal cell layer regions CA1 and CA3 (Table 1). In both Q54 (n = 18) and WT (n = 18) slices the average width of the CA1 pyramidal cell layer ranged from 90 to 110 µm, while the average width of the CA3 pyramidal cell layer ranged from 70 to 110 µm in WT slices and 90 – 130 µm in Q54 slices. Regardless of this variation in average ranges, no significant difference was found between WT and Q54 measurements in either CA1 or CA3 pyramidal cell layers during any age period.
Figure 3. Morphology of the hippocampus in Q54 hippocampal slices.

Nissel bodies stained with cresyl violet exhibit regular neuronal development of the hippocampus in both Q54 (n = 18) and WT (n = 18) slices at ten weeks. Scale bar for A and B is shown in A (1 mm). Scale bar for C and D is shown in C (200 µm).
Table 1. Quantitative Histology.
Quantitative measurement of cresyl violet stained hippocampal slices demonstrate no significant difference in CA1 or CA3 pyramidal cell layer between Q54 (n = 18) and WT (n = 18) littermates at four, six, eight, and ten weeks of age. Data presented rounded to nearest tens ± SD.
 |
Cerebral spinal fluid of Q54 mice
Changes in the ionic concentrations of the extracellular space can have a significant impact on neuronal membrane potentials and excitability. Electrolytes ([Na+], [K+], [Cl−]) and osmolarity of isolated CSF samples (< 5% contamination) were analyzed to evaluate the similarities between Q54 CSF, WT CSF, and conventional in vitro ACSF (nACSF). CSF was extracted from Q54 (n = 6) and WT (n = 6) mice using a microdisection technique, then ran on a chemical analyzer against normal artificial cerebral spinal fluid (nACSF) (n = 11). While extracellular chloride concentration was found to be comparable among all groups, extracellular sodium concentrations were found to be significantly higher and extracellular potassium was found to be significantly lower in both Q54 and WT when compared to nACSF (p < 0.05, Table 2). Osmolarity was also found to be significantly higher in Q54 only, when compared to both WT and nACSF samples (p < 0.05, Table 2).
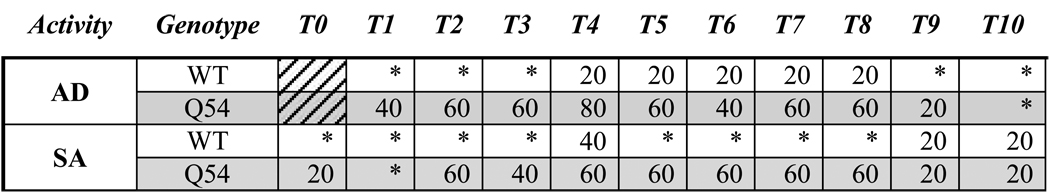
Table 2. Slices with CA1 activity following tetanic stimulus.
Both CA1 after discharge (AD) and spontaneous activity (SA) responses were detected less often in WT than in Q54 slices following the application of a high frequency tetanic stimulus trains (100 Hz, 100 µs, 75 – 250 µA) to Schaffer collaterals (* = 0%). Mice were 8–10 weeks old.
 |
Paired pulse test of recurrent inhibition
To evaluate the contribution of local inhibitory networks on CA1 pyramidal cell abnormal activity, a paired pulse stimulus paradigm was applied to the Schaeffer collaterals of Q54 (n = 5) and WT (n = 5) brain slices. Interpulse interval between two consecutive stimuli was varied by 10 ms steps from 10 ms to 100 ms, and the resultant PS amplitudes were recorded from CA1 (Fig. 4). A ratio of the amplitude of the second evoked response to the amplitude of the first evoked response gives an indication of local facilitation (>1) or inhibition (<1). Q54 slices reach their peak facilitation at 20 ms, while WT peak at 30 ms. It is also interesting to note that neither Q54 nor WT demonstrated a significant level of inhibition, which is normally expected when interpulse interval is close to 10 ms.
Figure 4. Reponse to paired pulse stimuli in Q54 hippocampal slices.

