

Abstract
The mechanistic details of aerobic alcohol oxidation with catalytic Pd(IiPr)(OAc)2(H2O) (IiPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) are disclosed. Under optimal conditions, β-hydride elimination is rate-limiting supported by kinetic studies including a high primary kinetic isotope effect (KIE) value of 5.5 ± 0.1 and a Hammett ρ value of −0.48 ± 0.04. On the basis of these studies, a late transition state is proposed for β-hydride elimination, which is further corroborated by theoretical calculations using density functional theory. Additive acetic acid modulates the rates of both the alcohol oxidation sequence and regeneration of the Pd catalyst. With no additive [HOAc], turnover-limiting reprotonation of intermediate palladium peroxo is kinetically competitive with β-hydride elimination, allowing for reversible oxygenation and decomposition of Pd(0). With additive [HOAc] (>2 mol %), reprotonation of the palladium peroxo is fast and β-hydride elimination is the single rate-controlling step. This proposal is supported by an apparent decomposition pathway modulated by [HOAc], a change in alcohol concentration dependence, a lack of [O2] dependence at high [HOAc], and significant changes in the KIE values at different HOAc concentrations.
Introduction
The observation that alcohols are oxidized by Pd(II), or at least that Pd(II) is reduced to Pd(0) in the presence of an alcohol, was first made over 175 years ago when Berzelius observed Pd metal precipitate out of a heated alcoholic solution of Pd(II).1 While the use of stoichiometric Pd(II) as an oxidant is not a practical approach to oxidizing alcohols,2 the ability to use a catalytic amount of Pd(II), along with a stoichiometric amount of a simple oxidant, such as molecular oxygen, to reoxidize the Pd(0) is highly attractive.3 Moiseev was the first to report a Pd-catalyzed oxidation of an alcohol under aerobic conditions in the presence of CuCl2 in 1963.4 In 1967, Lloyd disclosed a study of Pd-catalyzed aerobic oxidations of alcohols focusing largely on the role of additives to facilitate the reoxidation of Pd.5 The most important observation made in this study was catalytic amounts of PdCl2, without any additive, could facilitate the oxidation of alcohols under aerobic conditions although with very low efficiency. In 1977, Blackburn and Schwartz were the first to develop a synthetically useful procedure for the oxidation of alcohols under aerobic conditions without a copper additive.6 In their procedure, 1 mol % PdCl2 and 5 mol % NaOAc are used under the relatively mild conditions of 1 atm of O2 at 38°C in ethylene carbonate as solvent for the oxidation of a variety of alcohols. The major drawback to this procedure is the long reaction times required for high conversions (typically ~100 h).
It was not until 1998 that two groups reported significant improvements in palladium-catalyzed aerobic oxidation of alcohols. The first was reported by Peterson and Larock,7 who found the use of 5 mol % Pd(OAc)2, in combination with an appropriate carbonate base, provides an effective catalyst system for the oxidation of benzylic and allylic alcohols when DMSO is used as solvent at 1 atm of O2 and 80 °C.8–10 The second is a Pd(OAc)2/pyridine catalyst system developed by Uemura and co-workers.11 In this system, 5 mol % Pd(OAc)2, 20 mol % pyridine, and activated 3 Å molecular sieves prove an effective combination for the oxidation of benzylic and aliphatic alcohols at 1 atm of O2 and 80 °C. For allylic alcohols, greater amounts of pyridine are required for efficient oxidation. In addition to displaying good substrate scope, high conversions are achieved in relatively short times (~2 h) for most substrates. Uemura and others have extended this oxidation system to a variety of other interesting transformations.12–16
Since the reemergence of Pd-catalyzed aerobic oxidations of alcohols, several other groups have reported new catalysts. Sheldon and co-workers have developed a system for the oxidation of alcohols in aqueous solutions using a water-soluble phenanthroline ligand.17 The reaction is carried out under forcing conditions, 100 °C and 30 atm of air pressure, but these conditions do allow the use of rather low palladium catalyst loadings (typically 0.1–0.5 mol %). Moberg and co-workers have developed a catalyst system using an oxazoline–pallada-cycle complex.18 While this reaction is not as efficient as some of the previous examples, it is important as the first example of a homogeneous Pd catalyst for alcohol oxidation using ambient air atmosphere instead of pure O2.19 We have developed the first method for Pd-catalyzed aerobic alcohol oxidation at room temperature using 3 mol % Pd(OAc)2 and 6 mol % triethylamine with a scope similar to that of Uemura’s system.20,21 While excellent progress has been made in catalyst development for palladium-catalyzed aerobic oxidation of alcohols, all suffer from high catalyst loadings, the use of high pressures of O2, and/or high temperatures.
With clear limitations in reported palladium-catalyzed aerobic oxidations of alcohols, we sought to develop a more active and robust catalyst. The basis of this effort was to take recently reported mechanistic information on the fundamental steps involved to design an improved catalyst. Key observations include (1) Pd(0) species decompose during regeneration of the Pd catalyst without adequate support by ligand and/or solvent, (2) excess amine ligand inhibits alcohol oxidation, and (3) base is mandatory for alcohol oxidation. Stahl’s investigations of Larock’s22 and Uemura’s23 systems highlighted the need for substantial support of Pd(0) species during regeneration of the catalyst. In Larock’s system, the soft, donating character of the solvent dimethyl sulfoxide was found to have a stabilizing effect to prevent aggregation of Pd(0) species. In Uemura’s Pd(OAc)2/pyridine and our Pd(OAc)2/triethylamine aerobic alcohol oxidations, excess amine ligand is necessary for a competent oxidation presumably through stabilization of Pd(0) intermediates. A caveat to this is excess amine ligand also inhibits alcohol oxidation, leading to the need for higher catalyst loadings (3–5 mol %) for effective oxidations. Investigations of these systems indicate that the inhibition is most likely due to either competitive binding of the amine with the alcohol substrate or the need to dissociate an amine ligand to allow for rate-limiting β-hydride elimination.20,23 An additional role of amine ligand is as an exogenous Brønsted base to deprotonate a palladium-bound alcohol to form a palladium alkoxide for subsequent β-hydride elimination.24
On the basis of this analysis, the two design elements for improved aerobic alcohol oxidation catalysts are (1) a monodentate ligand to support Pd species involved and to provide a readily accessible coordination site to lower the energy barrier for rate-limiting β-hydride elimination and (2) a base that does not inhibit the oxidation. For ligand selection, N-heterocyclic carbene (NHC) ligands meet the above criteria and have proven effective in various Pd(0)-catalyzed processes25 as well as an oxidative kinetic resolution using (−)-sparteine as the exogenous base.26 Acetate was also selected as a ligand for Pd(II) to serve both as a counterion and as a Brønsted base to deprotonate the Pd-bound alcohol.24b,c Since the acetate base would be masked as an anionic ligand, an intriguing possibility is acetate could facilitate an intramolecular deprotonation (Scheme 1).
Scheme 1.

Design of a New Pd(II) Catalyst for Aerobic Oxidation of Alcohols
To test the catalyst design, [Pd(IiPr)Cl2] dimer (IiPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) was submitted to AgOAc metathesis to form the monomeric complex Pd(IiPr)(OAc)2(H2O), 1. At 5 mol %, this complex was effective in oxidizing sec-phenethyl alcohol to acetophenone with >99% conversion in less than 2 h at 50 °C. Upon discovery of such an active catalyst, the system was optimized to lower catalyst loadings. The optimized catalytic system includes 0.5 mol % 1, 2 mol % HOAc, and 3 Å molecular sieves in toluene at 60 °C under a balloon pressure of O2 (Figure 1).27 For activated substrates, up to 1000 turnovers can be accomplished, representing one of the most effective Pd catalysts for aerobic alcohol oxidation reported to date. This system converts primary and secondary benzylic, allylic, and aliphatic substrates to aldehydes and ketones in high yields. The robustness of the catalyst is demonstrated by the effective oxidation of alcohols using an ambient air atmosphere.
Figure 1.

