

Abstract
Fas/Apo-1 signals through the FADD adaptor protein which recruits and activates the apical Caspase 8, and leads to apoptosis. cFLIP is a homologue of Caspase 8 and also capable of binding to FADD. Previous studies suggest that cFLIP could either enhance or inhibit apoptosis, and lead to NF-κB and Erk1/2 activation. Like FADD or Caspase 8 deficiency, a lack of cFLIP disrupts embryogenesis and T cell proliferation. It has been demonstrated that B cells lacking either FADD or Caspase 8 were defective in both Fas-induced apoptosis and TLR-induced proliferation, which indicates that these death-inducing proteins have an additional role in regulating innate immunity. To analyze the function of cFLIP in B cells, conditional deletion of cFLIP was induced by using CD19Cre. The resulting B cell-specific cFLIP-deficient mice were found to have reduced numbers of peripheral B cells which were hypersensitive to Fas-induced apoptosis, and impaired in proliferation induced by TLRs and the BCR. Furthermore, there was aberrant expression of co-stimulatory proteins and activation markers in cFLIP-deficient B cells. Whereas LPS-induced activation of NF-κB and Erk1/2 appears to be unaffected, p38 and Jnk were spontaneously activated and hyper-induced in cFLIP-deficient B cells. Therefore, these data revealed novel functions of cFLIP in B cells.
Keywords: cFLIP, B cells, TLR, apoptosis, proliferation
INTRODUCTION
Apoptosis plays an essential role in lymphocyte development and homeostasis, and can be initiated by death receptors (DRs), which include Fas/Apo-1, TNF-R1, and TRAIL receptors. Fas deficiency leads to lymphoproliferative and autoimmune diseases (1). All DRs contain an intracellular protein-protein interaction motif, the death domain (DD), which directly binds to the DD of the Fas-associated death domain (FADD) protein (2). FADD interacts with Caspase 8/FLICE/MACH through a second protein-protein interaction module, the death effector domain (DED) (3, 4). The proximity induced by clustering of DRs while engaged to their cognate ligands facilitates autoprocessing of pro-Caspase 8. Activated Caspase 8 can directly act on downstream Caspases including Caspases 3, 6 and 7 to induce apoptosis.
Deletion of any one of the DRs has no noticeable effect on mouse development; while a deficiency in either FADD or Caspase 8 leads to early embryonic lethality (5, 6). These results indicate that FADD and Caspase 8 have additional functions that are independent of DRs. Conditional gene deletion studies have previously been performed to understand the function of these two proteins in the immune system. In contrast to the phenotype of fas−/− mice in which there is a marked accumulation of T cells in the spleen and lymph nodes, T cell-specific fadd−/− mice contain reduced numbers of T cells in the periphery (7). fadd−/− T cells are not only defective in Fas-induced apoptosis but also impaired in TCR-induced proliferation. T cell-specific caspase 8−/− mice essentially phenocopy T cell-specific fadd−/− mice (8). These results indicate that FADD and Caspase 8 cooperate in both DR-induced apoptosis and TCR-induced signaling essential for proliferation.
In addition to FADD and Caspase 8, the apoptotic signaling complex also contains the cellular FLICE inhibitory protein (cFLIP) (9). cFLIP has two NH2-terminal DED domains which can bind to the DED of FADD, and a Caspase-like domain at the carboxy terminus. Overexpression of cFLIP could lead to either inhibition of or sensitization to Fas-induced apoptosis (9, 10). Similar to the effect of FADD or Caspase 8 ablation, cFLIP deficiency also results in early embryonic lethality (11). However, unlike T cell-specific fadd−/− and caspase 8−/− mice which contain substantial numbers of peripheral T cells, T cell-specific cflip−/− mice lack peripheral mature T cells (7, 8, 12). Peripheral T cells numbers were dramatically reduced in cflip−/− →*rag-1−/− chimeras (13). cflip−/− T cells are highly sensitive to Fas-induced apoptosis, which suggests an inhibitory role for cFLIP. In addition, cflip−/− T cells have a severe defect in TCR-induced proliferation responses, a phenotype analogous to that seen with fadd−/− and caspase 8−/− T cells. The function of FADD and Caspase 8 in B cells has been studied previously using B cell-specific fadd−/− and caspase 8−/− mice (14, 15). As expected, fadd−/− and caspase 8−/− B cells displayed impaired Fas-induced apoptosis. Surprisingly, these mutant B cells have normal BCR-induced proliferative responses, but are defective in proliferation induced by Toll-like receptors (TLRs).
There has been insufficient understanding of the function of cFLIP in B cells. Previous studies have suggested a role for cFLIP in inhibiting apoptosis in B cells (16, 17). A lack of cFLIP in embryonic stem cells resulted in a deficiency of B cells in cflip−/− →rag-1−/− chimeras (13). However, the temporal requirement of cFLIP during B lineage development has not been determined. Overexpression of cFLIP reportedly led to the activation of NF-κB and Erk1/2 in T cells (18). However, cflip−/− T cells have normal TCR-induced activation of NF-κB and Erk1/2 (12). cFLIP has also been shown to inhibit TLR-induced activation of NF-κB (19). In this study, we generated mice lacking cFLIP specifically in B cells, and examined the requirement for cFLIP during B cell development, apoptosis and proliferation. The data showed that while dispensable for the development of bone marrow precursor B cells, cFLIP is necessary for B cell maturation in the periphery. Furthermore, cFLIP is essential for proliferative responses induced by both TLRs and the BCR and plays a role in regulating the activity of p38 and Jnk stress MAPKs
Materials and Methods
Mice
cflipf/f and cd19+/cre mice have been described previously (12, 20). The cflipf/f mice were crossed withcd19+/cre mice. The resultingcflip+/f cd19+/cre and cflip+/f cd19+/+ mice were crossed to produce the B cell-specific cFLIP-deficient, cflipf/f cd19+/cre, mice which contain one allele of the endogenous cd19 gene. PCR-mediated genotyping for the cflip gene alleles and cd19cre allele was performed as described (12, 14). All animal studies were approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University.