Q54 mice (n = 5) do not show a significant difference in comparison to WT mice (n = 5), however both groups maintain facilitation despite a short paired pulse delay suggesting compromised local inhibitory network activity. Characteristic extracellular trace of paired pulse stimulus is shown in inset (scale is 1 mV, 40 ms; ▼ indicates stimulus artifact; error bars represent SEM).
Response to high-frequency tetanic stimulus
In order to assess the threshold for inducing primary ADs using electrical stimulation, a sequence of ten, two second subthreshold tetanic stimuli (100 Hz, 100 µs, 75–250 µA) were applied to the Schaeffer collaterals of Q54 (n = 5) and WT (n = 5) hippocampal slices. Although the stimulus amplitude was markedly decreased from previously established protocols (Rafiq et al., 1995; Jahromi et al., 2002), primary Ads were detected in the CA1 pyramidal neuronal layer in both Q54 and WT (Fig. 5A and B). Spontaneous activity (SA) following stimulation was also detected in several slices, primarily in Q54 slices (Fig. 5C and D). Analysis of evoked activity immediately following stimulus train application showed fewer occurrences of an AD event in WT (n = 6/50 trains) when compared to Q54 (n = 24/50 trains) (Table 2). SA also occurred more regularly following tetanic stimulus in Q54 slices (n = 19/50 trains) than in WT slices (n = 5/50 trains) (Table 2). The duration of ADs generated by each pulse are plotted in Fig. 6. The duration of ADs are significantly higher in the Q54 when compared to the WT slices. The greatest average duration of AD response in Q54 mice was 6.8 ms and occurred following the fifth stimulus train (T5) compared to a 0.6 ms maximum average value for WT following the second stimulus train (T2) (Fig. 6). The increased presence of ADs as well as SA indicates an increased excitability in Q54 slices.
Figure 5. Response to tetanic stimulus in Q54 hippocampal slices.

Ten, two second tetanic stimuli (100 Hz, 100 µs, 75 – 250 µA) were applied to the Schaeffer collaterals of Q54 (n = 5) and WT (n = 5) hippocampal slices from mice aged 8–10 weeks. A) Typical extracellular field potential after discharge following the application of a high-frequency stimulus train recorded in CA1 of Q54 brain slice, B) Expanded segment (bar) from A; C) Spontaneous activity recorded in CA1 of Q54 slice; D) expanded segment (bar) from C (▼ indicates stimulus artifact in A and B).
Figure 6. Duration of evoked after discharge in CA1.

Recorded after discharge (AD) durations were markedly increased in Q45 slices when compared to WT (* p<0.01), with maximum average duration following the fifth stimulus train (T5). Mice were 8–10 weeks old. Error bars represent SD.
Analysis of interspike intervals showed key frequency components in both AD and SA recordings (Fig. 7). Primary frequencies recorded in Q54 AD were between 0–10 Hz, with additional secondary components in the 98–102 Hz range. Comparatively, WT AD data contained a significant amount of low frequency 0–10 Hz spiking, but lacked any significant high-frequency activity. The analysis of the frequency content of the SA induced by the stimulation revealed the presence of a high-frequency component (210–220 Hz) only in the Q54 mice. Furthermore, CA1 PS responses were recorded prior to the first stimulus train (T0) and prior to each additional train (T1–T10). A plot of CA1 PS amplitudes over all stimulus trains reveals a significant difference between linear regressions of the magnitudes of WT and Q54 over stimulus trains, with Q54 slices also having significantly higher amplitudes at individual trains, T0–T6 (Fig 8A, top). Additionally, the linear regression of CA1 PS duration also shows a significantly higher magnitude in the Q54 when compared to WT, with significantly different train activity at T0, T1, T2, and T8 (Fig 8B, bottom). The linear regression of evoked PS amplitude is significantly different between Q54 and WT with a p-value < 0.05, but the slope of each linear regression is strikingly similar: Q54 amplitude (m = 0.5 mV/train) and duration (m = 0.07 ms/train); WT amplitude (m = 0.3 mV/train) and duration (m = 0.06 ms/train). The presence of higher frequency components in bursting activity of Q54 slices in addition to increased amplitude of evoked PSs further demonstrate an increased response of Q54 slices to tetanic stimulation.
Figure 7. Frequency analysis of tetanically induced activity.

