

Abstract
The allosteric enzyme aspartate transcarbamoylase (ATCase) exists in two conformational states. The enzyme, in the absence of substrates is primarily in the low-activity T state, is converted to the high-activity R state upon substrate binding, and remains in the R state until substrates are exhausted. These conformational changes have made it difficult to obtain structural data on R-state active-site complexes. Here we report the R-state structure of ATCase with the substrate Asp and the substrate analogue phosphonactamide (PAM) bound. This R-state structure represents the stage in the catalytic mechanism immediately before the formation of the covalent bond between the nitrogen of the amino group of Asp and the carbonyl carbon of carbamoyl phosphate. The binding mode of the PAM is similar to the binding mode of the phosphonate moiety of N-(phosphonoacetyl)-L-aspartate (PALA), the carboxylates of Asp interact with the same residues that interact with the carboxylates of PALA, although the position and orientations are shifted. The amino group of Asp is 2.9 Å away from the carbonyl oxygen of PAM, positioned correctly for the nucleophilic attack. Arg105 and Leu267 in the catalytic chain interact with PAM and Asp and help to position the substrates correctly for catalysis. This structure fills a key gap in the structural determination of each of the steps in the catalytic cycle. By combining these data with previously determined structures we can now visualize the allosteric transition through detailed atomic motions that underlie the molecular mechanism.
Keywords: Cooperativity, allosteric transition, domain closure, X-ray crystallography, enzyme mechanism
Introduction
Allostery and cooperativity are important mechanisms for regulating the function of a variety of proteins.1 The majority of allosteric proteins are enzymes which act as control valves altering the flux of small molecules through metabolic pathways. Direct control of protein function via allosteric regulation is usually achieved via conformational changes induced by effectors that bind at sites remote from the active sites. Escherichia coli aspartate transcarbamoylase (EC 2.1.3.2, aspartate carbamoyltransferase, ATCase) catalyzes the committed step in de novo pyrimidine nucleotide biosynthesis, the formation of N-carbamoyl-L-aspartate and phosphate (Pi) from carbamoyl phosphate (CP) and L-aspartate (Asp).2 Substrates bind to the enzyme and induce a tertiary domain closure that triggers a quaternary conformational change resulting in the observed homotropic cooperativity.3 CTP and ATP, the end products of the pyrimidine and purine nucleotide biosynthetic pathways respectively, can inhibit / activate the enzyme heterotropically.4,5,6 Understanding the detailed atomic motions that underlie the molecular mechanism of domain closure and allosteric regulation in ATCase not only provides a more accurate description of how this enzyme functions in the regulation of metabolism, but allows a more general understanding of the function of other allosteric enzymes.7,8
E. coli ATCase is composed of six catalytic chains (Mr 34 000), organized as two catalytic trimers (catalytic subunits, C3), and six regulatory chains (Mr 17,000), grouped into three regulatory dimers (regulatory subunits, R2). Each catalytic chain is composed of a CP domain (residues 1-135) and an Asp domain (residues 136-291) that are primarily responsible for the binding of the two substrates, CP and Asp, respectively. Each of the three active sites in the catalytic subunit are composed of residues from both the CP and Asp domains as well as residues from the 80's loop of an adjacent catalytic chain. The binding of substrates is ordered with CP binding before Asp.9 Asp binding triggers domain closure and a dramatic quaternary structural change from the T to the R state, as evident by the 11 Å expansion of the separation between the two catalytic subunits, as well as relative rotations of the catalytic and regulatory subunits.10 Macol et al.11 showed that the binding of the bisubstrate analogue N-(phosphonacetyl)-L-aspartate (PALA) to just one of the six active sites of the enzyme is sufficient to induce the T- to R-state transition, and time-resolved small-angle X-ray scattering (SAXS) experiments have shown that as long as substrates are available, the enzyme remains in the R state, and returns to the T state only when the supply of the substrates is exhausted.12
X-ray structures of the enzyme in the T and R states have been used to identify residues involved in selectively stabilizing each state. An T-state X-ray structure of the enzyme•CP complex has revealed the molecular details utilized by the enzyme to achieve an ordered-binding mechanism by creating the Asp binding site only after CP binds.13 However, no R-state structure of the enzyme with substrates bound immediately before the nucleophilic attack has been determined. In order to obtain this structure we utilized a mutant version of ATCase (D236A) in which one critical T-state interaction, between Asp236 on the catalytic chain and Lys143 on the regulatory chain, was eliminated. Small-angle X-ray scattering experiments in solution indicated that the D236A enzyme does not exist in the T state in the absence of ligands, but rather in a T to R equilibrium with about an approximately equal distribution of molecules in the T and R states.14 Although the addition of the first substrate, CP, in the ordered-binding mechanism, does not alter significantly the SAXS pattern of the wild-type enzyme, the binding of CP is sufficient to convert the entire population of D236A molecules to the R state.14
Trapping the reactive enzyme•CP•Asp complex in the R-state was accomplished here by using both the T-state destabilized D236A enzyme to more easily attain the R state, and substituting a non-reactive analog of CP, phosphonacetamide (PAM), to prevent the actual chemical step. In this work we report a structural snapshot of the reaction immediately before the formation of the covalent bond between the nitrogen of the amino group of aspartate and the carbonyl carbon of CP in the R-allosteric state. This structure helps to define the catalytic cycle of ATCase, helps to explain the differences in substrate affinity and catalytic activity of the T and R states and helps to delineate the importance of domain closure for both formation of the R-state active site and for the facilitation of catalysis.
Results and Discussion
Structure of D236A ATCase in the presence of PAM and Asp
The catalytic mechanism of ATCase is ordered with CP binding before Asp and carbamoyl aspartate leaving before phosphate.15 The molecular mechanism responsible for the ordered binding of Asp, which takes place only after CP binds in the active site has been proposed based upon structures of ATCase in the absence and presence of CP.13 The binding of CP induces local conformational changes resulting in the formation of a new highly electropositive binding pocket for Asp.13 To understand the binding of Asp to the enzyme•CP complex and the concurrent conformational changes induced by Asp binding, X-ray crystallography was employed. We were able to obtain data quality crystals of the R state of the enzyme with substrates bound by utilized a mutant version of the enzyme with the D236A mutation. We did obtain similar crystals with the wild-type enzyme, unfortunately they diffracted very poorly. Using the D236A enzyme, crystals were obtained with PAM and Asp bound that diffracted to a maximal resolution of 2.6 Å in the P321 space group (Table 1). The unit cell dimensions (a = b = 121.08 Å, c = 155.24 Å) were very similar to those observed for crystals of both the wild-type enzyme•PALA complex (RPALA, PDB code: 1D09), and the D236A•PAM complex (R236_P, PDB code: 2A0F) in the same space group (a = b = 122.24 Å, c = 156.36 Å for RPALA and a = b = 120.45 Å, c =155.24 Å for R236_P). The similarity in unit cell dimensions suggested that the D236A•PAM•Asp complex was in an R-like quaternary structure. The structure of the D236A•PAM•Asp complex (R236_PA) was refined to an Rfactor/Rfree of 20.9%/25.4% using Refmac516 and CNS.17
Table 1.