Flow cytometry
The tibia, femur and humeri were isolated from cflip+/+ control mice and cflip−/− mutant mice, and bone marrow cells prepared. The spleen and lymph nodes were surgically isolated. Single cell suspensions were prepared, and red blood cells were depleted by hypotonic lysis. Peritoneal lavage was performed with 3-5 ml PBS containing 3% BSA and 1 mM EDTA. For cell surface protein staining, appropriate fluorochrome-conjugated Abs were added to single cell suspensions (106) in PBS containing 3% BSA, 0.5 mM EDTA, and 0.1% sodium azide, and samples incubated on ice for 20-30 min. Cells were washed once with PBS, and analyzed using a Coulter Epics XL analyzer (Beckman Coulter). The Abs used for flow cytometric analyses are as follows. CD21/35-FITC (7G6, 553818), CD23-PE (B3B4, 553139); IgD-PE (11-26C.2A 558597), Mac-1/CD11b-PE (M1/70), and biotinylated anti-CD19 (1D3), HSA-FITC(M1/69), Fas-PE (554258), and CD3-PE (145-2C11) were from BD-Pharmingen; IgM-FITC from Jackson ImmunoResearch; CD5-PE (53-7.3), TLR4-PE(MTS510,12-9924-81) and AA4.1-PE(12-5892-82) from eBiosciences; Tri-Color-B220 and streptavidin-Tri-color from Caltag. Data were analyzed with FlowJo software (Treestar).
Immunohistology
Spleen and lymph nodes cryostat sections (5-6 μm) were prepared and immunohistology was performed as previously described (14). Cryo-scetions were stained with HRP-anti-CD4 Abs (GK1.5, prepared in house) and biotin-anti-B220 Abs (eBioScience). The Vector Blue Streptoavidin-alkaline-phosphatase substrate kit III and the Vector NovaRed kit for peroxidase (Vector Laboratories) were then used the color development. The stained sections were analyzed using a light microscope (Leitz Diaplan; Axioplan Universal Microscope; Carl Zeiss MicroImaging, Inc.), and digital images captured using an Eastman Kodak Co. camera.
B cell preparation and culture
Lymph node and splenic B cells were purified by magnetic depletion of the CD43+ population using anti-CD43 Abs conjugated to MACS beads (Miltenyi Biotec), per the manufacturer';s instruction. The purity of these negatively selected cells was determined to contain >95% of CD19+ B cells by FACS analysis. The resulting B cells were cultured in RPMI 1640 (Mediatech) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 5 μM β-mercaptoethanol (Sigma-Aldrich), and 10% FBS (HyClone). Cells were kept at 37°C in a 5% CO2 incubator.
In vitro cell survival and apoptosis assay
B cells were seeded in 96-well round-bottom plates in triplicate (105/well) in 100 μl complete RPMI 1640. After incubation at 37°C in 5% CO2 for 3, 6 and 9 h, cells were incubated with propidium iodide (PI, 1 μg/ml) for 5 min at room temperature, and analyzed using a flow cytomter. The PI− populations were viable cells. To induce apoptosis, sFasL (Alexis Biochemical) was added to the culture along with anti-FLAG Abs (M2; 1 μg/ml; Sigma-Aldrich), in order to crosslink FasL which contains a FLAG tag. After a 3 h-incubation, cells were incubated with PI (1 μg/ml) for 5 min at room temperature, and analyzed using a flow cytomter. Apoptotic cells were the PI+ population. To isolate immature B cells, single cell suspensions were prepared as described above and stained with Tri-color-anti-B220, PE-anti-AA4.1 and FITC-anti-HAS Abs. B220+AA4.1hiHSAhi immature B cells were isolated using a MoFlo high-speed cell sorter (DakoCytomation). Apoptosis was induced by stimulation with anti-IgM F(ab′)2 of goat-anti- mouse IgM (μ; chain, Jackson ImmunoResearch) for 16 h. To analyze apoptosis in activated B cells, splenic B cells were purified using MACS beads, and stimulated for 48 h with anti-CD40 antibodies (10 μg/ml; FGK45, Alexis Biochemical). These activated B cells were resuspended in fresh medium, and treated with either FasL (50 ng/ml) for 6 h or TRAIL (100 ng/ml; Alexis) for 16 h. Cell death was determined by PI uptake.
B cell proliferation and activation marker analysis
B cells were plated in triplicate at 1×105cells/well in 96-well round-bottom plate in 100 μl of complete RPMI 1640. LPS, poly (I:C) (Sigma-Aldrich), CpG ODN 1826 (Coley Pharmaceutical), F(ab′)2 of goat-anti- mouse IgM (μ chain, Jackson ImmunoResearch), or anti-CD40 Abs (FGK45, Alexis Biochemical) was added, followed by for 40 h incubation. Cells were incubated in the presence of [3H] thymidine (2 μCi/well, ICN Biochemicals) for an additional 8 h. Cells were transferred to a 96 well Printed Filtermat A glass fiber filter (Wallac) using Harvester 96R (Tomtec). Radioactivity was determined using a 1205 Betaplate™ liquid scintillation counter (PerkinElmer).