Optimized alcohol oxidation conditions using catalyst 1.
Considering the unique activity of this catalyst and the mechanistic approach used for the discovery, we set out to confirm the rationale of the catalyst design through mechanistic studies. In addition, one anomalous additive, acetic acid, was necessary for consistent oxidation. Herein, we describe a detailed investigation of the nature of β-hydride elimination through experiment and theory as well as identification of the dynamic role of acid/base chemistry.
Results and Discussion
A general proposed mechanism for the aerobic oxidation of alcohols with 1 is depicted in Scheme 2. Dissociation of water to form A initiates the catalytic cycle with the water presumably trapped by molecular sieves. Alcohol substrate 2 binds to A, forming Pd-bound alcohol B. Intramolecular deprotonation of B forms palladium alkoxide C followed by subsequent β-hydride elimination to expunge carbonyl product and form palladium hydride D. This sequence will be referred to as the alcohol oxidation realm of the catalytic cycle. Palladium hydride D undergoes reductive elimination of acetic acid to yield Pd(0) species E. E is oxygenated to Pd(II) by molecular oxygen to produce palladium peroxo F, which is protonated by acetic acid to re-form A and 1 equiv of H2O2. This sequence will be referred to as the Pd(II) catalyst regeneration realm of the catalytic cycle.
Scheme 2.

A Proposed Mechanism for the Pd(II)-Catalyzed Oxidation of Alcohols Using 1
A key preliminary step in elucidating the mechanistic details was structural characterization of 1. A single crystal was grown from dichloromethane/hexanes. X-ray crystallographic analysis revealed a square planar structure with the carbene ligand bound monodentate and an acetate counterion on each side (Figure 2).27 Interestingly, the oxygen of a water molecule is datively bound to fill the last coordination site on Pd trans to the carbene. Additionally, the hydrogen atoms on the water molecule are bound to the carbonyl oxygen of each acetate ligand. This hydrogen-bonding phenomenon supports the proposal of an intramolecular deprotonation of the Pd-bound alcohol through the assistance of hydrogen bonding to the acetate carbonyl oxygen.
Figure 2.

Ortep representation of 1.
To determine whether the water molecule was an artifact of crystallization or if it was incorporated as the complex formed, the isolated crystalline complex was examined by 1H NMR. The 1H NMR spectrum of the crystallized material in CDCl3 was identical to that of the initial complex used. This suggests that they are the same complex; however, the expected peak of a bound water molecule was not observed in either spectrum. To address this discrepancy, the crystallized complex was studied from −60 to 0 °C by variable-temperature 1H NMR at 20 °C intervals (Figure 3). This set of spectra clearly displays a broad peak emerging as the temperature is decreased below 0 °C. The peak sharpens with a further decrease in temperature until it appears as a singlet at ~8.6 ppm at −60 °C, demonstrating the inclusion of a water molecule in solution.
Figure 3.

Overlaid 1H NMR spectra from −60 to 0 °C of 1 in CDCl3.
Kinetic Characterization of Optimized Conditions
Using in situ Fourier transform infrared spectroscopy to measure oxidation rates, initial kinetic studies revealed the alcohol oxidation sequence is rate limiting under slightly modified optimal conditions28 with first-order dependencies on [1]29 and sec-phenethyl alcohol concentration. Large primary kinetic isotope effects (KIEs) of 5.5 ± 0.1 for sec-phenethyl alcohol and 5.5 ± 0.5 for 2-decanol oxidation were observed, which are consistent with rate-limiting β-hydride elimination. Due to the unusual magnitude of these KIE values (vide infra), a Hammett plot was constructed to evaluate the electronic effects at the benzylic center (eq 1).30 A linear free energy relationship was observed with a calculated ρ value of −0.48 ± 0.04 (Figure 4). The negative ρ value signifies a positive charge buildup in the transition state, further supporting rate-limiting β-hydride elimination.
Figure 4.

log kobs vs σ for the oxidation of benzylic alcohols at 50 °C. Conditions: 0.5 mol % 1, 0.45 M alcohol, 2 mol % AcOH in benzene, with 3 Å molecular sieves and a balloon charged with O2.
 |
(1) |
Nature of β-Hydride Elimination
The initial kinetic evidence establishes β-hydride elimination as rate determining. However, the substantial magnitude of the measured KIE value (5.5 ± 0.1 for sec-phenethyl alcohol oxidation) implicates the nature of β-hydride elimination is unique compared to that of rate-limiting β-hydride elimination in other Pd-catalyzed alcohol oxidations. Significantly smaller KIE values have been measured for these other systems, which include a KIE of 1.31 ± 0.04 we reported for the oxidation of (S)-sec-phenethyl alcohol with Pd(II)/(−)-sparteine,24b,c a KIE of 1.3 ± 0.1 reported by Stahl et al. in evaluating Uemura’s Pd(II)/pyridine-catalyzed oxidation of benzyl alcohol (0.1 M),23 and a KIE of 1.4 reported by Sheldon et al. for the Pd(II)/bathophenanthroline disulfonate-catalyzed oxidation of sec-phenethyl alcohol.17c
This considerable disparity in KIE values measured for the monodentate carbene system and other Pd-catalyzed aerobic alcohol oxidation systems can provide insight into the modulation of β-hydride elimination. The catalyst was designed predicting the monodentate ligand leaves an accessible coordination site for β-hydride elimination, thereby altering the reactivity of the catalyst and the nature of the transition state. The high KIE value for this system indicates a transition state with minimal vibrational energy differences, maximizing ΔΔG‡ between α-H and α-D sec-phenethyl alcohol. Traditionally, linear, symmetric transition states are the models for systems with high KIE values because bond vibrations are minimal in the transition state.31 However, it is difficult to imagine a linear transition state for β-hydride elimination which leads to another model to describe the observed large KIE values.32
A high KIE value can be measured when the vibration in the transition state is minimal and/or the difference in vibrational energy levels in the transition state is small.33 Both criteria for a high KIE are met when the transition state is late in this transformation with almost complete C–H (C–D) bond cleavage and Pd–H (Pd–D) bond formation. The magnitude of vibration and the difference in vibrational energy levels in the transition state are minimized when the weaker bond of hydrogen with a significantly heavier atom, Pd–H (bond stretching frequency 1953 cm−1),34 is more fully formed and the stronger bond of hydrogen to a lighter atom, C–H (bond stretching frequency 2900 cm−1), is nearly cleaved (Figure 5). A readily accessible coordination site on Pd(II) should facilitate a late transition state. This contrasts with other Pd-catalyzed alcohol oxidations where a ligand saturates the coordination sphere. As a comparison, (−)-sparteine binds datively to Pd(II) in two coordination sites; the other two sites are occupied by anionic ligands (either chloride or alkoxide), rendering the square planar coordination sphere saturated (Figure 6). Two limiting scenarios can be envisioned for β-hydride elimination: (1) a dissociative pathway where a ligand/counterion dissociates completely prior to rate-limiting β-hydride elimination or (2) an associative pathway where the β-hydride and the ligand/counterion are partially coordinated to Pd(II) in the transition state. In the former case, a KIE similar to that of the Pd(IiPr)(OAc)2(H2O) system where a late transition state is proposed would be expected if β-hydride elimination remained rate limiting. For the latter case, there is not a readily accessible coordination site for β-hydride elimination; thus, the carbonyl product and palladium hydride are formed through a 5-coordinate transition state. In this scenario, the transition state is earlier, where H(D) is bound more to the carbon atom than to Pd(II), leading to a greater separation of the vibrational energy levels in the transition state and lowering the observed KIE values. Therefore, this associative substitution (5-coordinate transition state) scenario is most consistent with the smaller observed KIE values for both the Pd-catalyzed oxidative kinetic resolution of alcohols using (−)-sparteine and Uemura’s Pd(II)/pyridine alcohol oxidation system.35
Figure 5.