Data collection and refinement statistics for the R236_PA structure
| Data collection | |
| Space Group | P321 |
| Cell Dimensions | |
| a, b, c (Å) | 121.076, 121.076, 155.235 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å)a | 30 − 2.60 (2.69 − 2.60) |
| Rmerge(%)a,b | 9.7 (57.9) |
| Average (I/sigma) | 9.7 |
| Completeness (%)a | 99.9 (99.8) |
| Total Reflections | 787,608 |
| Unique reflections | 41,498 |
| Redundancya | 19.0 (15.3) |
| Refinement | |
| Resolution (Å) | 30 − 2.60 |
| Sigma cutoff (σ) | 0 |
| Reflections | 40,109 |
| Rfactor/Rfree | 0.209/0.254 |
| Total number waters | 354 |
| RMS deviations | |
| Bonds (Å) | 0.008 |
| Angles (°) | 1.58 |
Values in parentheses are for the highest resolution shell
To evaluate the quaternary structure of the R236_PA structure, the vertical separation between the upper and lower catalytic subunits was computed and compared to the vertical separations of known T and R state structures. The vertical separation for the T and the R structures are 45.2 Å and 56.1 Å respectively. The difference between the R- and T-state vertical separations corresponds to the vertical expansion of the enzyme observed during the T to R transition (56.1 − 45.2 = 10.9 Å). For the R236_PA structure, the vertical separation was 55.7 Å, indicating that the D236A•PAM•Asp complex was in the R conformation. Figure 1 shows the backbone RMS deviation of the C1 catalytic chain between the RPALA and R236_PA structures. The only significant shift in the backbone occurs at the N terminus and the area around the site of the mutation. The shift in the backbone around position 236 is most likely due to the loss of the interactions involving the side chain of Asp236 which stabilize this portion of the structure.
Figure 1.
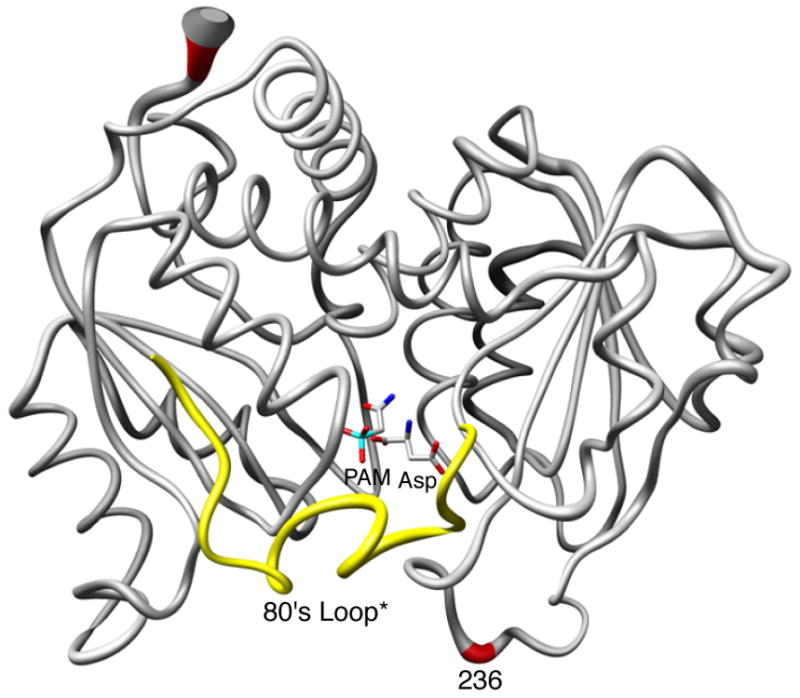
Comparison of one catalytic chain (grey, red) of the R236_PA and RPALA structures along with the 80's loop of the adjacent catalytic chain (yellow, 80's loop*) The width of the tube is proportional to the RMS deviation between the α-carbon positions of the two structures. PAM and Asp are shown. This figure was drawn using Chimera.40
The active site of the R236_PA structure
In the P321 space group, the asymmetric unit of the crystal contains one catalytic chain from the upper trimer (C1), two regulatory chains that comprise one regulatory dimer (R1-R6) and one catalytic chain from the lower trimer (C6). PAM and Asp are bound at 100% occupancy in both the C1 and C6 active sites (Figure 2). The position of the amino group of Asp in the C1 and C6 chains was confirmed by an extra round of refinement starting with the amino group flipped in the opposite direction. Analysis of the resulting Fo−Fc electron density maps revealed negative density on the amino group and positive density at the original position of the amino group, confirming the orientation of Asp in the active site. There are approximately 16 hydrogen-bonding interactions between the enzyme and PAM, and approximately 8 hydrogen-bonding interactions between the enzyme and Asp. In addition, there are hydrophobic interactions between the enzyme and both PAM and Asp. The C1 and C6 chains in the asymmetric unit were very similar with an overall RMS deviation of 0.41 Å for all α-carbon atoms. Although the interactions of PAM and Asp in C1 and C6 were similar, there are differences between these two active sites. In both the C1 and C6 active sites, the phosphate portion of PAM is stabilized and neutralized by interactions with Ser52, Thr53, Arg54, and Ser80 from the adjacent catalytic chain (Figure 3). In addition, Arg105 and Lys84c2 in the C1 chain (Figure 3(a)) and Thr55 in the C6 chain (Figure 3(b)) are involved in interactions with the phosphate portion of PAM. The amino group of PAM forms backbone interactions with Pro266 and Leu267 in both chains and with Gln137 in C6. The carbonyl oxygen of PAM interacts with Arg105 and His134 in both chains and with Thr55 in the C6 chain. Furthermore, the α-carboxylate of Asp is neutralized by Arg105 and Arg167, whereas the β-carboxylate is neutralized by Arg229 and Gln231 in both the C1 and C6 chains. The α-carboxylate has an additional interaction with Lys84c2 in the C1 chain. Finally, the amino group of Asp exhibits only one interaction with the backbone carbonyl oxygen of Leu267 in both the C1 and C6 chains.
Figure 2.