Histograms of frequency components found in WT (A, B) and Q54 (C, D) slices during afterdischarge (AD) or spontaneous activity (SA) following tetanic stimulation. Mice were 8–10 weeks old.
Figure 8. CA1 population spike between tetanic stimulus trains.

Orthodromic population spike (PS) was recorded prior to the first (0) and following each subsequent tetanic stimulus train (1–10). Linear regressions of PS amplitude (A) and duration (B) are significantly different between Q54 and WT, p<0.01 (* per train, p<0.05). Mice were 8–10 weeks old. Error bars represent SEM.
DISCUSSION
The most direct way to study basic physiological mechanisms underlying epilepsy is to explore the properties of human epileptic neuronal tissue in vivo. Recent developments in epilepsy surgery have led to an increase interest in the study of epileptic brain slices in vitro, unfortunately, human epileptic tissue is limited, difficult to maintain, and often damaged during removal. In addition, experimental control tissue for these studies is often inadequate (Kohling and Avoli, 2006). Recent developments in genetic mutations have led to numerous mouse models of human disease that match mutations found in human orthologs and result in similar disease phenotypes. In the case of heterozygous mutation models, age-matched littermate WT controls are ideal and readily available in their nontransgenic siblings. In addition, murine tissue is in greater supply and easier to harvest. For these reasons, genetically modified murine tissue is an ideal alternative to human tissue when studying the underlying mechanisms of disease, its development, and potential treatment strategies. The Q54 transgene, contains a missense mutation resulting in a three amino acid substitution, GAL879–881QQQ, of sodium channel gene, Scn2a. While this specific missense mutation has not been identified in humans, numerous mutations of both Scn1a and Scn2a have been discovered in families with inheritable epilepsy syndromes, including GEFS+, SMEI, and BFNIS (Meisler and Kearney, 2005), highlighting the importance of sodium channel function in the development of these seizure phenotypes. Moreover, several of these missense mutations, primarily Scn1a, are gain-of-function mutations resulting in increased persistent sodium current (Stafstrom, 2007), as was found in the Q54 hippocampal neurons (Kearney et al., 2001). Because the Q54 mouse model of epilepsy stems from a persistent sodium channel gain-of-function mutation that results in seizures within the temporal lobe, it is a useful model for the study of epilepsy mechanisms, primarily those involved in idiopathic or genetically inherited epilepsies of the temporal lobe and the transition from genotype to phenotype.
Our Q54 studies were conducted on mice outcrossed to SJL/J which has shown an increased expression of the Scn2a transgene and severity of the epilepsy phenotype, which could be a result of the heterozygosity of two modifier loci which are known to differ between B6 and SJL strains (Bergren et al., 2005). Q54 mice on (B6 × SJL)F2 or F3 background can be homozygous for these modifier loci, but are known to exhibit spontaneous focal seizures within 2–3 months, and EEG recordings from these animals demonstrated seizure activity primarily in the hippocampus (Kearney et al., 2001). We suspected that hippocampal tissue from Q54 mice would demonstrate spontaneous seizure activity in vitro. To test this hypothesis, unstimulated extracellular recordings were conducted in Q54 and WT hippocampal brain slices perfused with nACSF, from mice with a (B6 ×SJL)F1 background. While spontaneous epileptiform activity was not detected, interictal-like SA was observed in both the CA1 and CA3 regions of the hippocampal pyramidal cell layer of Q54 brain slices. This SA was not found in any of the WT control slices. Similarly, human epileptic brain slice studies have shown little evidence to support any alterations in the intrinsic properties of cortical or hippocampal neurons, yet experimental data does support a decrease in functional inhibition and/or an increase in synaptic excitation (Schwartzkroin, 1994).