Cell division kinetics was analyzed by the CFSE dilution assay. B cells (3 × 106) were resuspended in 1 ml PBS-5% FBS, and mixed with 0.1 ml PBS-5% FBS containing 50 μM CFSE (Molecular Probe). After 5 min incubation in the dark at 37°C, cells were washed 3 × with 5 ml of RPMI-10% FBS, and resuspended in 1.5 ml complete RPMI. These labeled cells were seeded to round-bottom 96-well plates (1 × 105/well in 50 μl), and 50 μl of LPS (2 μg/ml in RPMI) or anti-IgM Abs (20 μg/ml in RPMI) were then added to each well. After various times in culture, cells were analyzed using a flow cytometer. To detect activation marker expression, purified B cells were cultured with LPS (1 μg/ml) or anti-CD40 (10 μg/ml) in 96-well round-bottom plates as indicated above. After the indicated time, cells were washed once with 1 ml of staining buffer, and then stained on ice for 30 min with CD86-PE (GL1), CD54-PE (YN1/1.7.4; eBioSciences), I-Ab-FITC (AF6-120.1; BD Bioscience), CD69-PE, and CD62L-FITC (Caltag). The cells were then washed once in PBS, and analyzed by flow cytometry.
Ab responses
Pre-sera were collected from unimmunized mice. LPS-trinitrophenol (TNP) (25μg, Biosearch Technologies) was dissolved in 200 μl PBS, and injected intraperitoneally into mice. OVA-NP(15) (100μg in 100 μl PBS; Biosearch Technologies) was emulsified in 100 μl of Complete Freund's adjuvant (CFA; Sigma-Aldrich), and injected intraperitoneally into mice. Anti-sera were collected at days 3 and 6 from LPS-TNP-immunized mice or at days 7 and 14 from OVA-NP-immunized mice. ELISA was performed using goat-anti-mouse IgM-HRP, IgG2a-HRP or IgG2b-HRP (Southern Biotech) according to a protocol provided by the manufacturer with minor modifications.
Western blot analyses
Purified B cells (5-10 × 106) were resuspended in complete RPMI 1640 (2 × 106/0.1 ml), mixed with an equal volume of RPMI containing LPS (20 μg/ml), and incubated for 0, 15 or 30 min at 37°C. Cells were washed once in ice cold PBS and cell lysates prepared in ice-cold Triton lysis buffer (1% Triton X-100,50 mM Tris pH8.0, 150 mM NaCl, 20 mM EDTA, 1 mM PMSF, 0.7μg/ml pepstatin and completer protease inhibitor cocktail). Proteins (40 μg) were separated on 10% SDS/PAGE. Blots were incubated with antibodies specific for phospho-IκB, IkB, phospho-Erk1/2, Erk1/2, phospho-p38, phospho-Jnk, p65, p38, Jnk, and β-actin (Cell Signaling) overnight at 4°C followed by horseradish peroxidase-conjugated goat anti-rabbit Abs (1:2000; Pierce) according to manufacturer's standard procedures. All Abs were from Cell Signaling and used at 1:1000 dilution. For nuclear and cytoplasmic protein extraction, LPS-stimulated B cells (1.4 × 107) were resuspended in 150 μl cytosol extraction buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 × complete protease inhibitor (Roche), 1 mM DTT, and 0.2 mM PMSF), followed by 15 min incubation on ice. NP-40 was then added (0.5%). After centrifugation, supernatant (cytoplasmic extract) was transferred to a clean tube, and the pellet of nuclei was resuspended in 40 μl of ice-cold nuclear extraction buffer (10 mM HEPES pH 7.9, 400 mM NaCl, 0.1 mM EDTA, 1 × complete protease inhibitor, 1 mM DTT, 0.2 mM PMSF). After centrifugation, the supernatant (nuclear extract) was transferred to a clean tube. Proteins (20-40μg) were separated by 10% SDS/PAGE, and western blotting was performed using anti-NF-κB p65 and NF-kB p100/50 Abs (Cell Signaling).
Results
The effect of cFLIP deficiency on B cell development
B cell development is initiated in the bone marrow and continues in peripheral lymphoid organs. Western blotting showed that in cflip+/+ mice, bone marrow cells express lower levels of cFLIP than thymocytes and splenocytes (Fig. 1 A). Deletion of cFLIP results in early embryonic lethality (11). To investigate the function of cFLIP in B cells, conditional knockout (cflipf/f) mice containing floxed alleles of cflip were used. Previously, the cflipflf alleles were efficiently excised in T cells (12). In this study, B cell-specific deletion of cflipf/f was induced using cd19cre mice (20). In the resulting cflip−/− cd19cre mice (designated as b-cflip−/− mice hereafter), there is a significant reduction of the cFLIP protein level in the spleen, when compared with cflip+/+ mice (Fig. 1 A). Purified b-cflip−/− B cells contain minimal cFLIP protein, indicating efficient deletion of the cflip gene in B cells induced by cd19cre.
Figure 1.

(A) Western blot analyses of cFLIP protein expression in cflip−/− mutant and cflip+/+ control mice. (B) Cellularity of lymphoid organs. Lines indicate the average numbers. (C) B lineage cells in the bone marrow were analyzed by flow cytometry. The numbers indicate the percentages of each subset.