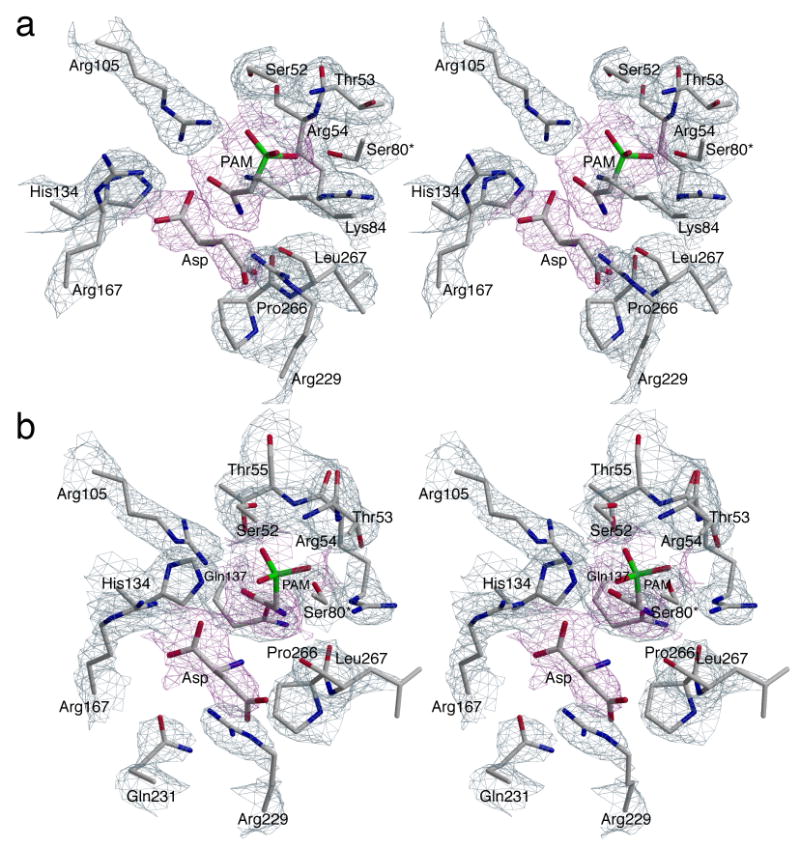
Stereoview of the active sites in ATCase complexed with PAM and Asp: (a) chain C1 and (b) chain C6. The refined coordinates of the side chains are overlaid on the 2Fo−Fc electron density map (grey) shown contoured at 1.5σ. PAM and Asp are overlaid on the Fo−Fc electron density map (magenta) shown contoured at 1.0σ.
Figure 3.
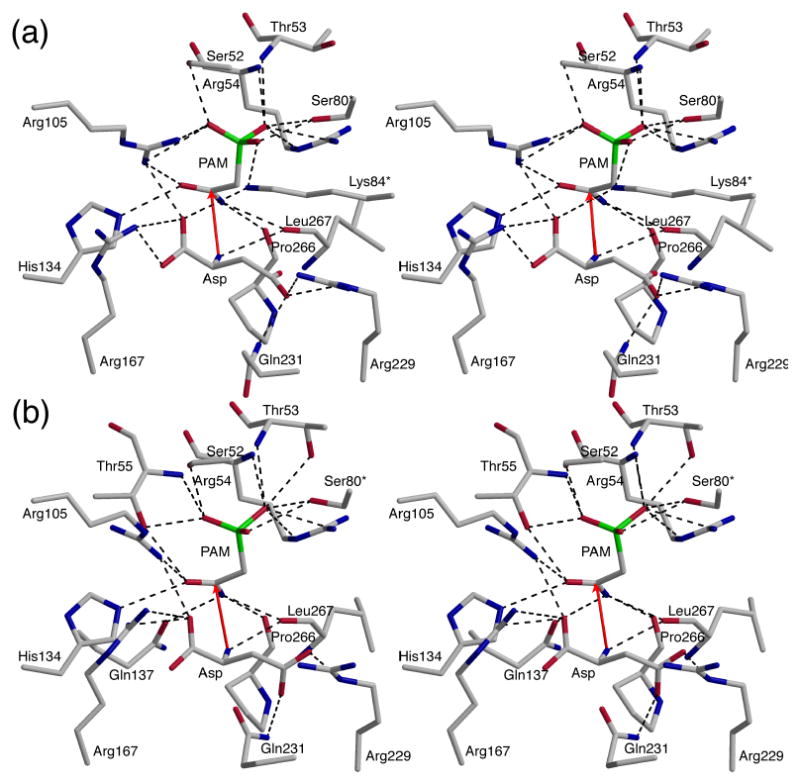
Stereoview of a portion of the active site of ATCase in (a) chain C1 and (b) chain C6. A and B, shown in the same orientation. Interactions with PAM and Asp are shown with black dashed lines. Red arrows indicate the proposed attack of the nitrogen of the α-amino of Asp onto the carbonyl carbon of PAM (CP).
As has been proposed, the catalytic mechanism of the ATCase reaction involves the nucleophilic attack of the amino group of Asp on the carbonyl group of CP, forming a tetrahedral intermediate.18 In the structure reported here, the nitrogen of the amino group of Asp is ∼2.9 Å away from the carbonyl carbon of PAM. The position and orientation of Asp are both correct for nucleophilic attack (Figure 3). Thus, this structure gives us a picture of the active site in molecular detail immediately prior to the nucleophilic attack.
Comparison of the R236_PA structure with the structure of wild-type ATCase in the presence of PAM and malonate
The R236_PA structure reported here, was compared to the structure of wild-type ATCase in the presence of PAM and malonate (RPM structure, PDB code: 2AT1).19 The backbone positions of these two structures are similar with a RMS deviation of 0.85 Å. The positions of all active site residues in the RPM structure are similar to those in the RPALA structure except for the position of Lys84.19 In the RPALA structure the amino group of Lys84 interacts with a phosphonate oxygen, an α-carboxylate oxygen and a β-carboxylate oxygen. In the RPM structure, the amino group of Lys84 does not interact with PAM, since the phosphonate oxygen of PAM is 4.0 Å away from Lys84. However, in the R236_PA structure, Lys84 interacts with PAM in the C1, although not in the C6 chain. Lys84 is 2.6 Å and 3.9 Å away from the closest phosphonate oxygen of PAM in the C1 and C6 chains respectively (compare Figure 3(a) and 3(b)).
A comparison of the positions of PAM and Asp in the active site of the R236_PA structure with the positions of PAM and malonate in the RPM structure is shown in Figure 4(a). PAM is located in the same position in the two structures while malonate has a different orientation, and has different interactions with the protein as compared to those of Asp. The reason that malonate can promote the T to R transition in the presence of PAM lies in the lack of an α-amino group.19 The two domains of the catalytic chain can attain the domain closed conformation with PAM and malonate, since there is less repulsion between them than there is between PAM and Asp. For the T-state destabilized D236A enzyme, PAM alone is sufficient to shift the enzyme to the R state. Therefore, the binding of Asp to the D236A•PAM complex does not induce the T to R transition since the D236A•PAM complex is already in the R state. In order to evaluate differences in the architecture between the R236_PA active site and other R-state structures, the planar angle between the CP and Asp domains were compared. The planar angle is defined as the angle formed between the centers of gravity of the two domains and a hinge point.20 The planar angles of the RPM, RPALA and R236_PA structure (average of C1 and C6 chain) were 127.8°, 128.0°, 128.6° respectively. The increased angle between the CP and Asp domains results in an active site cavity that is more open in the R236_PA structure than in the other R-state structures.
Figure 4.