Transition-state model for β-hydride elimination using catalyst 1.
Figure 6.

Limiting pathways for β-hydride elimination with 4-coordinate Pd.
Other experimental evidence also highlights the differences between the oxidation of alcohols with 1 and Pd[(−)-sparteine]-Cl2. Although a ρ value of −0.48 ± 0.04 measured for the oxidation of substituted benzyl alcohols with 1 supports rate-limiting β-hydride elimination, it differs significantly from the value measured using Pd[(−)-sparteine]Cl2 (ρ = −1.41 ± 0.15).24b The smaller ρ value of −0.48 ± 0.04 observed for oxidation of alcohols with 1 indicates that electronics have a relatively subtle influence on β-hydride elimination. This is consistent with the proposed late-transition-state model (Figure 5 and Figure 6) where positive charge buildup stabilization is less important for facile oxidation. For the Pd(II)/(−)-sparteine system, electron-donating groups clearly play a more important role by stabilizing the positive charge generated in the transition state, increasing the nucleophilicity of the hydride.
To further understand the unusually high KIE values and to gain a more detailed understanding of β-hydride elimination, computational studies using density functional theory were undertaken. The geometries of Pd(IiPr)(OAc) species 2C, 2C‡, and 2D36 (Figure 7) were fully optimized in the Gaussian37 program using B3LYP38,39 and the LANL2DZ40,41 basis set, which includes relativistic effective core potentials.42,43 The geometry of 2C‡ was elucidated using the QST244 search method. Frequency calculations were used to determine ΔGH‡ and ΔGD‡. All energies reported include zero-point and entropy corrections for the experimental temperature and pressure (323.15 K and 1 atm).45 Examination of key bond lengths and angles revealed the ground-state palladium alkoxide 2C has an optimized geometry very similar to that of the solved crystal structure of 1 (Figure 2).
Figure 7.

Optimized ground-state and transition-state structures for the oxidation of sec-phenylethyl alcohol with catalyst 1 using the B3LYP and the LANL2DZ basis sets.
The calculated transition state, 2C‡, correlates well with our proposed product-like transition-state model. Comparison of the bond lengths Pd–H1 and C2–H1 in the three structures reveals that the transition state is more comparable to product 2D than to palladium alkoxide ground state 2C (Table 1). The Pd–H1 distance is 2.70 Å in the ground state 2C, while the Pd–H1 bond length in 2D is 1.57 Å. The Pd–H1 distance is only 1.60 Å in 2C‡, indicating the hydride is nearly completely bonded to Pd. The trend is similar for the C2–H1 distances in the Pd-bound alkoxide. The bond length of C2–H1 is 1.10 Å in the ground state 2C, while the corresponding distance in 2D is 3.01 Å. The bond distance of 1.97 Å in the transition state indicates the hydrogen–carbon bond is nearly cleaved. The KIE was determined from the difference in the calculated free energies of the transition state. The theoretically determined KIE value is 3.8.46 This large value is reasonably consistent with our experimentally determined KIE value of 5.5 ± 0.1, indicating our model is relevant with the caveat that error values in these calculations are difficult to estimate.
Table 1.
Selected Lengths and Angles of 2C, 2C ‡, and 2D
| 2C | 2C‡ | 2D | |
|---|---|---|---|
| Lengths (Å) | |||
| Pd–O1 | 2.00 | 2.17 | 2.14 |
| Pd–O2 | 2.18 | 2.07 | 2.06 |
| Pd–O3 | 2.23 | 3.11 | 3.14 |
| Pd–H1 | 2.70 | 1.60 | 1.57 |
| C2–H1 | 1.10 | 1.97 | 3.01 |
| C2–O1 | 1.48 | 1.31 | 1.28 |
| Angles (deg) | |||
| Pd–O1–C2 | 110.2 | 89.0 | 123.3 |
| Pd–H1–C2 | 87.5 | 89.3 | 75.8 |
| C1–Pd–O3 | 164.1 | 131.2 | 120.2 |
| O1–Pd–O3 | 106.8 | 116.3 | 123.5 |
| O2–Pd–O3 | 60.9 | 47.9 | 47.4 |
| O1–Pd–C1 | 89.0 | 100.4 | 98.6 |
| C2–Pd–C1 | 103.2 | 95.0 | 92.4 |
Further examination of the computational models reveals finer detail of this β-hydride elimination. The computational studies depict the acetate counterion in the Pd square plane weakly chelating the carbonyl oxygen in 2C (Figure 8). Transition state model 2C‡ shows the acetate counterion rotated out of plane during β-hydride elimination. This change from the ground state to the transition state is supported experimentally by comparison of the activation parameters at 50 °C for the oxidations of sec-phenethyl alcohol, benzyl alcohol, and 2-decanol (Figure 9, Table 2). Interestingly, benzyl alcohol and 2-decanol have positive ΔS‡ values, while the ΔS‡ for sec-phenethyl alcohol is close to unity. The positive values indicate there is less organization in the transition state than in the ground state. This is consistent with the computational model with a gain in rotational degrees of freedom when acetate is rotated out of plane in the transition state compared to the ground state, where the acetate is chelated in the square plane of Pd. It is important to note that the experimental ΔG‡ of 21.2 ± 0.7 at 50 °C correlates well with the calculated ΔG‡ of 22.6 kcal/mol.
Figure 8.

Translation of the acetate counterion from the ground state to the transition state. Dihedral angles (C1–Pd–O2–C3) of 2C and 2C‡ are 181.1° and 145.1°, respectively.
Figure 9.

Eyring plots of benzyl alcohol, sec-phenethyl alcohol, and 2-decanol oxidation using catalyst 1. Conditions: 0.5 mol % 1, 0.45 M alcohol, 2 mol % AcOH in benzene, with 3 Å molecular sieves and a balloon charged with O2, temperature range from 40 to 55 °C.
Table 2.
Activation Parameters for Alcohol Oxidation at 50 °C Using Catalyst 1
| Substrate | Activation Parameters |
|---|---|
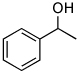 |
ΔG‡ = 21.2 ± 0.7 kcal/mol |
| ΔH‡ = 20.1 ± 0.5 kcal/mol | |
| ΔS‡ = −3.5 ± 1.6 eu | |
| ΔG‡ = 22.2 ± 1.4 kcal/mol | |
| ΔH‡ = 25.6 ± 0.95 kcal/mol | |
| ΔS‡ = −10.5 ± 3.0 eu | |
| ΔG‡ = 21.1 ± 2.5 kcal/mol | |
| ΔH‡ = 24.9 ± 1.8 kcal/mol | |
| ΔS‡ = 12.0 ± 5.5 eu | |
Alternative Scenarios for a Large KIE Value
The experimental data and literature precedent thus far point toward rate-limiting β-hydride elimination. However, alternative scenarios should be considered that may account for the large KIE. One possibility involves direct β-hydride elimination to the acetate counterion to form Pd(0), acetic acid, and ketone product through a possible linear transition state (Scheme 3). APT charge calculations on each atom of 2C indicate O(3) and O(2) to be electron rich.47 These calculations indicate hydride nucleophilic attack on either acetate oxygen is highly unlikely. In fact, all attempts to optimize a direct transition state failed, with the acetate oxygen strongly repulsing the hydride. Experimental evidence considering the electronics of different carboxylates (vide infra) further contradicts this proposal.
Scheme 3.