The average total cell numbers in the bone marrow were slightly reduced in b-cflip−/− mice, when compared with cflip+/+ mice (Fig. 1 B). Flow cytometric analyses showed that the percentages of total bone marrow B lineage cells (B220+) in b-cflip−/− mice were somewhat lower than in cflip+/+ mice (Fig. 1 C). The percentages of pro/pre B cells (B220loIgM−) and immature B cells (B220+IgM+) were not significantly changed in b-cflip−/− mice. However, the number of re-circulating mature B cell (B220hiIgM+) were lower in b-cflip−/− mice than in cflip+/+ mice (Fig. 1 C). This result was confirmed by double-staining for IgM and IgD, as there were fewer IgM+IgD+ re-circulating mature B cells in the bone marrow of cflip−/− mice than in cflip+/+ mice (Fig. 1 C). These data suggest that cd19cre-mediated deletion of cFLIP does not significantly affect B cell development in the bone marrow, but may play a role in peripheral mature B cells.
The average total cell numbers in the spleen and lymph nodes of cflip−/− mutant mice appear to be lower than in cflip+/+ control mice (Fig. 1 B). Splenocytes and lymph node cells were stained for the T cell marker CD3 and the B cell marker B220. The percentages of B220+CD3− B cells in the spleen and lymph nodes of b-cflip−/− mutant mice tend to be lower than in control cflip+/+ control mice (Fig. S1). On average, the numbers of B cells purified from the spleen and lymph nodes of b-cflip−/− mice are significantly reduced, when compared with control cflip+/+ mice (Fig. 2 A). Immunohistochemistry were performed on spleen and lymph node cryo-sections by staining with anti-B220 and anti-CD4 Abs and measurement of B cell follicles was analyzed. Although the lymphoid follicles in cflip−/− mice appeared to be smaller than that of control cflip+/+ mice (Fig. 2 B), the difference in the average size between the control and mutant mice is not significant (p=0.32, data not shown).
Figure 2. Analyses of peripheral B cells.
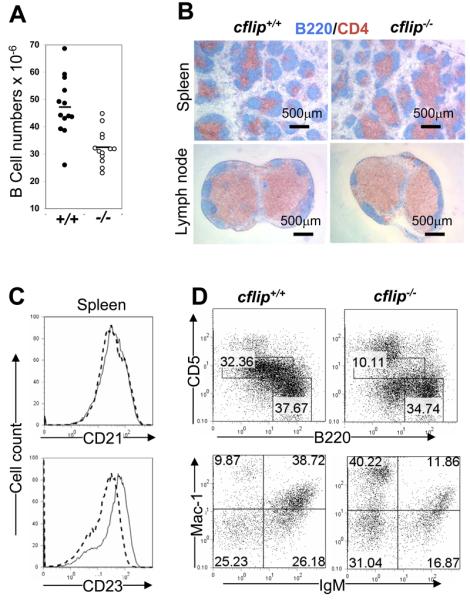
(A) Total numbers of purified B cells from cflip+/+ and b-cflip−/− mice. Lines indicate the average numbers. (B) Cryosections of the spleen and lymph nodes were stained for B cells with anti-B220 Abs (blue) and for T cells with anti-CD4 Abs (brown). The scale bars are shown. (C) Flow cytometric analysis of peripheral B cells (B220+) for CD21 and CD23 expression. Solid lines represent cflip−/− mice and dotted lines indicate cflip+/+ mice. (D) Peritoneal cells were stained with Abs for B220 and CD5 or IgM and Mac-1. The numbers represent the percentages of T cells and B cell subsets.
In subsequent studies, splenocytes and lymph node cells were stained for surface IgM and IgD and analyzed by flow cytometry. In the spleen, the percentages of IgD+IgMlo, IgD+IgM+, and IgDloIgM+ populations were not dramatically affected by a lack of cFLIP (Fig. S1). In b-cflip−/− lymph nodes, the percentage of mature IgD+IgMlo B cells is significantly decreased, as compared to control cflip+/+ mice. Conventional mature B cells are classified into two subsets in the mouse. Re-circulating follicular (FO) B cells are usually found in lymphoid follicles of the spleen and lymph nodes, while marginal zone (MZ) B cells reside primarily around the periphery of splenic lymphoid nodules. To analyze FO and MZ B cells, spleen cells were stained for B220, CD21 and CD23, and flow cytometric analyses indicated that cFLIP deficiency did not consistently affect the FO (B220+CD21+CD23+) and MZ (B220+CD21+CD23lo) B cell populations (Fig. S1). cflip−/− B cells express normal levels of CD21, but have higher levels of CD23 (Fig. 2 C).
The B1 population is a subset of peripheral B cells which express CD5, and are present mainly in the peritoneal and pleural cavities (21). The total number of peritoneal cells was significantly reduced in b-cflip−/− mice, as compared with cflip+/+ mice (Fig. 1B). Peritoneal B cell populations were analyzed by staining for B220 and CD5. The data showed that the percentages of B1 (CD5+B220lo) cells in b-cflip−/− mice were about 3 fold lower than in control cflip+/+ mice (top, Fig. 2 D). Peritoneal B1 cells also express Mac-1 (CD11b). Additional analyses were performed by staining peritoneal cells for IgM and Mac-1, and showed that the percentage of (IgM+Mac1+) B1 cells in b-cflip−/− mice was dramatically reduced, when compared with control cflip+/+ mice (bottom, Fig. 2 D).