Initially, local neuronal damage was suspected as a potential cause for the inability of pyramidal neurons to drive seizure activity in Q45 slices. Hippocampal sclerosis is a common feature of temporal lobe epilepsy, found in approximately 50–70% of surgical resections of the hippocampus from temporal lobe epilepsy patients (Honavar and Meldrum, 1997). In a study of the mechanisms of the pilocarpine rat model of temporal lobe epilepsy, maximal amplitude of orthodromically evoked response in epileptic slices was found to be reduced by about half that of control tissue (Sanabria et al., 2001). It was further suggested that the decreased PS amplitudes in epileptic mice were due to a reduction in neuronal density supported by prior evidence of status epilepticus-induced death of CA1 pyramidal cells (Mello et al., 1993; Liu et al., 1994). A similar reduction in PS amplitude was seen in our Q54 slices in comparison to WT, therefore we examined histological sections from a range of Q54 and WT mice across our experimental age window, 2–10 weeks, to determine the onset of cellular damage. Previous histological assessment of Q54 hippocampal slices from (B6 × SJL)F2 and F3 mice showed no histological abnormalities at 3 weeks, yet by 2 months of age extensive neuronal cell loss and gliosis were visible in animals with frequent seizures (Kearney et al., 2001). In contrast to the previous histological evaluation, our quantitative and visual interpretation of histological sections supports healthy development up to ten weeks in the Q54 hippocampus. One key difference from the previous histological study is that only “mice with frequent seizures” exhibited physiological neuronal damage in the hippocampus, indicating the possibility of a limited occurrence of damage within this subset of Q54 mice. Our statistical analysis suggests that neuronal damage is neither widespread nor significant prior to 10 weeks of age in (B6 × SJL)F1 Q54 mice, and supports that the abnormal PSs and SA recorded in nACSF of Q54 hippocampal slices are not necessarily due to pyramidal cell death or gliosis. Furthermore, data presented here supports previous evidence that gliosis and neuronal cell loss is a consequence of overt seizure activity, rather than the aging of genetically susceptible mice.
Another possible explanation for the lack of seizure activity in Q54 slices could be an electrolyte imbalance caused by the presence of increased persistent sodium current from the Scn2a mutation in Q54 mice. CSF electrolyte screening was conducted to evaluate differences in extracellular space composition. Electrolyte measurements ([Na+], [K+], [Cl−]) of CSF samples indicated no significant difference between Q54 and WT mice, yet both groups demonstrated a significantly higher sodium concentration and significantly lower potassium concentration when compared to nACSF solution. Moreover, the CSF samples from Q54 mice had a significantly higher osmolarity than nACSF. Such deviations of chemically imbalanced CSF from conventional ACSF electrolyte composition could affect the excitation state of brain slices during in vitro experiments. For example, osmotic pressure was determined to be critical determinant of endogenous firing patterns of CA1 pyramidal cells in rat hippocampal slices (Azouz et al., 1997). The same study also concluded that perfusion of hippocampal brain slices with elevated osmolarity suppressed endogenous burst firing, while reducing osmolarity converted nonbursting neurons to bursting neurons. Although, preliminary testing of modified ACSF with elevated sodium concentration (180 mM), did not result in significantly different behavior in the in vitro slice prep of Q54 mice (n = 2), changes in ionic balance due to perfusion with nACSF could facilitate increased excitability and resultant SA seen in Q54 slices. Experiments with decreased [K+]o or increased osmolarity were not carried out because both are known to decrease the susceptibility to seizure like events (Durand, 1993).
It has been hypothesized that a loss of inhibition among local circuit interactions is involved in the development of epileptiform activity within the hippocampus and several AEDs including diazepam are thought to act by exciting inhibitory interneurons (Knowles and Schwartzkroin, 1981). A recent study of Scn1a mutant mice found a significant decrease in action potential generation in dissociated hippocampal interneurons of Scn1a mutants and proposed the loss of inhibitory interneuron excitability as a possible mechanism for the resultant epilepsy phenotype in these mice (Yu et al., 2006). Recurrent inhibition describes the method by which local interneurons, excited by the firing of pyramidal cell axon collaterals, return inhibitory GABAergic processes back to pyramidal cell somas forming a negative feedback loop (Andersen, 1975). In this study, we tested the hypothesis that recurrent inhibition was reduced in hippocampal brain slices of Q54 mice with paired-pulse stimulation. Although we did not detect a significant difference in local inhibition when compared to WT, it is important to note that neither group demonstrated a significant amount of recurrent inhibition. Normally, as the delay between stimuli decreases, local inhibition is seen as a reduction of the second PS. In the case of both Q54 and WT, recurrent inhibition was rarely detected at all. Even more intriguing is that recurrent inhibition was found in a different strain of mice, namely C57Bl/6J (data not shown). These data support that the hyperexcitability observed in these slices is not mediated by a lack of inhibition alone. Therefore there is another factor, perhaps genetically strain-based, that could be involved in the development of seizure activity in these animals which is further supported by recent evidence in strain susceptibility to the Scn2a mutation (Bergren et al., 2005; Kearney et al., 2006a).