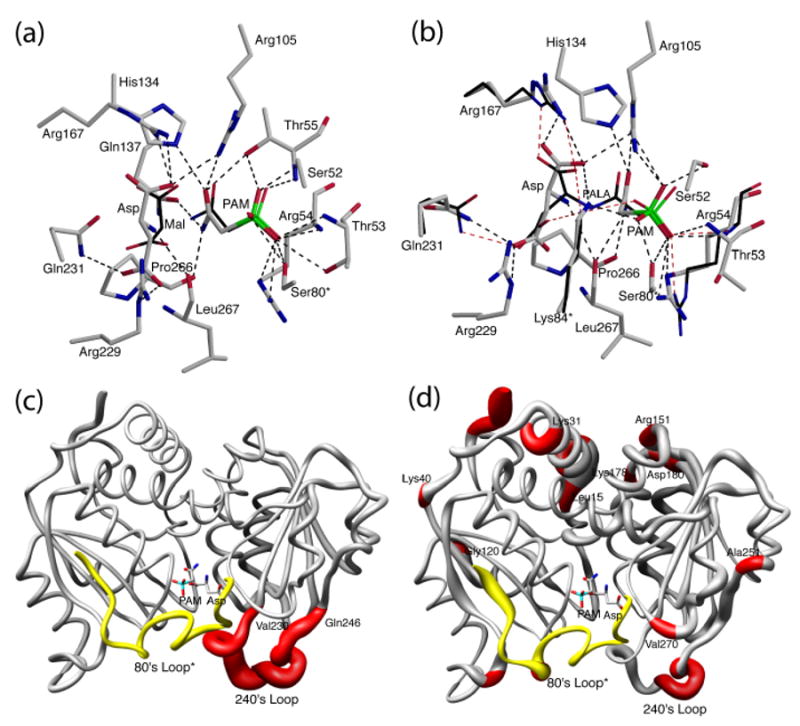
The positions of PAM and Asp (elemental color) in the active site of R236_PA structure compared to (a) the positions of PAM and malonate (thin, black) in the RPM structure and (b) the positions of PALA in the RPALA structure (thin, black, only the residues that are in different positions are shown). The two structures were first superimposed using SEQUOIA.41 The comparison of one catalytic chain (grey, red) of ATCase along with the 80's loop of the adjacent catalytic chain (yellow, 80's loop*) of the R236_PA structure (c) with the TCP structure, and (d) with the R236_P structure. The width of the tube is proportional to the RMS deviation between the α-carbon positions of the two structures. PAM and Asp are shown to indicate the active site position.
Comparison of the R236_PA structure with the RPALA structure
PALA inhibits ATCase with Ki = 27 nM and induces the conversion of the enzyme to the R state21. PALA exhibits competitive inhibition with respect to CP, whereas it shows noncompetitive inhibition with respect to Asp.21,22 Collins and Stark21 first suggested that this difference in inhibition is evidence for ordered-substrate binding, whereas Heyde23 questioned whether PALA really functions as a transition-state analog. To probe these questions, the positions of PAM and Asp were compared with that of PALA in the active site as shown in Figure 4(b). PAM and the CP part of PALA superimpose closely, as PAM and CP share similar interactions with side chain and backbone residues. Although Asp and the Asp portion of the PALA have differences in their position and orientation, they nevertheless interact with the same residues in the active site (Table 2). Further evaluation of these interactions indicates that although the side chains of Arg167, Arg105, Arg54, Gln231 and Lys84c2 are in somewhat shifted positions in the RPALA structure as compared to the R236_PA structure, these residues interact with the same functional group of the ligands and the number of hydrogen-bonding interactions is the same (Figure 4(b)). Since the CP and Asp portions of PALA are linked covalently, they are spatially closer than PAM and Asp can be because of the repulsive force between the amino group of Asp and PAM. As would be expected, PAM and Asp do not overlay exactly on the corresponding parts of PALA. However, PALA has almost all the same binding interactions observed for both substrates, explaining the high affinity of PALA for the enzyme and supporting the notion that PALA is a bisubstrate analog not a transition-state analog. This comparison also suggests that the structure of the D236A•PAM•Asp complex reported here is a good model for the enzyme just before the final repositioning of the domains that forces the two substrates together to create the tetrahedral intermediate.
Table 2.
Interactions between the active site residues and ligands in the R-state structures
| R236_Pa | R236_PAa | RPALAa | RCP_suca | RPMa | Rpi_cita | ||
|---|---|---|---|---|---|---|---|
| 1b | NH2 | Gln137 | Gln137c | N/A | Gln137 | Gln137 | N/A |
| Pro266 | Pro266 | Pro266 | |||||
| Leu267 | Leu267 | ||||||
| 2 | O- | Thr55 | Thr55 | Arg105 | Thr55 | Thr55 | Thr55 |
| Arg296 | Arg105 | His134 | Arg105 | Arg105 | Gln137 | ||
| His134 | His134 | His134 | His134 | His134 | |||
| 3 | ester O | N/A | N/A | N/A | Arg54 | N/A | N/A |
| 4 | amide NH | N/A | Leu267 | Leu267 | N/A | N/A | N/A |
| 5 | α-COO- | N/A | Arg105 | Arg105 | Arg167 | Arg167 | Arg167 |
| Arg167 | Arg167 | Lys84* | Lys84* | ||||
| Lys84* | Lys84* | ||||||
| 6 | β-COO- | N/A | Arg229 | Arg229 | Arg229 | Arg229 | Arg229 |
| Gln231 | Gln231 | Gln231 | Gln231 | ||||
| 7 | Pi | Ser52 | Ser52 | Ser52 | Ser52 | Ser52 | Ser52 |
| Thr53 | Thr53 | Arg54 | Arg54 | Thr53 | |||
| Arg54 | Arg54 | Arg54 | Arg105 | Thr55 | Arg54 | ||
| Thr55 | Thr55 | Thr55 | Ser80* | Arg105 | Thr55 | ||
| Arg105 | Arg105 | Lys84* | Ser80* | Arg105 | |||
| Ser80* | Ser80* | Ser80* | |||||
| Lys84* | Lys84* | Lys84* |
R236_P, the X-ray structure of D236A ATCase in the presence of PAM (PDB code:2A0F); R236_PA, the X-ray structure of D236A ATCase in the presence of PAM and Asp (PDB code:2HSE); RPALA, the X-ray structure of wild-type ATCase in the presence of PALA (PDB code: 1D0925); RCP_suc, the X-ray structure of wild-type ATCase in the presence of CP and succinate18; RPM, the X-ray structure of wild-type ATCase in the presence of PAM and malonate (PDB code: 2AT1)19; Rpi_cit, the X-ray structure of wild-type ATCase in the presence of phosphate and citrate (PDB code:1R0B)24. 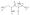
Number refers to the numbering of the reaction intermediate.
Italics indicates that the interaction is observed in only one of the two catalytic chains in the asymmetric unit.
