

The crystal structure of adenylate kinase from the psychrophile M. marinus has been determined at 2.0 Å resolution and the kinetic parameters of this cold-adapted enzyme have been examined.
Keywords: psychrophiles, adenylate kinases, phosphotransferases, adaptation
Abstract
Adenylate kinases (AKs; EC 2.7.4.3) are essential members of the NMP kinase family that maintain cellular homeostasis by the interconversion of AMP, ADP and ATP. AKs play a critical role in adenylate homeostasis across all domains of life and have been used extensively as prototypes for the study of protein adaptation and the relationship of protein dynamics and stability to function. To date, kinetic studies of psychrophilic AKs have not been performed. In order to broaden understanding of extremophilic adaptation, the kinetic parameters of adenylate kinase from the psychrophile Marinibacillus marinus were examined and the crystal structure of this cold-adapted enzyme was determined at 2.0 Å resolution. As expected, the overall structure and topology of the psychrophilic M. marinus AK are similar to those of mesophilic and thermophilic AKs. The thermal denaturation midpoint of M. marinus AK (321.1 K) is much closer to that of the mesophile Bacillus subtilis (320.7 K) than the more closely related psychrophile B. globisporus (316.4 K). In addition, the enzymatic properties of M. marinus AK are quite close to those of the mesophilic AK and suggests that M. marinus experiences temperature ranges in which excellent enzyme function over a broad temperature range (293–313 K) has been retained for the success of the organism. Even transient loss of AK function is lethal and as a consequence AK must be robust and be well adapted to the environment of the host organism.
1. Introduction
Adenylate kinases (AKs) from a variety of organisms have proven to be excellent subjects for studies of molecular evolution (Counago et al., 2006 ▶, 2008 ▶) and protein folding (Henzler-Wildman et al., 2007 ▶; Whitford et al., 2007 ▶, 2008 ▶; Bae et al., 2008 ▶; Lu & Wang, 2008 ▶; Arora & Brooks, 2007 ▶; Bae & Phillips, 2004 ▶, 2006 ▶; Wolf-Watz et al., 2004 ▶; Criswell et al., 2003 ▶). AK homologs isolated from extremophiles have been used to elucidate the underlying basis of adaptation as well as to provide new insights for models of protein folding, dynamics and design (Bae et al., 2008 ▶; Whitford et al., 2007 ▶; Bae & Phillips, 2004 ▶; Criswell et al., 2003 ▶; Nguyen et al., 2008 ▶). AK (EC 2.7.4.3) regulates adenylate homeostasis (ATP + AMP ↔ 2ADP) and is essential for proper maintenance of the cellular energy charge ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]) (Atkinson, 1968 ▶). In addition to its metabolic role, AK is a prototypical phosphoryl-transfer enzyme and as such is a good model for the critically important family of kinases implicated in cellular signaling. AK can be broadly described as having a LID domain (residues 128–159) and a CORE (residues 1–30, 60–127 and 160–217) domain.
High-resolution structures exist for representative members of both monomeric and trimeric forms of AK (Bae & Phillips, 2004 ▶; Criswell et al., 2003 ▶; Vonrhein et al., 1998 ▶). Within the monomeric AK family, structures have been determined of AKs from the thermophile Geobacillus stearothermophilus (AKGS; Berry & Phillips, 1998 ▶), the mesophiles Bacillus subtilis (AKBS; Bae & Phillips, 2004 ▶) and Escherichia coli (AKEC; Muller & Schulz, 1998 ▶), and the psychrophile B. globisporus (AKBG; Bae & Phillips, 2004 ▶). While structures across a broad temperature range are represented, the enzyme kinetics for a psychrophilic AK have not been determined. We have solved the structure of Marinibacillus marinus AK (AKMM) at 2.0 Å resolution and have also determined its stability and temperature-dependent kinetic parameters. The structure and kinetic parameters suggest that AKMM has properties that are consistent with an organism that has optimized enzymatic performance over a broader temperature range than the related psychrophile B. globisporus.