In vitro cell survival and apoptosis analyses
Because of the reduced mature B cell numbers in b-cflip−/− mice, we determined whether cFLIP deficiency had an effect on B cell survival. Purified B cells were cultured in complete medium for various times, and percentage of live cells determined. The viability of cflip−/− B cells appeared to be similar to that of control cflip+/+ B cells (Fig. 3 A and B), which indicates that cFLIP deficiency does not affect in vitro survival of B cells. It has been proposed that cFLIP competes with Caspase 8 for binding to FADD, and thus interferes with apoptotic signaling. However, overexpression of cFLIP reportedly could also lead to apoptosis (10). To determine the effect of cFLIP deficiency on apoptotic responses, resting B cells were cultured in the presence of Fas ligand (FasL) in order to trigger Fas-induced apoptosis. cflip+/+ B cells are relatively resistant to various concentration of FasL (Fig. 3 C). In contrast, FasL induced a dose-dependent apoptotic response in mutant cflip−/− B cells, reaching a maximum of 70% apoptosis with 50 ng/ml of FasL. When stimulated with anti-IgM Abs to crosslink the BCR, both cflip−/− and control cflip+/+ immature B cells underwent apoptosis similarly (Fig. S2 A). We also performed Fas-induced apotposis in activated B cells, and the data showed that a lack of cFLIP rendered activated B cells more sensitive to FasL (Fig. 3 D). A lack of cFLIP did not affect the basal level expression of Fas in resting B cells (left, Fig. 3 E). The induction of Fas in activated cflip−/− B cells was lower than in activated control cflip+/+ B cells (right, Fig. 3E). B cell apoptosis induced by TRAIL was also examined. As shown in Figure S2 B, activated cflip−/− B cells are more sensitive to TRAIL-induced apoptosis than control cflip+/+ B cells. These results indicate that cFLIP plays an inhibitory role in both Fas- and TRAIL-induced signaling pathway in B cells.
Figure 3. B cell survival and apoptosis.

(A) B cells were cultured in complete medium. After incubation at 37°C for the indicated times, B cells were stained with PI, and the percentage of viable cells was scored as the PI− population. Filled circles: cflip+/+ mice and open circles cflip−/− mice. (B) Representative dot plots were shown to indicate the percentages of cell death (PI+) at 0 and 9 h of incubation. ss: side scatter. (C) To induce apoptosis, resting B cells were cultured with FasL. After 3 h incubation, the percentages of apoptotic B cells (PI+) were determined. Filled circles: cflip+/+ mice and open circles cflip−/− mice. (D) Activated B cell apoptosis induced by Fas was determined by stimulating B cells with anti-CD40, and the resulting activated B cells were treated with FasL (50 ng/ml) for 6 h. Filled bars: cflip+/+ mice and open bars: cflip−/− mice. (E) Fas expression in resting and LPS-activated B cells was determined by staining with anti-Fas antibodies and flow cytometry. Fas mutant (Fas−/−) mice were used as negative controls.
Analyses of proliferation and antibody responses in b-cflip−/− mice
fadd−/− and caspase 8−/− B cells are defective in TLR-induced proliferation (14, 15, 22). To evaluate a requirement for cFLIP in TLR-induced proliferation, B cells were stimulated with dsRNA, LPS, or CpG containing DNA, which are the ligands for TLR3, TLR4, and TLR9, respectively. A dose-dependent proliferation was induced by treatment of cflip+/+ control B cells with LPS, whereas this response was severely defective in cflip−/− mutant B cells (Fig. 4 A). B cell proliferation induced by the dsRNA mimetic poly (I:C) was also impaired in the absence of cFLIP. In contrast, cflip−/− B cells had normal proliferation when stimulated with CpG-containing oligo DNA (Fig. 4 A). fadd−/− and caspase 8−/− B cells retain normal proliferation responses to BCR and CD40 stimulation (14, 15, 22). B cells were stimulated with the F(ab′)2 fragments of anti-IgM Abs to crosslink the BCR. As shown in Figure 4A, proliferation in response to BCR stimulation was reduced in cflip−/− B cells as compared to cflip+/+ B cells. In contrast, stimulation of B cells using anti-CD40 Abs induced normal proliferation responses in cflip−/− B cells (Fig. 4 A). Proliferation was also analyzed by determining cell division kinetics. B cells were labeled with the fluorescent dye, carboxyfluorescein succinimidyl ester (CFSE). At 48 h after stimulation with LPS, 91.59% of cflip+/+ B cells divided one or more times (Fig. 4 B). In contrast, 43.68% of cflip−/− B cells had only divided once. When stimulated with anti-IgM Abs for 48 h, 82.51% of cflip+/+ B cells have divided three or more times. A smaller fraction (41.61%) of cflip−/− B cells had only divided one or two times (Fig. 4 B). To further analyze these proliferation defects, CFSE-labeled B cells were stained with 7-amino actinomycin D (7AAD) to measure both cell division and apoptosis. At 48 h post stimulation with LPS, there was 75.64% cell death (7AAD+) in cflip−/− mutant B cells, which was much higher than in cflip+/+ B cells (12.85%, Fig. 4 B). B cells were also stained with propidium iodide (PI) at additional times following LPS stimulation, and cell death curves generated. Higher percentages of cell death were detected in cflip−/− B cells than in cflip+/+/ B cells as early as 3 h after LPS stimulation (Fig. 4 C). By 24 h, there was 80% cell death in cflip−/− B cells, which is much higher than the 30% death in cflip+/+ B cells. Therefore, cFLIP appears to play an essential anti-apoptotic role in B cells activated through LPS stimulation. A lack of cFLIP did not affect the expression of TLR 3 and 4 in B cells (Fig. 4 D and E).