Conformational changes induced by the binding of Asp to the TCP structure
The binding of the first substrate, CP, to the T-state enzyme induces local conformational changes mainly involving the 50's and 80's loops, along with some comparatively small shifts in the 240's loop.13 A comparison of the R236_PA structure with the T-state enzyme•CP structure (TCP) indicates that the binding of Asp induces large backbone movements of 240's loop closing in on the two substrates (Figure 4(c)). Some residues that point away from the active site in the TCP structure, such as the guanidinium group of Arg229 and the side chain of Gln231, move into the active site and interact with Asp as a result of the movement of the 240's loop (Figure 3). A number of residues in the 240's loop are involved in stabilizing the T state of the enzyme, such as the interactions between Glu239 in the C1 chain with Lys164 and Tyr165 in the C4 chain. The repositioning of the 240's loop breaks these T-state stabilizing interactions allowing the molecule to transit to the R state.
Comparison of the R236_PA structure with the R236_P structure
Macol et al.11 found that the binding of PALA to one of the six active sites of the holoenzyme is sufficient to promote the full T to R quaternary conformational change. Time-resolved SAXS experiments have also shown that once the enzyme is converted to the R state, it remains in the R state until the substrates are exhausted.12 Therefore, when the enzyme is converted to the R state, some of the active sites are either empty or have only the first substrate, CP, bound. A backbone comparison of the R236_P and R236_PA structures reveals the conformational change induced by the binding of Asp in the R-state (Figure 4(d)). Besides the large movements of the 80's and 240's loops, the helix from Leu15 to Lys31 shifts, and there are further positional alterations at Lys40, Gly120, Arg151, Lys178-Asp180 and Ala251. The difference in the two structures reflects different steps in the catalytic mechanism. In the R236_P structure, the active site is organized for Asp binding, and it is the binding of Asp that causes domain closure. In the R236_PA structure, as well as in the RPM structure, the catalytic chain domains and 240's loop are in their fully closed conformation. The closure of the active site accelerates catalysis by approximation. However, this closed active site conformation is not conducive for either product release or substrate binding.
Implications of the structure for the catalytic mechanism
The catalytic mechanism of ATCase involves the attack by the amino group of Asp onto the carbonyl carbon of CP and formation of the tetrahedral intermediate. In the R236_PA structure, His134, Arg105 and Thr55 interact with the carbonyl oxygen and thereby assist the reaction by polarizing the carbonyl (Figure 3). Arg105 and Leu267, which are the only two residues that interact with both PAM and Asp, position the two substrates in the proper orientation and proximity. The guanidinium group of Arg105 interacts with the carbonyl oxygen of PAM and with the α-carboxylate of Asp, while the backbone carbonyl of Leu267 interacts with the NH2 of both PAM and Asp (Figure 3). These interactions correctly position Asp for the nucleophilic attack. An alternate position of Pro266 and Leu267 was observed in the R-state structure of ATCase in the presence of citrate (an analog of carbamoyl aspartate) and phosphate (PDB code:1R0B).24 Both residues are flipped out of the active site in the enzyme•citrate•Pi structure, no longer interacting with citrate or Pi. This observation suggests that Pro266 and Leu267 are important for the correct positioning of the substrates as well as for product release.
To form the covalent bond between the amino nitrogen and the carbonyl carbon, two protons must be removed from the amino group of Asp. The positive electrostatic field in the active site will lower the pKa of the amino group of Asp so it should initially bind in the deprotonated form. Two routes are possible for the loss of the second proton from the amino group in conjunction with the formation of the tetrahedral intermediate. One possibility, as suggested by Jin et al.,25 is that Lys84 from the adjacent chain acts as a general base capturing the proton from the amino group of Asp as the tetrahedral intermediate is formed. In the C1 chain of the R236_PA structure, Lys84 interacts with the α-carboxylate group of Asp and a phosphonate oxygen of PAM, whereas in the C6 chain, Lys84 does not interact with either substrate. The lack of an interaction with the amino group of Asp in both the C1 and C6 chains suggests that Lys84 may not be acting as a general base in the reaction. The second possibility, proposed by Gouaux et al.,26 is that the proton is directly transferred to the leaving phosphate group by formation of a six-membered ring in a chair conformation (Figure 5(a)). To predict the interactions of a tetrahedral intermediate in a chair conformation with the enzyme, it was docked into the active site using AUTODOCK.27 Both PAM and Asp were removed from the R236_PA structure, and the cyclic model of the tetrahedral intermediate was docked into the empty active site. The position of the cyclic model of the tetrahedral intermediate overlaps with the positions of PAM and Asp (Figure 5(b)), indicating that there is a binding pocket for the intermediate and the formation of the six-membered ring intermediate is feasible. This modeling supports the proposal of Gouaux et al.26 that the second proton can be directly transferred to the leaving phosphate group.
Figure 5.
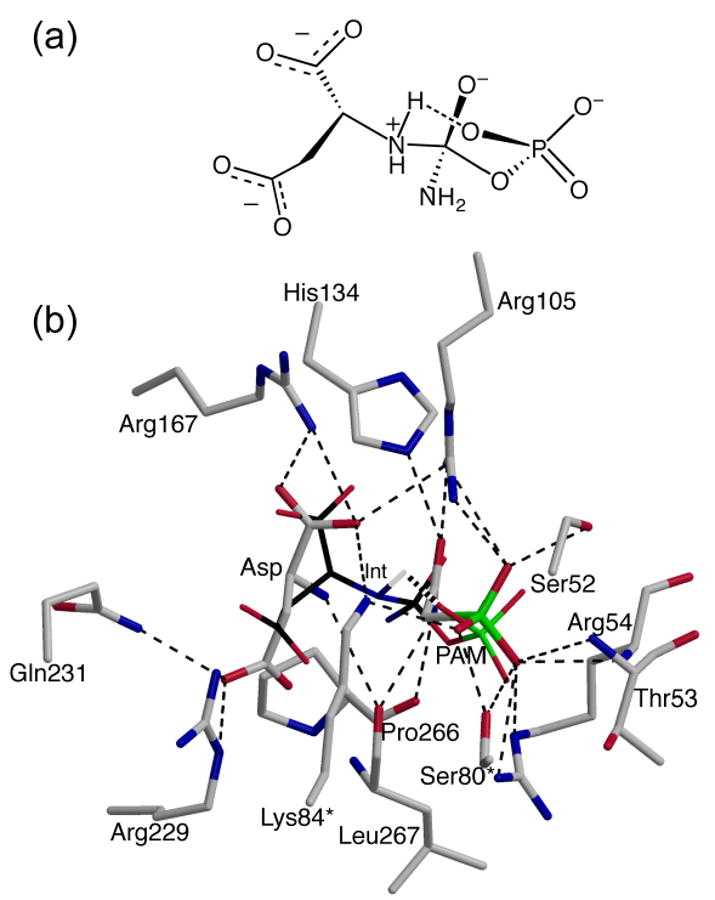
(a) The proposed conformation of the tetrahedral intermediate of the reaction between CP and Asp as catalyzed by ATCase.26 The postulated proton transfer between a terminal phosphate oxygen and the amino group of Asp is indicated by the long dashed line. (b) The active site of the R236_PA structure showing the position of the cyclic model of the tetrahedral intermediate (Int, thin, black) determined using AUTODOCK.27 For comparison, PAM from the R236_PA structure is also shown. The hydrogen that is part of the six-membered ring of the tetrahedral intermediate is shown.