M. marinus is a subspecies of B. globisporus (their AKs are 72.8% identical) that was originally isolated from sediment in the North East Atlantic as an obligate halophile with an optimum growth range of 285–296 K (Yoon et al., 2001 ▶; Rüger & Richter, 1979 ▶; Rüger et al., 2000 ▶). Structure–function studies on psychrophilic AKs are quite limited, but comparative studies of mesophilic and thermophilic enzymes suggest strong correlations between protein dynamics and function that are tuned adaptively towards optimal enzyme performance within the environmental niche of the host organism (Counago et al., 2006 ▶; Henzler-Wildman et al., 2007 ▶; Bae & Phillips, 2004 ▶). While no kinetic parameters have been measured for AKBG, the total enzyme activity as a function of temperature has been examined and shows a distinct maximum at 308 K, while AKMM has a much broader activity range that is more similar to those of the mesophilic AKs. Although M. marinus AK has kinetic characteristics that are similar to those of the mesophilic AKBS, the K M for both ATP and AMP shows less temperature-dependence, making this a robust enzyme over temperature ranges below the denaturation temperature of the protein and over the entire growth range of the host organism.
2. Materials and methods
Pyruvate kinase, l-lactate dehydrogenase, phosphoenolpyruvate, β-nicotinamide adenine dinucleotide, reduced disodium salt (NADH), adenosine 5′-triphosphate sodium salt (ATP), adenosine 5′-monophosphate disodium salt (AMP) and P1,P5-di(adenosine-5′)-pentaphosphate pentasodium salt (AP5A) were purchased from Sigma–Aldrich.
2.1. Cloning, expression and purification of AKMM
The gene encoding M. marinus adenylate kinase (AY690426) was amplified from genomic DNA (ATCC 29841) and subcloned into pET11a as pET11a-AKMM. E. coli Rosetta cells containing plasmid pET11a-AKMM were grown at 310 K in LB medium containing 50 µg ml−1 carbenicillin and 25 µg ml−1 chloramphenicol to mid-log phase (OD600 = 0.6–0.7). Prior to induction, the temperature was reduced to 303 K. Expression of AKMM was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 1 mM and the culture was incubated for an additional 17 h. Cells were harvested by centrifugation and the cell lysate was cleared and loaded onto a HiTrap Q-XL Sepharose anion-exchange chromatography column and eluted with a gradient from 0 to 0.5 M NaCl. The pooled fractions were concentrated, dialyzed against 50 mM Tris pH 7.5, 50 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, 0.3 mM DTT and passed over a column containing Affi-Gel Blue resin (Bio-Rad). The absorbed protein was eluted using a gradient from 0.1 to 2.5 M NaCl. Following concentration, AKMM was passed over a HiLoad 16/60 Superdex-200 column (Amersham Biosciences). The sample purity was assessed by SDS–PAGE and found to be >95%.
2.2. AKMM activity assays
Kinetic parameters for the formation of ATP by AKMM were determined at 293, 303 and 313 K by a continuous assay (Girons et al., 1987 ▶; Vieille et al., 2003 ▶). The reaction mixture consisted of reaction buffer (25 mM phosphate buffer pH 7.2, 5 mM MgCl2, 65 mM KCl, 0.06 mM NADH, 0.1 mM phosphoenolpyruvate, five units of both lactate dehydrogenase and pyruvate kinase, 1.4 mM AMP) and various ATP concentrations (0.1, 0.5, 1, 5, 10, 50, 100 and 500 µM). The mixture was kept at the desired temperature for 5 min in a water bath prior to addition of AKMM to a final concentration of 9 nM. A sample of the reaction buffer without the addition of AKMM was used as a control at each temperature. The amount of ADP produced was estimated from the conversion of NADH to NAD+ as indicated by the absorbance at 340 nm (molar extinction coefficient ∊340nm = 6200 M −1 cm−1), subtracting the amount of NADH consumed from the NADH concentration for the control reaction. The kinetic parameters (K M and V max) were estimated in an initial velocity versus [ATP] plot by fitting the data to the Michaelis–Menten equation using Kaleidagraph v.3.51 (Synergy Software). The data shown are the average of two experiments performed in triplicate at each temperature. The enzyme activity of AKMM was also determined at 293, 303 and 313 K for the formation of AMP by varying the AMP concentration (0.1, 0.5, 1, 5, 10, 50, 100 and 500 µM) and maintaining a constant ATP concentration (1.4 mM) using the continuous assay described previously.
2.3. Thermal unfolding (T m) of AKMM
The thermal stability of AKMM and of the complex of AKMM with the transition-state analog AP5A were measured by changes in ellipticity (Jasco J-815 CD) at 220 nm as a function of temperature (0.5° min−1). To minimize the error arising from baseline corrections, the thermal unfolding midpoint (T m) was calculated using the first derivative of the CD signal versus temperature (John & Weeks, 2000 ▶). The first derivative was obtained using a simple differential of the CD signal [f(T) = ΔCD/ΔT]. The position of the peak maximum was used to determine the thermal unfolding midpoint (T m). Data were acquired in 10 mM potassium phosphate pH 7.2, 65 mM KCl and then converted into a plot of fraction unfolded versus temperature. Scans were performed from 293 to 363 K for AKMM and from 293 to 358 K for the complex with AP5A (100 µM). The data shown are the average of two independent experiments performed in triplicate.