It is possible that a critical mass is required to develop full-blown seizure activity in the absence of pharmacological or stimulatory intervention. This critical mass theory is further supported by the fact that the removal from the host environment, and the slicing of brain tissue leads to physical damage that may affect distal as well as local networks and their ability to trigger and maintain synchronous seizure waveforms, primarily a lack of recurrent inhibitory network loops. Because of this and the apparent reduction of local inhibition in Q54 slices demonstrated in our paired-pulse study, we decided to evaluate the response of Q54 slices to high-frequency stimulation. Highfrequency tetanic stimulus trains have been shown to drive epileptiform primary and secondary ADs in the entorhinal cortex-hippocampal slice (Rafiq et al., 1993). In addition, self-sustained, or SA in addition to ADs has been shown in response to tetanic stimulus in the hippocampal-parahippocampal slice (Rafiq et al., 1995). In this study, we applied a series of titanic stimulus trains with minimized amplitude to evaluate the sensitivity of Q54 hippocampal neurons to drive seizure-like activity. In our study, the stimulus amplitude was set to that required to drive 75% of the CA1 pyramidal cell response, and commonly found to be less than five volts (current injection less than 150 µA).
To evaluate the viability of the neural tissue during tetanic recordings, orthodromically evoked CA1 PSs were measured periodically. While PS amplitude and duration increased at a similar rate, both variables were consistently significantly higher in Q54 slices suggesting an elevated level of synaptic excitation. High-frequency stimulus trains triggered both ADs as well as SA in Q54 slices nearly four times as often as in WT slices, further supporting hyperexcitability and increased synaptic function in the hippocampus due to the expression of altered Scn2a sodium channels in Q54 mice. SA recorded was similar to that recorded in normal ACSF without stimulation, with the exception that it contained a much higher interburst frequency close to 200 Hz.
In the hippocampus, synchronized neuronal population activity is categorized according to their frequency as theta (4–12 Hz), gamma/beta (15–70 Hz) or high-frequency/very fast oscillations (>70 Hz) (Buzsaki, 2002; Traub et al., 2004). While theta oscillations are thought to represent the “on-line” or “ready” state of the hippocampus (Vertes and Kocsis, 1997), high-frequency oscillations are associated with epileptiform activity in both humans and rats (Bragin et al., 2002; Bragin et al., 2004) and have been found to be nonsynaptic in nature (Draguhn et al., 1998; Hormuzdi et al., 2001). An evaluation of frequency components during spontaneous and AD activity revealed highfrequency oscillations of 100 and 200 Hz, in Q54 slices that were not detected in WT slices, supporting the presence of a nonsynaptic component to Q54 hyperexcitability. Theta band oscillations were found to be present in SA as well as AD responses of both WT and Q54 hippocampal slices.
In conclusion, electrophysiological in vitro studies were conducted to study abnormal activity in the hippocampus of Q54 mice expressing modified Scn2a sodium channels. A significant amount of spontaneous firing was found in both CA1 and CA3 regions of Q54 hippocampal slices, but absent in WT slices. Spontaneous interictal like activity was seen more commonly following brief periods of high-frequency titanic stimulus in both Q54 and WT slices. Q54 had four times the amount of spontaneous as well as AD activity during tetanic stimulus application suggesting increased sensitivity to external input. The hypothesis that neural tissue in Q54 mice was hyperexcitable was further supported by significantly higher PS amplitude and duration in Q54 slices throughout tetanic stimulus testing. Paired pulse analysis did not show evidence of increased inhibition in Q54 mice, yet both Q54 and WT mice demonstrated substantial facilitation. Cresyl violet histology did not reveal any obvious cell loss, and CSF samples suggest the need for correction of perfusion solutions as well as a seemingly compensatory elevation of osmolarity in Q54 mice. Finally, high-frequency oscillations unique to spontaneous and AD activity of Q54 slices following tetanic stimulus were observed. These data support the hypothesis that modified Scn2a channels in Q54 mice result in hyperexcitability of the hippocampus as well as an increased response to orthodromic input most likely involved in the development and/or maintenance of temporal lobe seizures.