An Unlikely Alternative Scenario That May Lead to a Large KIE
Another possibility could be that reductive elimination is rate limiting, where a large KIE could be observed through a linear-type transition state. A linear transition state could only occur through an intermolecular deprotonation of the hydride. While this type of process cannot be completely ruled out, the concentration of exogenous base (acetate) is very low under the acidic reaction conditions, which renders this an unlikely alternative. In this case, a highly organized transition state would be predicted, leading to a large negative ΔS‡, which is not observed. Additionally, under acidic conditions, reductive elimination has been shown to be reversible, which leads to insignificance in the measurement of a KIE.48 Although this circumstantial evidence does point toward rate-limiting β-hydride elimination, it is impossible to rule out a scenario where the large KIE value is derived from the combination of reversible β-hydride elimination (an equilibrium isotope effect) and rate-limiting reductive elimination (a primary KIE).
Roles of Acetic Acid
While the initial goal of the catalyst design was to avoid additives, it was found empirically that additive acetic acid was mandatory for consistent oxidation reactions. However, the selection of acetic acid as an additive in the optimization process was based on the proposed mechanism where acetic acid is a byproduct or functional reagent in several steps. Additionally, by increasing the amount of acetic acid in the reaction, air can be substituted for pure O2, showcasing the ability of acetic acid to modulate the catalysis. Therefore, identifying the role(s) of acetic acid should provide insight into the robust features of this catalyst system.
Considering the initial kinetic evidence strongly suggests rate-limiting β-hydride elimination, a predicted role of additive acetic acid is reprotonation of the palladium alkoxide, thus inhibiting the alcohol oxidation and decreasing the oxidation rates. A simplified rate equation describing this kinetic scenario shows the rate should have an inverse first-order dependence on acetic acid concentration (eq 2). To determine whether acetic acid
| (2) |
simply inhibits alcohol oxidation or has a more profound role within the catalytic system, the dependence of oxidation rate on acetic acid concentration was evaluated from 0 to 67.5 mM (from 0 to 15 mol %). Interestingly, an initial rate increase was observed at low concentrations of additive acetic acid (Figure 10). A maximal rate was achieved at 2.79 mM (0.62 mol %) acetic acid followed by inhibition as more acetic acid was added. When the data ranging from 6.75 to 67.5 mM (from 1.5 to 15 mol %) were fit to a simple inverse first-order equation using nonlinear least-squares analysis, the agreement was excellent (Figure 11). The results under these conditions are consistent with a simple rate equation similar to eq 2. However, the observations from 0 to 1.5 mol % allude to a more complicated scenario with acetic acid playing multiple roles within the catalytic system.
Figure 10.

Rate dependence of sec-phenethyl alcohol oxidation using various HOAc concentrations at 50 °C. Conditions: 0.5 mol % 1, 0.45 M alcohol in benzene, with 3 Å molecular sieves and a balloon charged with O2. The range of [AcOH] is 0–67.5 mM (0–15 mol %).
Figure 11.

Inverse first-order rate dependence on [HOAc] for the oxidation of sec-phenethyl alcohol using 1. The plotted range of [AcOH] is from 6.75 to 67.5 mM (from 1.5 to 15 mol %).
To understand how acetic acid enhances the rates of alcohol oxidation under low concentrations, the rates of alcohol disappearance were examined at different HOAc concentrations (Figure 12). Analysis of these data using a pseudo-first-order plot showed distinct linearity for the reactions carried out with 2 and 3 mol % HOAc. This confirms the first-order rate dependence on sec-phenethyl alcohol concentration and the relative simplicity of the oxidation when >2 mol % HOAc is added. The plots also show the relative retardation of oxidation rate by adding 3 mol % HOAc compared to 2 mol % HOAc predicted by eq 2. However, analysis of the data for the reactions run at 0 and 0.75 mol % HOAc shows a significant deviation from linearity. The oxidation at 0 mol % HOAc did not proceed to completion, while the reaction at 0.75 mol % had a much greater intial oxidation rate and overall conversion of alcohol, demonstrating that low concentrations of additive HOAc increase oxidation rates. These data support a time-dependent process, presumably a catalyst decomposition pathway, is occurring at low acid concentration.
Figure 12.

Natural logarithm of sec-phenethyl alcohol concentration vs time at various HOAc concentrations at 50 °C (every 10 time points displayed). Conditions: 0.5 mol % 1, 0.45 M alcohol in benzene, with 3 Å molecular sieves and a balloon charged with O2.
Previous studies indicate Pd-catalyzed oxidation of alcohols involves generation of Pd(0) species which are oxidized by molecular oxygen.49 A proposed pathway of catalyst decomposition is the aggregation of Pd(0) species to form inactive Pd.50 Additive acetic acid can decrease catalyst decomposition by retarding the rate of alcohol oxidation through reprotonation of C, thus preventing the buildup of Pd(0) species. This alcohol oxidation rate retardation was clearly demonstrated in Figure 11 from 6.75 to 67.5 mM (from 1.5 to 15 mol %) acetic acid. The assumption of catalyst decomposition with no additive acetic acid suggests that a step involved in Pd(II) catalyst regeneration also influences the rate. By adding acetic acid, Pd(0) concentration can be decreased with concomitant slowing of the decomposition rate through two possible turnover steps. With rate-limiting oxygenation, oxidative addition of acetic acid to Pd(0) species can re-form palladium hydride D.48 Additive acetic acid can also accelerate the protonation of palladium peroxo F, preventing formation of Pd(0) through reversible oxygenation.
To probe these possible scenarios, the rate dependence on sec-phenethyl alcohol concentration was evaluated by measuring initial rates with no additive acetic acid. A log–log plot was constructed (Figure 13), and a partial order of 0.50 ± 0.02 was observed, which is consistent with two transition states of comparable free energy.51 That is, β-hydride elimination within the manifold of alcohol oxidation is competitive with the turnover-limiting step of Pd(II) catalyst regeneration. To further explore this, a KIE value of 1.7 ± 0.2 was measured with no additive acetic acid. This value is much smaller than the KIE value measured (5.5 ± 0.1) when 9 mM (2 mol %) acetic acid is included. Although not large, the KIE value of 1.7 ± 0.2 is a significant primary KIE, which implies that β-hydride elimination influences the rate when no additive acetic acid is present, but the considerable diminishment of the KIE and a partial order dependence on alcohol concentration suggest a step in Pd(II) catalyst regeneration is competitive. To explore this hypothesis, the KIE values for sec-phenethyl alcohol oxidation were measured at acetic acid concentrations ranging from 0 to 9 mM (from 0 to 2 mol %) (Table 3). The KIE values gradually increase as the concentration of additive acetic acid is increased. This is consistent with a change in rate-influencing steps from a combination of β-hydride elimination and a turnover-limiting step to only β-hydride elimination at 9 mM (2 mol %) acetic acid.
Figure 13.