Figure 4. Proliferation and Ab responses in b-cflip−/− B cells.

(A). Splenic and lymph node B cells were stimulated with the indicated agonists. Proliferation was determined by the incorporation of radioactivity of [3H] thymidine (cpm, counts per minute). (B). B cell division kinetics were analyzed by CFSE dilution assays. At 48 h after stimulation, CFSE-labeled B cells were stained with 7AAD and analyzed by flow cytometry. The percentages of cell death are indicated by the 7AAD+ populations. (C) Cell death was determined at various times following LPS stimulation and death curves generated. (D) TLR3 expression in B cells was determined by western blotting. (E) TLR4 expression in B cells was analyzed by flow cytometry.
Given the in vitro proliferation defect in cflip−/− B cells, Ab responses in b-cflip−/− mice were analyzed. After immunization with LPS conjugated with trinitrophenol (TNP) or ovalbumin (OVA) conjugated with nitrophenol (NP), serum Ab titers were determined. The levels of TNP-specific IgM in b-cflip−/− mice were lower than in cflip+/+ mice immunized with either the T cell-independent Ag LPS-TNP or the T cell-dependent Ag OVA-NP (Fig. 5 A and C, respectively). In addition, the levels of IgG2b specific for LPS-TNP and IgG2a specific for OVA-NP were also lower in b-cflip−/− mice as compared with control cflip+/+ mice, indicating Ab class switching is affected by cFLIP deficiency (Figure 5 B and D). Therefore, cFLIP plays a role during in vivo antibody responses.
Figure 5. Antibodies responses.

Mice were immunized with LPS-TNP (A and B) or with OVA-NP (C and D), and at the indicated times, relative titers of IgM (A and C), IgG2b (B), and IgG2a (D) were measured by ELISA.
Activation marker expression on cflip−/− B cells
Activation of B cells by LPS leads to the up-regulation of several cell surface proteins including CD69, the adhesion molecule CD54 (ICAM-1), the costimulatory molecule CD86 (B7.2), and MHC class II proteins. To determine the impact of cFLIP deficiency on these protein expressions, B cells were stimulated with LPS for various times, and analyzed by flow cytometry. The induction of CD54 and MHC II on cflip−/− B cells was slightly lower than on cflip+/+ B cells (Fig. 6 A). A clear defect was seen in CD69 and CD86 induction on cflip−/− B cells particularly at 12 h and 24 h after LPS stimulation. LPS-induced downregulation of CD62L was dramatically impaired in cflip−/− B cells (Fig. 6 A). In contrast, the induction of activation markers through CD40 stimulation was normal in cflip−/− B cells (Fig. S3).
Figure 6.
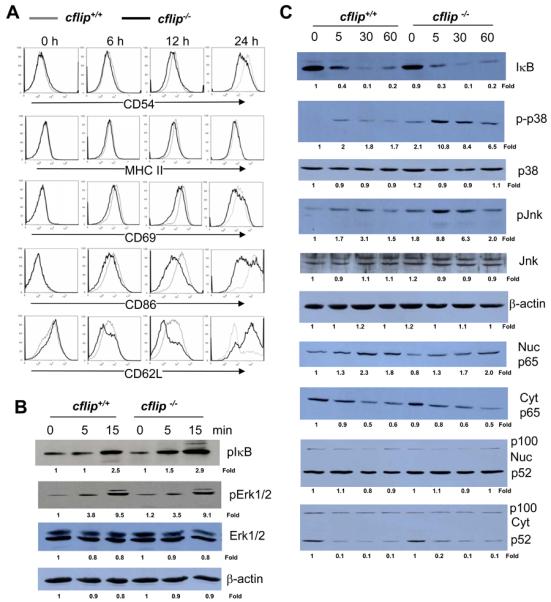
(A) Analysis of LPS-induced expression of activation markers and costimulatory proteins by flow cytometry. (B and C) B cells were stimulated with LPS, and western blotting was performed using Abs specific for pIκB, IκB, pErk1/2, Erk1/2, p65, p-p38, p38. pJnk, Jnk, and NF-κB p100/52. The values shown below each gel indicate folds of changes normalized against unstimulated sample (0 h) and loading controls (β-actin). Cyt: cytomplasmic; Nuc: nuclear.
NF-κB and MAPK activation in cflip−/− B cells
Stimulation of TLRs leads to the activation of NF-κB and Erk1/2 MAPKs (23). Previous reports have shown that overexpression of cFLIP resulted in the activation of NF-κB and Erk1/2 (18, 24). To analyze whether cFLIP deficiency has an effect on the NF-κB activation pathway, IκB phosphorylation and degradation was determined by western blotting. In cflip+/+ B cells, phosphorylation of IκB was evident within 15 min after stimulation with LPS, and similar LPS-induced IκB phosphorylation kinetics were present in cflip−/− B cells (Fig. 6 B). At 30 min after stimulation with LPS, IκB levels were dramatically decreased in cflip+/+ B cells (Fig. 6 C). Similar IκB degradation kinetics were detected in cflip−/− B cells. Activation-induced nuclear translocation of NF-κB was also determined by western blotting with anti-p65 NF-κB Abs. Increasing accumulation of nuclear p65 NF-κB was detected within 5 min after LPS stimulation, and this event was not consistently affected by a lack of cFLIP in B cells (Fig. 6 C). The noncanonical NF-κB pathway was also analyzed by western blotting with anti-p100/52 Abs, which revealed no obvious defect in cflip−/− B cells (Fig. 6 C). These results suggest that cFLIP is not essential for TLR4 induced NF-κB activation.