The allosteric transition of ATCase
Over the past few years work in our laboratory has concentrated on obtaining structural snapshots of ATCase at various steps in the chemical reaction and the allosteric transition leading to product formation. The necessary structural snapshots include: the T-state enzyme in the absence of ligands (Tapo, PDB code 1ZA1),13 the T-state enzyme in the presence of the first substrate to bind, CP (Tcp, PDB code 1ZA2),13 the R-state enzyme in the absence of ligands (PDB code: 2A0F);28 the R-state enzyme in the presence of the first substrate (PDB code: 2A0F),28 which was accomplished using PAM to substitute for CP and the D236A T-state destabilized mutant ATCase, and finally the R-state enzyme with CP and Asp. This structural snapshot was determined here by using the D236A T-state destabilized mutant ATCase with PAM and Asp bound.
Based upon this series of structures we can now visualize the allosteric transition through detailed atomic motions that underlie the molecular mechanism. Because of the ordered-binding mechanism the unliganded T-state enzyme (Figure 6(a), Tapo, PDB code 1ZA1)13 binds CP first. The binding of CP induces local conformational changes of the 50's, 240's and 80's loops. Important T-state stabilization interactions such as the interaction between Glu239c1–Lys164c4 and Glu239c1–Tyr165c4 are weakened and the interaction between Arg105-Glu50 is broken allowing these residues to participate in substrate binding (Figure 6(b), Tcp, PDB code 1ZA2).13 The net result of CP binding is to destabilize the T state of the enzyme.
Figure 6.
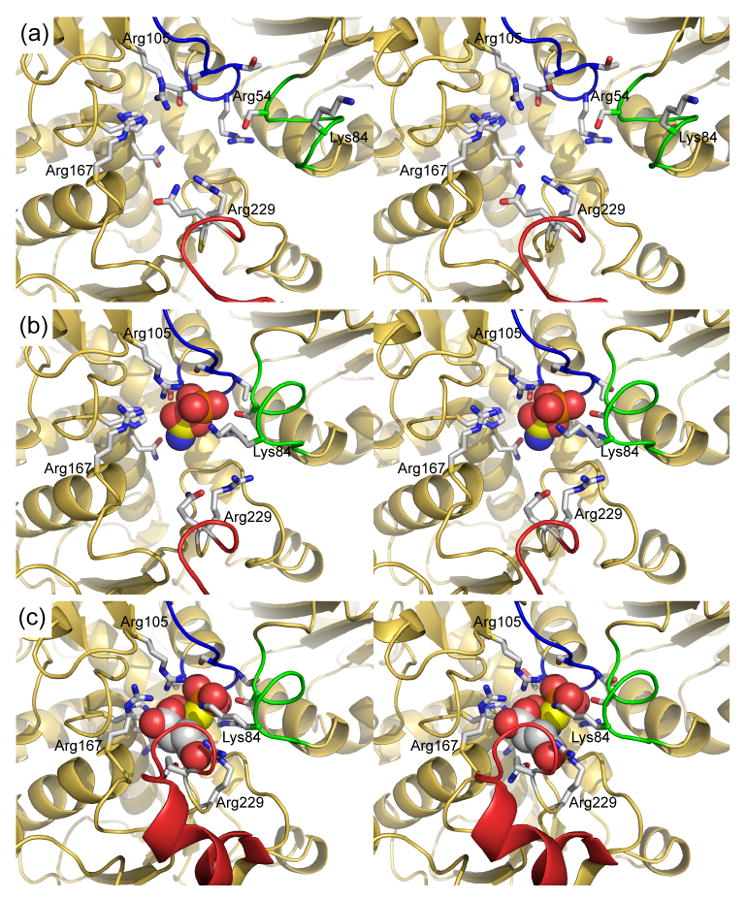
The conformational changes associated the reaction between CP and Asp in the active site of ATCase. In each panel the 50's, 80's and 240's loops are shown in blue, green and red, respectively. The 80's loop is donated into the active site from the adjacent catalytic chain. The positively charged residues that interact with the substrates are labeled. Labels for other side chains have been omitted for clarity. (a) The active site of ATCase in the absence of substrates (Tapo, PDB code 1ZA1).13 The conformation the side chains of active site residues such as Ser80, Lys84, Arg105, Arg229, and Gln231 are directed away from the active site in this structure. (b) The active site of ATCase in the presence of the first substrate to bind in the ordered-binding mechanism, CP (TCP, PDB code 1ZA2).13 The single carbon of CP is shown in yellow to distinguish it from the carbons of Asp in panel (c). The binding of CP induces a reorganization of the 50's and 80's loops which dramatically changes the electrostatics of the active site creating the binding site for Asp.13 The side chains of residues such as Ser80, Lys84, Arg54, Arg105 and Gln137 reorientation to create the binding pocket for CP. (c) The active site of ATCase in the presence of PAM, an analog of CP, and Asp (RPA, PDB code 2HSE). The binding of Asp induces a major conformational change in the 240's loop (red) that initiates the quaternary conformational change to the R state. A number of side chains reorient to stabilize the transition state including Lys84, Arg105 and Arg229. The domain closure also forces the substrates together accelerating the reaction by approximation.
The movement of side chains within the active site when CP binds, as observed in the structure of the Tcp structure (Figure 6(b)), also creates a very electropositive binding pocket for Asp, providing a structural explanation for the ordered-binding of substrates in ATCase. The binding of Asp to the enzyme•CP complex induces the closure of the CP and Asp domains within the particular chain to which Asp binds. The closure of the domains and the repositioning of the 240's loop breaks the already weakened T-state stabilizing interactions (Figure 6(c)). However, because of steric clashes domain closure and the reorientation of the 240's loop cannot be completed without the enzyme undergoing the quaternary conformational change. The elongation of the holoenzyme by approximately 11 Å along the three-fold axis of the molecule overcomes the steric constraints of the T structure, and allows the high-activity high-affinity active site to form.
As shown by Macol et al.11 the transition from the T to the R state only requires the binding of PALA to one of the six active sites, therefore, the other active sites in the molecule will be in the R-quaternary structure. These active sites will either contain no ligands; CP, the first substrate; CP and Asp, immediately before the chemical reaction; the tetrahedral intermediate; Pi and CA, the products immediately before release; or Pi, after CA has left the active site.