log–log plot of the rate of sec-phenethyl alcohol oxidation vs sec-phenethyl alcohol concentration. Conditions: 2.25 mM 1, 0% AcOH in benzene, with 3 Å molecular sieves and a balloon charged with O 2. The range of sec-phenethyl alcohol concentration is from 0.03 to 0.45 M.
Table 3.
KIE Values at Various HOAc Concentrations
| [HOAc] | KIE | [HOAc] | KIE | [HOAc] | KIE |
|---|---|---|---|---|---|
| (mM) | value | (mM) | value | (mM) | value |
| 0 | 1.7 | 2.79 | 3.1 | 6.75 | 5.2 |
| 2.12 | 2.7 | 4.50 | 3.4 | 9.00 | 5.5 |
The two scenarios in which a turnover-limiting step can influence Pd catalyst decomposition are (1) turnover-limiting oxygenation with reversible reductive elimination and/or (2) reversible oxygenation with protonation of palladium peroxo F to form A and evolution of H2O2 (Scheme 4). If oxygenation (k5[E][O2]) is turnover limiting at 0 mM HOAc, a rate dependence on molecular oxygen concentration should be observed. The alcohol oxidation rate dependence on percent molecular oxygen was measured from 0 to 100% (molecular oxygen and nitrogen mixtures) at balloon pressure.52 At 0 mol % acetic acid, a zeroth-order rate dependence is observed from 29% to 100% O2/N2 (Figure 14). However, significant decomposition was noted immediately upon alcohol addition at 23% O2/N2. These results indicate oxygenation is not turnover limiting under these conditions, but a minimal concentration of oxygen must be present for Pd(II) catalyst regeneration. This also is congruent with being unable to effectively oxidize alcohols using air under these conditions. Additionally, oxygen dependence was evaluated at 3 and 7 mol % (13.5 and 31.5 mM) HOAc from 5% to 100% O2/N2. At 3 mol % acetic acid, there is no dependence on oxygen concentration from 23% to 100% O2/N2. From 5% to 23% O2/N2, significant decomposition is observed. At a higher acetic acid concentration, 7 mol %, the rate dependence is zeroth order in oxygen concentration from 5% to 100% oxygen with no observed decomposition. These observations demonstrate how air can be used as an effective O2 source by using increased concentrations of acetic acid. From 0 to 7 mol % acetic acid, decomposition occurs at progressively lower percentages of oxygen until it is not detected at 7 mol % HOAc. It is important to note that this phenomenon corresponds to a significant decrease in alcohol oxidation rate due to inhibition through reprotonation (C to B), which regulates the concentration of O2 needed for efficient catalyst regeneration.
Scheme 4.

Revised Proposed Mechanism: Possible Rate-Influencing Steps of Pd-Catalyzed Aerobic Oxidation
Figure 14.

Initial rates of the sec-phenethyl alcohol oxidation at 50 °C using various AcOH concentrations and oxygen/nitrogen mixtures. Conditions: 0.5 mol % 1, 0.45 M sec-phenethyl alcohol, with 3 Å molecular sieves and a balloon charged with O2.
Competitive turnover-limiting protonation of peroxo F is most consistent with the above kinetic data and the observation of a partial positive order acetic acid concentration dependence from 0 to 2.79 mM (from 0 to 0.62 mol %). With this proposal, the observation of decomposition at 0 mM acetic acid suggests reversible oxygenation. Therefore, as more acetic acid is added, the rate of protonation of F increases while the rate of alcohol oxidation decreases from inhibition through reprotonation of C. When >2 mol % acetic acid is added, protonation of F is fast relative to alcohol oxidation, leading to observed rate-limiting β-hydride elimination under these conditions. This scenario rationalizes the change in alcohol dependence from partial order at low concentrations of acetic acid to first order at higher concentrations of acetic acid. It is also consistent with the change in KIE values from 1.7 at 0 mM acetic acid when there is a combination of rate-influencing steps to 5.5 at 9 mM (2 mol %) acetic acid when only β-hydride elimination is rate limiting.
Using the King–Altman method, a rate law can be derived to mathematically describe Scheme 4, where β-hydride elimination and the protonation of F are assumed as irreversible (eq 3).52,53
| (3) |
In eq 3, kprot is all terms in the denominator that k6[AcOH] can be factored from, kalc is the remaining terms in the denominator with k1[2] factored out, and [PdT] is the total concentration of all Pd species. When no acetic acid is added, the full derived rate law describes the kinetic events. The terms in the denominator, k6[AcOH]kprot and k1[2]kalc, must be comparable because of the observation of a partial order dependence on alcohol concentration and a partial order dependence on acetic acid concentration. As more acetic acid is added to the reaction, the magnitude of k6[AcOH]kprot increases dramatically compared to that of k1[2]kalc. Therefore, k6[AcOH]kprot completely governs over the other terms in the denominator at higher [HOAc], leading to a simplification of the rate law (eq 4). Two observations allow for further simplification to eq 5: There is no dependence on oxygen concentration, and the rate is limited by β-hydride elimination at high [HOAc]. This equation describes rate-limiting β-hydride elimination in the steady-state form. If equilibrium conditions are assumed, the rate simplifies to eq 2.
| (4) |
| (5) |
pKa Dependence
Considering the significance of acetic acid in Pd(IiPr)(OAc)2(H2O)-catalyzed oxidation of alcohols, it is logical that a pKa effect could be observed when the carboxylate ligand and additive carboxylic acid are changed. Several catalysts were synthesized by submitting Pd(IiPr)Cl2 dimer complex to salt metathesis with different silver carboxylates. These new Pd(IiPr)(carboxylate)2 complexes were tested for catalytic activity with a 2 mol % concentration of the corresponding carboxylic acid (eq 6). A Brønsted-type plot was constructed by comparing the measured initial oxidation rates to the pKa of the corresponding carboxylic acid in H2O (Figure 15).54,55 A linear free energy relationship was observed with a calculated α value of 1.44 ± 0.13. An α value of 1.44 ± 0.13 is very large, indicating a significant pKa effect. By definition, α values should not exceed 1.00 if describing only one step. Since the anionic ligand was changed as well as the added acid, two influences are manifested in this α value: the basicity of the anionic ligand/base for deprotonation of Pd-bound alcohol B and the acidity of the acid for reprotonation of palladium alkoxide C. With such a large α value, reprotonation is probably the dominant step, with a weaker carboxylic acid leading to higher alcohol oxidation rates.56
Figure 15.

Brønsted-type plot of alcohol oxidation with a 2 mol % concentration of the corresponding acid at 50 °C. Conditions: 0.5 mol % Pd catalyst, 0.25 M sec-phenethyl alcohol in benzene, with 3 Å molecular sieves and a balloon charged with O2.
 |
(6) |
To differentiate the influence of carboxylate/carboxylic acid pKa on deprotonation of B and on reprotonation of C, the Pd(IiPr)(carboxylate)2 complexes were assessed with no additive carboxylic acid, where reprotonation of C should be negligible.52 However, performing these experiments at 0 mol % acetic acid conditions changes the scenario of rate-limiting steps. Under these conditions, previous experimental observations show turnover-limiting protonation of F is competitive with β-hydride elimination. Therefore, the acid/base effects most likely being evaluated under these conditions are deprotonation of Pd-bound alcohol B by carboxylate and protonation of peroxo F by carboxylic acid. A carboxylate from a higher pKa carboxylic acid provides for a better base for deprotonation of B. In contrast, a carboxylic acid with a lower pKa facilitates protonation of peroxo F. A positive linear free energy relationship was observed with a calculated α value of 0.31 ± 0.04 (Figure 16). This value is significantly smaller than the value of 1.44 ± 0.13 at 2 mol % carboxylic acid conditions and within the typical range from 0 to 1.00. The positive slope indicates a stronger conjugate base facilitates the overall reaction. This corresponds with deprotonation of the Pd-bound alcohol being more sensitive to pKa effects than protonation of peroxo F with no added acid. These results showcase the importance of the basicity of the carboxylate ligands for an efficient Pd catalyst for oxidation of alcohols.
Figure 16.