Western blotting was performed to analyze MAPK activation in cflip−/− B cells. Activation of Erk1/2 was evident in cflip+/+ control B cells, as indicated by Erk1/2 phosphorylation induced within 15 min by LPS stimulation (Fig. 6 B). Similar Erk1/2 activation kinetics was observed in cflip−/− B cells. Interestingly, there appears to be spontaneous activation of p38 and Jnk MAPKs, as shown by higher levels of p-p38 and p-Jnk in un-stimulated cflip−/− B cells than in cflip+/+ B cells (Fig. 6 C). Furthermore, LPS stimulation led to hyper-induction of p-p38 and pJnk in cflip−/− B cells. These results indicate that cFLIP is dispensable in LPS-induced activation of Erk1/2 MAPKs, and plays a role in the downregulation of p38 and Jnk.
Discussion
Gene ablation has helped understand the diverse functions of the FADD-Caspase 8-cFLIP signaling complex. A previous study has shown that a lack of cFLIP in ES cells blocked B cell development in cflip−/−→rag-1−/− chimeras (13). We demonstrated that deletion of cFLIP in pro-B cells had no obvious effect on bone marrow B cell development (Fig. 1B and C). Therefore, cFLIP may be essential in hematopoietic stem cells (HSC), and becomes dispensable following lineage commitment. Similarly, FADD or Caspase 8 in pro-B cells has no major function in bone marrow B cells, but may play a critical role in HSC (25). A lack of FADD or Caspase 8 resulted in impaired Fas-induced apoptosis, which leads to increased numbers of peripheral mature B cells (14, 15, 22). In contrast, b-cflip−/− mice contain reduced numbers of mature B cells (Fig. 1, 2, and S1). This phenotype may be due in part to the hypersensitivity of cflip−/− B cells to Fas-induced apoptosis (Fig. 3 C and D). Thus, cFLIP is critical for inhibiting erroneous apoptosis in B cells possibly induced in vivo by circulating and cell surface FasL.
In additional to the role of inhibiting apoptosis in primary B cells, this study has helped reveal a novel function for cFLIP required in TLR-mediated innate immune responses, as indicated by the defective proliferation in response to LPS and dsRNA (Fig. 4). This phenotype of cflip−/− B cells is reminiscent of that seen in fadd−/− and caspase 8−/− B cells. Perhaps, FADD, Caspase 8, and cFLIP participate in the same pathways in TLR-mediated signaling. While a lack of FADD or Caspase 8 has little effect on B cell proliferation in response to BCR signaling, cflip−/− B cells are impaired in BCR-induced proliferation (Fig. 4 A). This result implies that cFLIP interacts with additional proteins in a novel pathway that is independent of FADD and Caspase 8. The proliferation defects in cflip−/− B cells may be partly responsible for the reduced mature B cell pool in b-cflip−/− mice (Fig. 1 and 2). Whereas a lack of FADD and Caspase 8 has no effect on the expression of activation markers and co-stimulatory proteins in B cells, this was impaired in cflip−/− B cells (Fig. 6 A). It has been shown previously that LPS-induced expression of CD54 and CD86 is dependent on the TLR4-TRIF pathway (26). Therefore, cFLIP deficiency likely affects TRIF-mediated signaling. These collective defects in cflip−/− B cells had an impact on in vivo Ab responses, as b-cflip−/− mice produced significantly reduced levels of Abs and were defective in class switching in response to immunization with either T cell-independent or T cell-dependent Ag (Fig. 5).
A role for FADD, Caspase 8, and cFLIP in NF-κB activation was initially suggested by in vitro over-expression of these proteins in cell lines (18). Later, transgenic overexpression of cFLIP in mice resulted in activation of NF-κB and Erk1/2 in T cells (24). However, no apparent defect in the activation of NF-κB and Erk1/2 were detected in fadd−/− B cells stimulated with LPS (14). In an initial study, caspase 8−/− B cells were shown to be capable of NF-κB and Erk1/2 activation (15). A recent report suggested that defective proliferation of caspase 8−/− B cells is due to impaired NF-κB activation (22). We showed that a lack of cFLIP did not affect IκB phosphorylation, IκB degradation, or NF-κB p65 nuclear translocation in B cells stimulated with LPS (Fig. 6 B and C). In addition, LPS-induced phosphorylation of Erk1/2 was unaltered in cflip−/− B cells. These data indicate that cFLIP is not essential for the activation of NF-kB and Erk1/2 MAPKs in LPS stimulated B cells, which is in agreement with a previous study showing that cFLIP deficiency does not affect the NF-κB and MAPK pathways in T cells (12).
Interestingly, resting cflip−/− B cells appeared to have some characteristics of activated B cells, such as elevated CD23 expression and spontaneous activation of p38 and Jnk (Fig 2 C and 6 C). CD23/FcεRII has been characterized as an early B cell activation marker, and is regulated in a p38-dependant manner (27). It is possible that the proliferation defect in cflip−/− B cells is due in part to the initiation of activation-induced cell death (Fig. 4). It is known that p38 and Jnk MAPKs could be pro-apoptotic (28). cflip−/− B cells have both spontaneous activation and hyperinduction of p38 and Jnk (Fig. 6 C). Crosslinking of CD23 has also been shown to cause apoptosis in B lymphocytes (29). The aberrant activation of p38 and Jnk and CD23 upregulation induction may contribute to the enhanced apoptosis seen in cflip−/− B cells (Fig. 4 B and C). In support of this notion, previous studies have suggested that cFLIP could inhibit p38 and Jnk activation during apoptosis induced by bile acid and TNFα (30, 31). Taken together, our results indicate that cFLIP plays a role in modulating the activation of both Caspases and stress MAPKs.