Our structure of the D236A•PAM complex in the R state (PDB code: 2A0F)28 is a structural analog of an R-state structure with just CP in the active site awaiting the binding of Asp. In this structure half of the active sites have PAM bound, the other half do not. The half of the structure without PAM therefore is a model for the unliganded R-state enzyme awaiting the binding of CP. Particularly noteworthy about this structure is that the two domains of the catalytic chains are in a more open conformation in the unliganded R-state then the PAM-liganded R-state active sites. However, in both types of active sites, the positions of the domains are closed relative to their T-state positions. These results are consistent with the observation from time-resolved X-ray scattering that as long as substrates are available the enzyme remains in the R state.12 However, once all substrates are exhausted, the enzyme returns to the T state.
The structure reported here, a R-state structure with an analogue of CP (PAM) and Asp bound (Figure 6(c)) provides a representation of the enzyme immediately before the formation of the bond between the two substrates. The structure of the D236A•PAM•Asp complex mimics the R-state active site after Asp binds and before the formation of the tetrahedral intermediate. A comparison of this structure with that of the structure of the enzyme•PALA complex reveals that the CP and Asp domains in this structure are not in their fully closed position, yet the interactions that stabilize the two substrates in the active sites are nearly identical to the corresponding interactions in the enzyme•PALA structure. These data suggest that the final domain closure is directly linked to the bond formation in the tetrahedral intermediate. Upon collapse of the tetrahedral intermediate the domains are no longer “locked” in the reactive position and open sufficiently for ordered-product release from the R-state active site, thus allow another catalytic cycle to begin while the enzyme is still in the R-allosteric state.
Methods
Enzyme preparation
The D236A aspartate transcarbamoylase was overexpressed utilizing E. coli strain EK110429 containing plasmid pEK114.30 The isolation and purification were as described previously29. The purity of the enzyme was checked by SDS-PAGE31 and nondenaturing PAGE.32,33 The concentration of purified enzyme was determined by the Bio-Rad version of Bradford's dye-binding assay using wild-type enzyme as the standard.34
Synthesis of phosphonacetamide
PAM was prepared using a two-step reaction starting from commercially available 2-chloroacetamide and triethylphosphite at 150 °C to afford diethylphosphonoacetamide.35 Deprotection of the phosphonate was achieved with trimethylsilyl bromide followed by water.19
Crystallization and freezing of crystals
The D236A ATCase was crystallized by microdialysis, using 50 μL wells. The enzyme solution, at ∼18 mg/mL, was dialyzed against a solution of 50 mM maleic acid, 3 mM sodium azide, 10 mM PAM, and 20 mM Asp (pH 5.8). One large crystal (1.5 × 1.2 × 0.8 mm) was cut into eight pieces, which were transferred into a solution of crystallization buffer containing 30% 2-methyl-2,4-pentanediol as the cyroprotectant for ∼1 min prior to freezing in liquid nitrogen.
X-ray data collection and processing
The data for D236A ATCase in the presence of PAM and Asp were collected at the National Synchrotron Light Source (Beamline X29) at Brookhaven National Laboratory. The diffraction data were integrated, scaled and averaged with HKL2000.36
Structural refinement
The initial model for the R236_PA structure was derived from the coordinates of the previously published D236A ATCase structure in the presence of PAM (R236_P, PDB code: 2A0F).28 Prior to refinement, all of the waters and ligands were removed. After initial rigid body, simulated annealing and B-factor refinement in CNS,17 the electron density maps were checked. A large amount of negative density was observed between adjacent catalytic chains in the Fo−Fc electron density map. Although the R236_PA and the R236_P structures have the same mutation and the crystals are in the same space group, there are significant differences near the subunit interfaces as well as in the crystal packing. To improve the initial model, automated molecular replacement (AmoRe)37 was performed using the combination of a catalytic and the adjacent regulatory chain (C1-R1) as the starting model.
After molecular replacement, a round of refinement was performed with CNS. The Rfactor dropped 3.2% (Rfactor: 25.3%) compared to the refinement without AmoRe (Rfactor: 28.5%). The structure was further refined with one round of Refmac516 followed by another round of refinement in CNS. The Rfactor dropped to 24.6% with no ligands in the active site. Since the N-terminus of the regulatory chain was disordered, the first seven amino acid residues were removed from the model. PAM and Asp were introduced into the structure using XtalView38 and were confirmed by omit maps calculated in CNS. Several rounds of manual rebuilding of both the catalytic and regulatory chains were performed to improve the quality of the electron density maps. When the Rfactor fell below 23%, waters were added to the structure using COOT39 on the basis of Fo−Fc electron density maps at or above the 2.5σ level, and were checked and retained only when they could be justified by hydrogen bonds. The details of data processing and refinement statistics of the structure are given in Table 1.
Automated docking procedure
The program AUTODOCK27 was used for the docking of the six-membered ring intermediate to the active site. In order to have one complete active site, two adjacent catalytic chains were used for the docking procedure. The charge on the six-membered ring intermediate was set at -3. The grid box was set at 30 Å3 centered at the middle of the active site with a grid spacing of 0.275 Å between grid points. In each case, 25 docking runs were performed using the Genetic Algorithm with a maximum of 500,000 energy evaluations. Analysis of the results was carried out with AutoDockTools.
Protein Data Bank accession code
The atomic coordinate of the structure of D236A ATCase in the presence of PAM and Asp (R236_PA) has been deposited in the Protein Data Bank (PDB entry: 2HSE).
Acknowledgments
This work was supported in part by Grant GM26237 from the National Institutes of Health. Data for the crystals with CP bound were measured at Beamline ×29 of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health. We thank Howard Robinson of Brookhaven National Laboratory for data collection and assistance with data processing.