Brønsted-type plot of alcohol oxidation with 0 mol % additive acid at 50 °C. Conditions: 0.5 mol % Pd catalyst, 0.25 M sec-phenethyl alcohol in benzene, with 3 Å molecular sieves and a balloon charged with O2.
Of note is the log(kobs) value for Pd(IiPr)(pivalate)2 has a standard residual value of 1.6. Pivalate was the largest counterion tested in this experiment. Relief of steric strain through formation of the alkoxide as well as the higher basicity of pivalate provides an explanation for the increased catalytic activity relative to those of other carboxylate catalysts. These studies have led to the development of a highly active Pd(IiPr)(pivalate)2 catalyst for oxidation of alcohols at room temperature using air, which will be reported in due course.57
Overall Mechanistic Implications
The details for the aerobic oxidation of alcohols with Pd(IiPr)(OAc)2(H2O) have been elucidated, confirming much of the mechanistic rationale used to design it. Using a monodentate dative ligand and an anionic ligand as an internal base yields a highly effective catalyst system for alcohol oxidation. Initially, kinetic experiments revealed a relatively simple system, but as the alcohol oxidation was evaluated at other conditions, it became clear the entire scenario is much more complicated. Two results highlight the unique nature of the catalytic system: a large KIE value and the dynamic dependence on [HOAc].
The observed large KIE value leads to a proposed late transition state with significant Pd–H character. Theoretical studies support this late-transition-state model for β-hydride elimination from the Pd(IiPr)(OAc) alkoxide, and the calculated KIE value of 3.8 correlates with the experimentally determined KIE value of 5.5 ± 0.1. As demonstrated by large differences in KIE and ρ values, the nature of the ligand has a significant influence on β-hydride elimination. A single monodentate ligand provides an open coordination site leading to the proposed late transition state, while a bidentate ligand (or two monodentate ligands) most likely involves a 5-coordinate early transition state. These results reveal the ability to modulate β-hydride elimination by careful selection of ligand and lead to significant implications in the design of asymmetric catalysts.
Acetic acid plays multiple roles in aerobic oxidation of alcohols with Pd(IiPr)(OAc)2(H2O). Acetic acid is vital to the system to prevent decomposition of the catalyst and maintain consistent reactions. A key observation is increasing acetic acid concentration modulates the steps that influence the alcohol oxidation and Pd catalyst regeneration rates. When no acetic acid is present, β-hydride elimination works synergistically with turnover-limiting protonation to influence the overall reaction rate. Addition of acetic acid increases the rate of protonation of F until it is no longer rate influencing, rendering β-hydride elimination the single rate-controlling step. The discovery of Pd catalyst decomposition when protonation of the peroxo species F influences the overall rate suggests oxygenation is reversible, allowing for a decomposition pathway through Pd(0). Key studies of rate dependence on oxygen percent reveal additive acetic acid decreases alcohol oxidation rates and prevents deoxygenation through reversible peroxo formation. This ability to balance alcohol oxidation with Pd catalyst regeneration events allows for the use of air as a viable oxygen source.
The balance of alcohol oxidation and Pd catalyst regeneration events with additive acetic acid has allowed for insight into both realms of the catalytic cycle. This insight allows for the analysis of other mechanistic pathways for regeneration. It has been proposed for Pd-catalyzed aerobic alcohol oxidations that a palladium hydroperoxide species can be converted directly to the palladium alkoxide by alcohol especially under basic conditions.3,11a The need of acetic acid for efficient catalyst turnover implies a Pd(II) species such as A is formed. This can bind the alcohol as a Lewis acid to increase the acidity of the hydroxyl proton, allowing for effective deprotonation and formation of palladium alkoxide B. If the palladium peroxo F can be converted directly to B when exposed to alcohol in the presence of base, addition of NBu4OAc should promote turnover of the Pd catalyst and prevent decomposition. Instead, significant decomposition is observed with addition of NBu4OAc to the extent that initial rates can only be measured to 3 mol % NBu4OAc in the oxidation of sec-phenethyl alcohol.52 These observations contradict base-promoted turnover of the palladium peroxo species directly to the palladium alkoxide C, but strongly support the regeneration of A, followed by substrate binding and subsequent deprotonation to C.
Conclusions
The development of a well-defined catalyst, Pd(IiPr)(OAc)2(H2O), represents a clear example where catalyst design was significantly enhanced by detailed structural and kinetic information. The study of this catalyst has led to a fascinating picture of β-hydride elimination and how it can be modulated by careful selection of ligand and counterion. For other Pd-catalyzed aerobic oxidations of alcohols, only one realm of the catalytic cycle has been studied kinetically. Elucidation of the roles of acetic acid allowed for the concomitant study of Pd-catalyzed alcohol oxidation and catalyst regeneration by molecular oxygen. Further studies are aimed at understanding the precise mechanism of decomposition and using these studies as a foundation to design new catalysts for other Pd-catalyzed aerobic oxidations.
Acknowledgment
This work was supported by the National Institutes of Health (NIGMS Grant No. RO1 GM63540) and supported by a Research Innovation Award sponsored by Research Corp. We thank the University of Utah Research Foundation, Merck Research, Rohm and Haas Research, and Invenux Inc. for partial support of this research. We thank the donors of the Petroleum Research Fund, administered by the American Chemical Society, for partial support of this research (PRF Grant No. 37536-G1). We thank Johnson Matthey for supplies of various palladium salts. The X-ray crystal structure analysis was performed by Atta Arif. We also thank David R. Jensen and Professor Thanh N. Truong for insightful discussions.
Footnotes
Supporting Information Available: Statistical analyses, rate tables, rate law derivation, preparation of catalyst derivatives, DFT calculation data, and NMR spectra (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Berzelius J. J. Ann. Phys. Chem. 1828;13:435–488. [Google Scholar]
- 2.(a) Moreno-Manas M, Pleixats R, Villarroya S. Organometallics. 2001;20:4524–4528. [Google Scholar]; (b) Bellosta V, Benhaddou R, Czernecki S. Synlett. 1993:861–863. [Google Scholar]; (c) Benhaddou R, Czernecki S, Ville G, Zegar A. Organometallics. 1988;7:2435–2439. [Google Scholar]; (d) Zaw K, Lautens M, Henry PM. Organometallics. 1983;2:197–199. [Google Scholar]; (e) Tomioka H, Takai K, Oshina K, Nozaki H. Tetrahedron Lett. 1981;22:1605–1608. [Google Scholar]