Acknowledgement
We thank Xiaohui Zhou, Matthew Farabaugh, Susan A. Shinton, and James F. Oesterling for technical assistance, Nu Zhang for providing a cflipf/f mouse genotype protocol, Dr. Klaus Rajewsky for providing cd19cre mice, and Dr. R. C. Rickert for providing the protocol for PCR typing of cd19cre mice, and Jim Martin for critical reading of the manuscript.
Footnotes
This study was supported in part by NIH grants (CA95454 and AI076788) and a Thomas Jefferson University Pilot grant (920012) to J.Z.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1455. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Winoto A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell. Biol. 1996;16:2756–2763. doi: 10.1128/mcb.16.6.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Kramer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 4.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Absence of Fas-mediated apoptosis and T cell receptor-induced proliferation in FADD-deficient mice. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- 6.Varfolomeev EE, Schuchmann M, Luria V, Chainnilkulchai N, Beckmann SJ, Mett I, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham k. B., Goncharov T, Holtman H, Lonai P, Wallach D. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Rosenberg S, Wang H, Imtiyaz HZ, Hou YJ, Zhang J. Conditional Fas-Associated Death Domain Protein (FADD):GFP Knockout Mice Reveal FADD Is Dispensable in Thymic Development but Essential in Peripheral T Cell Homeostasis. J. Immunol. 2005;175:3033–3044. doi: 10.4049/jimmunol.175.5.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, McGlade JC, Ohashi PS, Hakem R. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J-L, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 10.Shu H-B, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 11.Yeh W-C, Ite A, Elia AJ, Ng M, Shu H-B, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW. Requirement for Capser (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 12.Zhang N, He YW. An essential role for c-FLIP in the efficient development of mature T lymphocytes. J. Exp. Med. 2005;202:395–404. doi: 10.1084/jem.20050117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chau H, Wong V, Chen NJ, Huang HL, Lin WJ, Mirtsos C, Elford AR, Bonnard M, Wakeham A, You-Ten AI, Lemmers B, Salmena L, Pellegrini M, Hakem R, Mak TW, Ohashi P, Yeh WC. Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J. Exp. Med. 2005;202:405–413. doi: 10.1084/jem.20050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imtiyaz HZ, Rosenberg S, Zhang Y, Rahman ZS, Hou YJ, Manser T, Zhang J. The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells. J. Immunol. 2006;176:6852–6861. doi: 10.4049/jimmunol.176.11.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beisner DR, Ch'en IL, Kolla RV, Hoffmann A, Hedrick SM. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J. Immunol. 2005;175:3469–3473. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 16.Hennino A, Berard M, Krammer PH, Defrance T. FLICE-inhibitory Protein Is a Key Regulator of Germinal Center B Cell Apoptosis. J. Exp. Med. 2001;193:447–458. doi: 10.1084/jem.193.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Lobito AA, Shen F, Hornung F, Winoto A, Lenardo MJ. Inhibition of Fas-mediated apoptosis by the B cell antigen receptor through c-FLIP. Eur. J. Immunol. 2000;30:155–163. doi: 10.1002/1521-4141(200001)30:1<155::AID-IMMU155>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. J. Biol. Chem. 2000;275:10838–10844. doi: 10.1074/jbc.275.15.10838. [DOI] [PubMed] [Google Scholar]
- 19.Bannerman DD, Eiting KT, Winn RK, Harlan JM. FLICE-like inhibitory protein (FLIP) protects against apoptosis and suppresses NF-kappaB activation induced by bacterial lipopolysaccharide. Am J Pathol. 2004;165:1423–1431. doi: 10.1016/s0002-9440(10)63400-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardy RR. B-1 B Cell Development. J. Immunol. 2006;177:2749–2754. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- 22.Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, Hakem A. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J. Biol. Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- 23.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 24.Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr. Biol. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- 25.Pellegrini M, Bath S, Marsden VS, Huang DC, Metcalf D, Harris AW, Strasser A. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood. 2005;106:1581–1589. doi: 10.1182/blood-2005-01-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 27.Pesu M, Aittomaki S, Takaluoma K, Lagerstedt A, Silvennoinen O. p38 Mitogen-activated protein kinase regulates interleukin-4-induced gene expression by stimulating STAT6-mediated transcription. J. Biol. Chem. 2002;277:38254–38261. doi: 10.1074/jbc.M201427200. [DOI] [PubMed] [Google Scholar]
- 28.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 29.Campbell KA, Studer EJ, Kilmon MA, Lees A, Finkelman F, Conrad DH. Induction of B cell apoptosis by co-cross-linking CD23 and sIg involves aberrant regulation of c-myc and is inhibited by bcl-2. Int. Immunol. 1997;9:1131–1140. doi: 10.1093/intimm/9.8.1131. [DOI] [PubMed] [Google Scholar]
- 30.Grambihler A, Higuchi H, Bronk SF, Gores GJ. cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J. Biol. Chem. 2003;278:26831–26837. doi: 10.1074/jbc.M303229200. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima A, Komazawa-Sakon S, Takekawa M, Sasazuki T, Yeh WC, Yagita H, Okumura K, Nakano H. An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J. 2006;25:5549–5559. doi: 10.1038/sj.emboj.7601423. [DOI] [PMC free article] [PubMed] [Google Scholar]