REFERENCES
- Andersen P. Isolated brain slices: A new preparation for theoretical and clinical research. Tidsskr Nor Laegeforen. 1975;95:349–351. [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R, Kohling R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol. 2005;77:166–200. doi: 10.1016/j.pneurobio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol. 1997;502(Pt 1):175–187. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergren SK, Chen S, Galecki A, Kearney JA. Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm Genome. 2005;16:683–690. doi: 10.1007/s00335-005-0049-4. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, Gambardella A, Steinlein OK, Grinton BE, Dean JT, Bordo L, Hodgson BL, Yamamoto T, Mulley JC, Zara F, Scheffer IE. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–557. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Almajano J, Mody I, Engel J., Jr High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis. Epilepsia. 2004;45:1017–1023. doi: 10.1111/j.0013-9580.2004.17004.x. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Staba RJ, Reddick M, Fried I, Engel J., Jr Interictal high-frequency oscillations (80–500 Hz) in the human epileptic brain: entorhinal cortex. Ann Neurol. 2002;52:407–415. doi: 10.1002/ana.10291. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol. 2004;30:236–243. doi: 10.1016/j.pediatrneurol.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Ceulemans B, Audenaert D, Smets K, Lofgren A, Del-Favero J, Ala-Mello S, Basel-Vanagaite L, Plecko B, Raskin S, Thiry P, Wolf NI, Van Broeckhoven C, De Jonghe P. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. 2003;21:615–621. doi: 10.1002/humu.10217. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229–236. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JG. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394:189–192. doi: 10.1038/28184. [DOI] [PubMed] [Google Scholar]
- Durand D. Ictal patterns in experimental models of epilepsy. J Clin Neurophysiol. 1993;10:281–297. doi: 10.1097/00004691-199307000-00004. [DOI] [PubMed] [Google Scholar]
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus--and prevalence of variants in patients with epilepsy. Am J Hum Genet. 2001;68:866–873. doi: 10.1086/319524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Fisher RS. Animal models of the epilepsies. Brain Res Brain Res Rev. 1989;14:245–278. doi: 10.1016/0165-0173(89)90003-9. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain. 2003;126:531–546. doi: 10.1093/brain/awg053. [DOI] [PubMed] [Google Scholar]
- Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–852. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- Honavar M, Meldrum BS. Epilepsy. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. 6th Edition. London: Arnold; 1997. pp. 931–971. [Google Scholar]
- Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron. 2001;31:487–495. doi: 10.1016/s0896-6273(01)00387-7. [DOI] [PubMed] [Google Scholar]
- Jahromi SS, Wentlandt K, Piran S, Carlen PL. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol. 2002;88:1893–1902. doi: 10.1152/jn.2002.88.4.1893. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24:2690–2698. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, Dejonghe P, Frankel WN. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006a;15:1043–1048. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Wiste AK, Stephani U, Trudeau MM, Siegel A, RamachandranNair R, Elterman RD, Muhle H, Reinsdorf J, Shields WD, Meisler MH, Escayg A. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr Neurol. 2006b;34:116–120. doi: 10.1016/j.pediatrneurol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Knowles WD, Schwartzkroin PA. Local circuit synaptic interactions in hippocampal brain slices. J Neurosci. 1981;1:318–322. doi: 10.1523/JNEUROSCI.01-03-00318.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Avoli M. Methodological approaches to exploring epileptic disorders in the human brain in vitro. J Neurosci Methods. 2006;155:1–19. doi: 10.1016/j.jneumeth.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Lerche H, Weber YG, Baier H, Jurkat-Rott K, Kraus de Camargo O, Ludolph AC, Bode H, Lehmann-Horn F. Generalized epilepsy with febrile seizures plus: further heterogeneity in a large family. Neurology. 2001;57:1191–1198. doi: 10.1212/wnl.57.7.1191. [DOI] [PubMed] [Google Scholar]