- 3.For a recent review on Pd-catalyzed oxidations of alcohols, see: Muzart J. Tetrahedron. 2003;59:5789–5816.
- 4.Nikiforova AV, Moiseev II, Syrkin YK. Zh. Obshch. Khim. 1964;33:3239–3242. [Google Scholar]
- 5.Lloyd WG. J. Org. Chem. 1967;32:2816–2819. [Google Scholar]
- 6.Blackburn TF, Schwartz JJ. Chem. Commun. 1977:157–158. [Google Scholar]
- 7.Peterson KP, Larock RC. J. Org. Chem. 1998;63:3185–3189. [Google Scholar]
- 8.This oxidative system has been extended to several transformations. For intramolecular cyclizations, heterocyclizations, and carbocylizations, see: Toyota M, Sasaki M, Ihara M. Org. Lett. 2003;5:1193–1195. doi: 10.1021/ol034027+. Toyota M, Rudyanto M, Ihara M. J. Org. Chem. 2002;67:3374–3386. doi: 10.1021/jo011169d. Toyota M, Ihara M. Synlett. 2002:1211–1222. Toyota M, Ilangovan A, Okamoto R, Masaki T, Arakawa M, Ihara M. Org. Lett. 2002;4:4293–4296. doi: 10.1021/ol020187u. Toyota M, Odashima T, Wada T, Ihara M. J. Am. Chem. Soc. 2000;122:9036–9037. Toyota M, Rudyanto M, Ihara M. Tetrahedron Lett. 2000;41:8929–8932. Toyota M, Wada T, Ihara M. J. Org. Chem. 2000;65:4565–4570. doi: 10.1021/jo000142b. Toyota M, Wada T, Fukumoto K, Ihara M. J. Am. Chem. Soc. 1998;120:4916–4925. Larock RC, Hightower TR. Synlett. 1998:522–524. Rönn M, Andersson PG, Bäckvall J-E. Tetrahedron Lett. 1997;38:3603–3606. Rönn M, Andersson PG, Bäckvall J-E. Acta Chem. Scand. 1997;51:773–777. Larock RC, Hightower TR, Hasvold LA, Peterson KP. J. Org. Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i. Rönn M, Bäckvall J-E, Andersson PG. Tetrahedron Lett. 1995;36:7749–7752. van Bentham RATM, Hiemstra H, van Leeuwen PWNM, Geus JW, Speckamp WN. Angew. Chem., Int. Ed. Engl. 1995;34:457–460. van Bentham RATM, Hiemstra H, Longarela GR, Speckamp WN. Tetrahedron Lett. 1994;35:9281–9284. van Bentham RATM, Hiemstra H, Michels JJ, Speckamp WN. J. Chem. Soc., Chem. Commun. 1994:357–359. Larock RC, Hightower TR. J. Org. Chem. 1993;58:5298–5300.
- 9.For dehydrosilylation of silyl enol ethers using Pd(II)/O2 in DMSO, see: Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423–2426.
- 10.For oxidative C–C bond cleavage of tertiary alcohols using Pd(II)/O2 in DMSO, see: Park S-B, Cha JK. Org. Lett. 2000;2:147–149. doi: 10.1021/ol991250r.
- 11.(a) Nishimura T, Onoue T, Ohe K, Uemura S. J. Org. Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734. [DOI] [PubMed] [Google Scholar]; (b) Nishimura T, Onoue T, Ohe K, Uemura S. Tetrahedron Lett. 1998;39:6011–6014. [Google Scholar]
- 12.For a review, see: Nishimura T, Ohe K, Uemura S. Synlett. 2004;2:201–216.
- 13.For ring opening of hydroxycyclopropanes, see: Park S-B, Cha JK. Org. Lett. 2000;2:147–149. doi: 10.1021/ol991250r.
- 14.For intramolecular oxidative amination, see: Fix SR, Brice JL, Stahl SS. Angew. Chem., Int. Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b.
- 15.For a Wacker cyclization, see: Trend RN, Ramtohul EM, Ferreira EM, Stoltz BM. Angew. Chem., Int. Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.
- 16.For annulation of indoles, see: Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y.
- 17.(a) ten Brink G-J, Arends IWCE, Hoogenraad M, Verspui G, Sheldon RA. Adv. Synth. Catal. 2003;345:497–505. [Google Scholar]; (b) ten Brink G-J, Arends IWCE, Sheldon RA. Adv. Synth. Catal. 2002;344:355–369. [Google Scholar]; (c) Sheldon RA, Arends IWCE, ten-Brink G-J, Dijksman A. Acc. Chem. Res. 2002;35:774–781. doi: 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]; (d) ten Brink G-J, Arends IWCE, Sheldon RA. Science. 2000;287:1636–1639. doi: 10.1126/science.287.5458.1636. [DOI] [PubMed] [Google Scholar]
- 18.Hallman K, Moberg C. Adv. Synth. Catal. 2001;343:260–263. [Google Scholar]; For a related catalyst, see: Paavola S, Zetterberg K, Privalov T, Csöregh I, Moberg C. Adv. Synth. Catal. 2004;346:237–244.
- 19.Heterogeneous and biphasic Pd catalyst systems have been reported to oxidize alcohols under air atmosphere, see: Gómez-Bengoa E, Noheda P, Echavarren AM. Tetrahedron Lett. 1994;35:7097–7098. Aït-Mohand S, Hénin F, Muzart J. Tetrahedron Lett. 1995;36:2473–2476.
- 20.Schultz MJ, Park CC, Sigman MS. Chem. Commun. 2002:3034–3035. doi: 10.1039/b209344h. [DOI] [PubMed] [Google Scholar]
- 21.For the use of catalytic Pd(II) and NEt3 in oxidative amination, see: Timokhin VI, Anastasi NR, Stahl Shannon S. J. Am. Chem. Soc. 2003;125:12996–12997. doi: 10.1021/ja0362149.
- 22.Steinhoff BA, Fix SR, Stahl SS. J. Am. Chem. Soc. 2002;124:766–767. doi: 10.1021/ja016806w. [DOI] [PubMed] [Google Scholar]
- 23.Steinhoff BA, Stahl SS. Org. Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e. [DOI] [PubMed] [Google Scholar]
- 24.For mechanistic studies and elucidation of the role of base in Pd[(−)sparteine]Cl2-catalyzed oxidative kinetic resolution of secondary alcohols, see: Mandal SK, Sigman MS. J. Org. Chem. 2003;68:7535–7537. doi: 10.1021/jo034717r. ; (b) Mueller JA, Sigman MS. J. Am. Chem. Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n. [DOI] [PubMed] [Google Scholar]; (c) Mueller JA, Jensen DR, Sigman MS. J. Am. Chem. Soc. 2002;124:8202–8203. doi: 10.1021/ja026553m. [DOI] [PubMed] [Google Scholar]; (d) Jensen DR, Pugsley JS, Sigman MS. J. Am. Chem. Soc. 2001;123:7475–7476. doi: 10.1021/ja015827n. [DOI] [PubMed] [Google Scholar]; Simultaneously and independently a related kinetic resolution was reported, see: Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2001;123:7725–7726. doi: 10.1021/ja015791z. ; (f) Bagdanoff JT, Ferreira EM, Stoltz BM. Org. Lett. 2003;5:835–837. doi: 10.1021/ol027463p. [DOI] [PubMed] [Google Scholar]; (g) Bagdanoff JT, Stoltz BM. Angew. Chem., Int. Ed. 2004;43:353–357. doi: 10.1002/anie.200352444. [DOI] [PubMed] [Google Scholar]
- 25.For a review, see: Herrmann WA. Angew. Chem., Int. Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y.
- 26.Jensen DR, Sigman MS. Org. Lett. 2003;5:63–65. doi: 10.1021/ol027190y. [DOI] [PubMed] [Google Scholar]
- 27.Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew. Chem., Int. Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997. [DOI] [PubMed] [Google Scholar]
- 28.The sec-phenethyl alcohol concentration was decreased to 0.45 M, with the relative mole percentages of all other reagents remaining the same These reactions were performed at 50 ± 0.1 °C in benzene, see: See the Supporting Information for details.
- 29.Determined by measurement of the initial oxidation rate of sec-phenethyl alcohol with 0–1 mol % Pd(IiPr)(OAc)2(H2O).
- 30.σ values were obtained from Lowry TH, Richardson KS. Mechanism and Theory in Organic Chemistry. 3rd ed. New York: Harper and Row; 1987. p. 144.
- 31.For a discussion on the magnitude of kinetic isotope effects, see: Westheimer FH. Chem. Rev. 1961;61:265–273.