Abbreviations used
- ATCase
aspartate transcarbamoylase (EC 2.1.3.2, aspartate carbamoyltransferase)
- C1/C6
the two catalytic chains of ATCase in the asymmetric unit of the crystal
- D236A
the mutant ATCase in which Asp236 in the catalytic chains have been replaced by Ala
- PALA
N-(phosphonoacetyl)-L-aspartate
- CP
carbamoyl phosphate
- PAM
phosphonoacetamide
- R236_P
the X-ray structure of D236A ATCase in the presence of PAM (PDB code 2A0F)
- R236_PA
the X-ray structure of D236A ATCase in the presence of PAM and Asp determined here (PDB code 2HSE)
- RPALA
the X-ray structure of wild-type ATCase in the presence of PALA (PDB code 1D09)
- RCP_suc
the X-ray structure of wild-type ATCase in the presence of CP and succinate
- RPM
the X-ray structure of wild-type ATCase in the presence of PAM and malonate (PDB code 2AT1)
- Rpi_cit
the X-ray structure of wild-type ATCase in the presence of phosphate and citrate (PDB code 1R0B)
- Tapo
the X-ray structure of wild-type ATCase in the absence of substrates and substrate analogs (PDB code 1ZA1)
- TCP
the X-ray structure of wild-type ATCase in the presence of CP (PDB code1ZA2)
- 80's loop
a loop in the catalytic chain of ATCase comprised of residues 73-88
- 240's loop
a loop in the catalytic chain of ATCase comprised of residues 230-245
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perutz MF. Mechanism of cooperativity and allosteric regulation in proteins. Quaterly Rev Biophys. 1989;22:139–236. doi: 10.1017/s0033583500003826. [DOI] [PubMed] [Google Scholar]
- 2.Jones ME, Spector L, Lipmann F. Carbamyl phosphate. The carbamyl donor in enzymatic citrulline synthesis. J Am Chem Soc. 1955;77:819–820. [Google Scholar]
- 3.Hervé G, Moody MF, Tauc P, Vachette P, Jones PT. Quaternary structure changes in aspartate transcarbamylase studied by X-ray solution scattering; signal transmission following effector binding. J Mol Biol. 1985;185:189–199. doi: 10.1016/0022-2836(85)90190-1. [DOI] [PubMed] [Google Scholar]
- 4.Gerhart JC, Pardee AB. The effect of the feedback Inhibitor CTP, on subunit interactions in aspartate transcarbamylase. Cold Spring Harbor Symp Quant Biol. 1963;28:491–496. [Google Scholar]
- 5.Gerhart JC, Schachman HK. Allosteric interactions in aspartate transcarbamylase II. Evidence for different conformational states of the protein in the presence and absence of specific ligands. Biochemistry. 1968;7:538–552. doi: 10.1021/bi00842a600. [DOI] [PubMed] [Google Scholar]
- 6.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamylase. Proc Natl Acad Sci U S A. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–49. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 8.Hayward S. Identification of specific interactions that drive ligand-induced closure in five enzymes with classic domain movements. J Mol Biol. 2004;339:1001–1021. doi: 10.1016/j.jmb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Wedler FC, Gasser FJ. Ordered substrate binding and evidence for a thermally induced change in mechanism for E. coli aspartate transcarbamylase. Arch Biochem Biophys. 1974;163:57–68. doi: 10.1016/0003-9861(74)90454-8. [DOI] [PubMed] [Google Scholar]
- 10.Ke HM, Lipscomb WN, Cho Y, Honzatko RB. Complex of N-phosphonacetyl-L-aspartate with aspartate carbamoyltransferase: X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J Mol Biol. 1988;204:725–747. doi: 10.1016/0022-2836(88)90365-8. [DOI] [PubMed] [Google Scholar]
- 11.Macol CP, Tsuruta H, Stec B, Kantrowitz ER. Direct structural evidence for a concerted allosteric transition in Escherichia coli aspartate transcarbamoylase. Nat Struc Biol. 2001;8:423–426. doi: 10.1038/87582. [DOI] [PubMed] [Google Scholar]
- 12.Tsuruta H, Sano T, Vachette P, Tauc P, Moody MF, Wakabayashi K, Amemiya Y, Kimura K, Kihara H. Structural kinetics of the allosteric transition of aspartate transcarbamoylase produced by physiological substrates. FEBS Lett. 1990;263:66–68. doi: 10.1016/0014-5793(90)80706-o. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2005;102:8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fetler L, Kantrowitz ER, Vachette P. Direct observation in solution of pre-existing structure equilibrium for a mutant of allosteric aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2007;104:495–500. doi: 10.1073/pnas.0607641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsuanyu Y, Wedler FC. Kinetic mechanism of native Escherichia coli aspartate transcarbamylase. Arch Biochem Biophys. 1987;259:316–330. doi: 10.1016/0003-9861(87)90498-x. [DOI] [PubMed] [Google Scholar]
- 16.Pannu NJ, Murshudov GN, Dodson EJ, Read RJ. Incorporation of prior phase information strengthen maximum-likelihood structure refinement. Acta Cryst. 1998;D54:1285–1294. doi: 10.1107/s0907444998004119. [DOI] [PubMed] [Google Scholar]
- 17.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 18.Gouaux JE, Lipscomb WN. Three-dimensional structure of carbamyl phosphate and succinate bound to aspartate carbamyltransferase. Proc Natl Acad Sci U S A. 1988;85:4205–4208. doi: 10.1073/pnas.85.12.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gouaux JE, Lipscomb WN. Crystal structures of phosphonoacetamide ligated T and phosphonoacetamide and malonate ligated R states of aspartate carbamoyltransferase at 2.8 Å resolution and neutral pH. Biochemistry. 1990;29:389–402. doi: 10.1021/bi00454a013. [DOI] [PubMed] [Google Scholar]
- 20.Stieglitz K, Stec B, Baker DP, Kantrowitz ER. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the E50A mutant in four distinct allosteric states. J Mol Biol. 2004;341:853–868. doi: 10.1016/j.jmb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Collins KD, Stark GR. Aspartate transcarbamylase: Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J Biol Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 22.Porter RW, Modebe MO, Stark GR. Aspartate transcarbamylase: Kinetic studies of the catalytic subunit. J Biol Chem. 1969;244:1846–1859. [PubMed] [Google Scholar]
- 23.Heyde E. A unifying concept for the active site region in aspartate transcarbamylase. Biochim Biophys Acta. 1976;452:81–8. doi: 10.1016/0005-2744(76)90059-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Lipscomb WN. Aspartate transcarbamylase (ATCase) of Escherichia coli: a new crystalline R-state bound to PALA, or to product analogues citrate and phosphate. Biochemistry. 2004;43:6415–21. doi: 10.1021/bi030213b. [DOI] [PubMed] [Google Scholar]
- 25.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 26.Gouaux JE, Krause KL, Lipscomb WN. The catalytic mechanism of Escherichia coli aspartate carbamoyltransferase: A molecular modeling study. Biochem Biophys Res Commun. 1987;142:893–897. doi: 10.1016/0006-291x(87)91497-5. [DOI] [PubMed] [Google Scholar]
- 27.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Stieglitz KA, Dusinberre KJ, Cardia JP, Tsuruta H, Kantrowitz ER. Structure of the E. coli aspartate transcarbamoylase trapped in the middle of the catalytic cycle. J Mol Biol. 2005;352:478–486. doi: 10.1016/j.jmb.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 29.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 30.Newton CJ, Kantrowitz ER. The regulatory subunit of Escherichia coli aspartate carbamoyltransferase may influence homotropic cooperativity and heterotropic interactions by a direct interaction with the 240s loop of the catalytic chain. Proc Natl Acad Sci U S A. 1990;87:2309–2313. doi: 10.1073/pnas.87.6.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Davis BJ. Disc electrophoresis-II Method and application to human serum proteins. Ann N Y Acad Sci. 1964;121:404–427. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 33.Ornstein L. Disc electrophoresis. I. Background and theory. Ann N Y Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 35.Balsiger RW, Jones DG, Montgomery JA. Synthesis of potential anticancer agents XVIII. Analogs of carbamoyl phosphate. J Org Chem. 1959;24:434–436. [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods Enzymol. Vol. 276. Academic Press; NY: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.Navaza J. AMoRe: an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 38.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 41.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]