- Liu Z, Nagao T, Desjardins GC, Gloor P, Avoli M. Quantitative evaluation of neuronal loss in the dorsal hippocampus in rats with long-term pilocarpine seizures. Epilepsy Res. 1994;17:237–247. doi: 10.1016/0920-1211(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Lombardo AJ, Kuzniecky R, Powers RE, Brown GB. Altered brain sodium channel transcript levels in human epilepsy. Brain Res Mol Brain Res. 1996;35:84–90. doi: 10.1016/0169-328x(95)00194-w. [DOI] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Ohmori I, Ouchida M, Ohtsuka Y, Oka E, Shimizu K. Significant correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy. Biochem Biophys Res Commun. 2002;295:17–23. doi: 10.1016/s0006-291x(02)00617-4. [DOI] [PubMed] [Google Scholar]
- Ohmori I, Kahlig KM, Rhodes TH, Wang DW, George AL., Jr Nonfunctional SCN1A is common in severe myoclonic epilepsy of infancy. Epilepsia. 2006;47:1636–1642. doi: 10.1111/j.1528-1167.2006.00643.x. [DOI] [PubMed] [Google Scholar]
- Rafiq A, DeLorenzo RJ, Coulter DA. Generation and propagation of epileptiform discharges in a combined entorhinal cortex/hippocampal slice. J Neurophysiol. 1993;70:1962–1974. doi: 10.1152/jn.1993.70.5.1962. [DOI] [PubMed] [Google Scholar]
- Rafiq A, Zhang YF, DeLorenzo RJ, Coulter DA. Long-duration self-sustained epileptiform activity in the hippocampal-parahippocampal slice: a model of status epilepticus. J Neurophysiol. 1995;74:2028–2042. doi: 10.1152/jn.1995.74.5.2028. [DOI] [PubMed] [Google Scholar]
- Sanabria ER, Su H, Yaari Y. Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol. 2001;532:205–216. doi: 10.1111/j.1469-7793.2001.0205g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE. Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res. 2002;135:139–148. doi: 10.1016/s0079-6123(02)35014-3. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Cellular electrophysiology of human epilepsy. Epilepsy Res. 1994;17:185–192. doi: 10.1016/0920-1211(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology. 2002;58:1122–1124. doi: 10.1212/wnl.58.7.1122. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Ito M, Nagafuji H, Fukuma G, Mitsudome A, Wada K, Kaneko S, Hirose S, Yamakawa K. Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology. 2001a;57:703–705. doi: 10.1212/wnl.57.4.703. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001b;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suls A, Claeys KG, Goossens D, Harding B, Van Luijk R, Scheers S, Deprez L, Audenaert D, Van Dyck T, Beeckmans S, Smouts I, Ceulemans B, Lagae L, Buyse G, Barisic N, Misson JP, Wauters J, Del-Favero J, De Jonghe P, Claes LR. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat. 2006;27:914–920. doi: 10.1002/humu.20350. [DOI] [PubMed] [Google Scholar]
- Tian LM, Otoom S, Alkadhi KA. Endogenous bursting due to altered sodium channel function in rat hippocampal CA1 neurons. Brain Res. 1995;680:164–172. doi: 10.1016/0006-8993(95)00258-r. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, LeBeau FE, Buhl EH, Whittington MA. Cellular mechanisms of neuronal population oscillations in the hippocampus in vitro. Annu Rev Neurosci. 2004;27:247–278. doi: 10.1146/annurev.neuro.27.070203.144303. [DOI] [PubMed] [Google Scholar]
- Vertes RP, Kocsis B. Brainstem-diencephalo-septohippocampal systems controlling the theta rhythm of the hippocampus. Neuroscience. 1997;81:893–926. doi: 10.1016/s0306-4522(97)00239-x. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Parasivam G, Barnett S, Wallace GB, Sutherland GR, Berkovic SF, Mulley JC. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology. 2002;58:1426–1429. doi: 10.1212/wnl.58.9.1426. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL, Jr, Mulley JC, Berkovic SF. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68:859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]