- 32.Other scenarios have been reported where reactions with nonlinear transition states give high KIE values. The high KIE values were attributed to significant intermolecular force constants. For discussion of this concept, see: Motell EL, Boone AW, Fink WH. Tetrahedron. 1978;34:1619–1626. ; Additionally, for discussion of proton-transfer reactions and KIE, see: O’Ferrall RAM. Chapter 8. In: Caldin EF, Gould V, editors. Proton-Transfer Reactions. NewYork: Chapman & Hall; 1975.
- 33.Collins CJ, Bowman NS. Isotope Effects in Chemical Reactions. New York: Van Nostrand Reinhold Co; 1970. [Google Scholar]
- 34.Andrews L, Wang X, Alikhani ME, Manceron L. J. Phys. Chem. A. 2001;105:3052–3063. [Google Scholar]
- 35.A similar proposal was recently reported, see: Trend RM, Stoltz BM. J. Am. Chem. Soc. 2004;126:4482–4483. doi: 10.1021/ja039551q. ; While the current paper was in review, a computational model of the Pd(II)-catalyzed oxidative kinetic resolution of secondary alcohols using (−)-sparteine was reported. In this study, the authors report that the chloride ion is closely associated with the Pd(II) center (3.0 Å) at the transition state for β-hydride elimination, see: Nielsen RJ, Keith JM, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2004;126:7967–7974. doi: 10.1021/ja031911m.
- 36.Initial calculations performed on both cis- and trans-Pd(IiPr)(OAc) alkoxides (the NHC ligand was truncated) revealed the Pd complex with the alkoxide cis to the carbene ligand was found to be significantly lower in energy than the trans geometric isomer. See the Supporting Information for details.
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Kudin KN, Burant JC, Millam JM, Stratmann RE, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Iyengar S, Petersson GA, Ehara M, Toyota K, Nakatsuji H, Adamo C, Jaramillo J, Cammi R, Pomelli C, Ochterski J, Ayala PY, Morokuma K, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox T, Keith DJ, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 01. Pittsburgh, PA: Gaussian, Inc; 2001. Development Version (Revision B.01) [Google Scholar]
- 38.(a) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; (b) Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]; (c) Becke AD. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 39.(a) Miehlich B, Savin A, Stoll H, Preuss H. Chem. Phys. Lett. 1989;157:200–206. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 40.(a) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270–283. [Google Scholar]; (b) Wadt WR, Hay PJ. J. Chem. Phys. 1985;82:284–298. [Google Scholar]; (c) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:299–310. [Google Scholar]
- 41.Dunning TH, Jr, Hay PJ. In: Modern Theoretical Chemistry. Schaefer HF III, editor. New York: Plenum; 1976. pp. 1–28. [Google Scholar]
- 42.(a) Frenking G, Antes I, Boehme M, Dapprich S, Ehlers AW, Jonas V, Neuhaus A, Otto M, Stegmann R, Veldkamp A, Vyboishchikov SF. Rev. Comput. Chem. 1996;8:63–143. [Google Scholar]; (b) Cundari TR, Benson MT, Lutz ML, Sommerer SO. Rev. Comput. Chem. 1996;8:145–202. [Google Scholar]
- 43.(a) Kohn W, Sham LJ. Phys. Rev. A. 1965;140:1133–1138. [Google Scholar]; (b) Hohenberg P, Kohn W. Phys. Rev. B. 1964;136:864–871. [Google Scholar]
- 44.Peng C, Schlegel HB. Isr. J. Chem. 1993;33:449–454. [Google Scholar]
- 45.No scaling factors were applied to frequency calculations. No solvent corrections were applied.
- 46.Zhang Shaowen, Truong Thanh N. VKLab version 1.0. University of Utah; 2001. [Google Scholar]
- 47.Mulliken charges and APT charges were both calculated The APT charges best correlated with a β-hydride elimination mechanism. Mulliken charges indicated the β-H to be more proton-like; therefore, this analysis was not used.
- 48.For the reversible formation of palladium(II) hydride species under acidic conditions, see: Amatore C, Jutand A, Meyer G, Carelli I, Chiarotto I. Eur. J. Inorg. Chem. 2000:1855–1859.
- 49.Two pathways have been suggested to describe how molecular oxygen re-forms the active Pd catalyst: (1) O2 insertion into the Pd–H bond or (2) reductive elimination of Pd(X)–H to form a Pd(0) species which is reoxidized by O2 to form a palladium peroxo species. The latter of these pathways is supported by the isolation of a palladium peroxo–bathocuproin (bc) complex by Stahl and co-workers formed by exposure of Pd(0)(bc) to molecular oxygen, see: Stahl SS, Thorman JL, Nelson RC, Kozee MA. J. Am. Chem. Soc. 2001;123:7188–7189. doi: 10.1021/ja015683c. ; They demonstrated the palladium peroxo–bc species yields Pd(OAc)2(bc) and hydrogen peroxide when exposed to acetic acid. A related Pd–bathocuproine complex was shown to be an excellent catalyst by Sheldon and co-workers for Pd-catalyzed aerobic oxidation of alcohols in aqueous conditions (see ref 17).
- 50.For a discussion on the nontriviality of Pd(0) turnover, see: Tsuji J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis. New York: John Wiley and Sons; 1995. p. 20. ; For an example of Pd(0) aggregation to precipitate Pd black in the Wacker oxidation, see: Tang HG, Sherrington DC. J. Catal. 1993;142:540, 551.
- 51.For recent examples, see: Dewanti AR, Xu Y, Mitra B. Biochemistry. 2004;43:1883–1890. doi: 10.1021/bi036021y. Dewanti AR, Mitra B. Biochemistry. 2003;42:12893–12901. doi: 10.1021/bi035349o. Waas WF, Rainey MA, Szafranska AE, Dalby KN. Biochemistry. 2003;42:12273–12286. doi: 10.1021/bi0348617. Kimura Y, Toyoshima N, Hirakawa N, Okamoto K, Ishijima A. J. Mol. Biol. 2003;328:939–950. doi: 10.1016/s0022-2836(03)00341-3. Fatin-Rouge N, Blanc S, Leize E, Van Dorsselaer A, Baret P, Pierre J-L, Albrecht-Gary A-M. Inorg. Chem. 2000;39:5771–5778. doi: 10.1021/ic000229f. Bebelis S, Zeritis AS, Tiropani C, Neophytides SG. Ind. Eng. Chem. Res. 2000;39:4920–4927.
- 52.See the Supporting Information for details.
- 53.For the King–Altman method, see: Cornish-Bowden A. Principles of Enzyme Kinetics. London: Butterworth; 1976. pp. 38–40.
- 54.Isaacs N. Physical Organic Chemistry. 2nd ed. Harlow, U.K: Prentice Hall; 1995. pp. 381–384. [Google Scholar]
- 55.For pKa values in water, see: Lide DR, editor. CRC Handbook of Chemistry and Physics. 75th ed. Boca Raton, FL: CRC Press Inc; 1995. ; For the pKa value of trifluoroacetic acid, see: Budavari s., editor. Merck Index. 12th ed. Whitehouse Station, NJ: Merck & Co., Inc; 1996.
- 56.Another consideration is the change in carboxylate ligand may also affect β-hydride elimination since this ancillary ligand is bound to palladium alkoxide C during rate-limiting β-hydride elimination. If the bound carboxylate has a significant effect on β-hydride elimination, electron-withdrawing groups on the carboxylate should increase the electrophilicity of the Pd(II) center. Thus, carboxylic acids with lower pKa values should lead to increases in rate. The Brønsted-type plot at 2 mol % additive carboxylic acid shows the opposite trend where carboxylate ligands with electron-donating groups show increased reaction rates. Thus, the influence of different carboxylate ligands on β-hydride elimination is probably minimal compared to the acid/base effects present.
- 57.Hamilton S, Schultz MJ, Sigman MS. Unpublished results [Google Scholar]