2.4. Crystallization and structure determination of AKMM
Preliminary AKMM crystallization trials were performed by the sitting-drop vapour-diffusion method using a sparse-matrix crystallization approach (Jancarik & Kim, 1991 ▶). 0.5 µl protein solution (15 mg ml−1) with 9 mM AP5A in 10 mM HEPES pH 7.0 and 0.5 µl mother liquor were mixed in a 96-well plate using a Hydra II Plus One crystallization robot (Matrix Technology) and incubated at 283 K. The first crystals of AKMM were obtained in 1.6 M sodium citrate pH 6.5. This condition was successively modified using streak-seeding with crushed crystals and additive screening, resulting in crystals that were suitable for data collection. The final crystallization condition was 1.5 M sodium citrate pH 6.5, 150 mM sodium chloride, 0.5% n-dodecyl-N,N-dimethylamine-N-oxide at 283 K.
Crystals of AKMM were passed briefly through cryoprotectant solutions consisting of 1.5 M sodium citrate pH 6.5, 150 mM sodium chloride, 0.5% n-dodecyl-N,N-dimethylamine-N-oxide supplemented with 5, 10, 15 and 20%(v/v) ethylene glycol. Diffraction data were collected to 2.0 Å resolution on a Rigaku R-AXIS IV++ image-plate detector system with Varimax HF optics. All data were collected at 100 K using a nitrogen cryostream. The crystals belonged to space group C2, with unit-cell parameters a = 92.6, b = 46.5, c = 62.7 Å, α = γ = 90.0, β = 98.7°. The data were processed and merged using the program d*TREK (Pflugrath, 1999 ▶).
The structure of AKMM was solved by molecular replacement with the program Phaser from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) using only the protein coordinates of AKBG (PDB code 1s3g) as a search model (Bae & Phillips, 2004 ▶). The initial solution suggested the presence of one monomer per asymmetric unit, consistent with a Matthews coefficient of 2.78 Å3 Da−1 (56% solvent). The molecular-replacement solution was further confirmed by the initial composite omit maps generated using CNS (Brünger et al., 1998 ▶), which clearly indicated strong electron density for AP5A and the structural zinc ion that were not included in the original search model. Refinement (20.0–2.0 Å) was carried out in REFMAC5 (Murshudov et al., 1997 ▶) using standard restraints. After iterative refinement and model building, water molecules were added using the Coot find water function in REFMAC5. The AKMM model was refined to convergence, resulting in an R factor of 18.0% and an R free of 22.6% to 2.0 Å resolution. Ramachandran plots and root-mean-square deviations from ideality for bond angles and lengths for AKMM were determined using PROCHECK v.3.5 (Laskowski et al., 1996 ▶) and the structure-validation program MolProbity (Davis et al., 2007 ▶). All figures containing molecular structures were generated using PyMOL (DeLano, 2002 ▶) and CCP4MG (Potterton et al., 2002 ▶, 2004 ▶). The structure of AKMM has been deposited in the RCSB Protein Data Bank with code 3fb4.
3. Results
3.1. Enzyme kinetics for AKMM
Kinetic parameters for the utilization of ATP and AMP were determined at 293, 303 and 313 K for AKMM using a continuous assay (Girons et al., 1987 ▶; Vieille et al., 2003 ▶). The temperature-dependence of the steady-state kinetic parameters (K M and V max) for ATP are shown in Fig. 1 ▶(a) and summarized in Table 1 ▶. AKMM displayed a fairly constant V max over the range 293–303 K but a modest twofold increase from 295 to 313 K. The K M values increased slightly over the same temperature range. The reaction patterns of AKMM for AMP at the indicated temperatures are shown in Fig. 1 ▶(b) and are summarized in Table 1 ▶. The temperature-dependence of K M for AMP utilization was less than that of ATP, but the overall K M for ATP (2–5 µM) is consistently lower than that for AMP (21–23 µM). The apparent difference in the observed k cat for ATP and AMP may be the result of AMP inhibition at higher concentrations, as AMP is able to bind at the ATP-binding site (Sheng et al., 1999 ▶).
Figure 1.

(a) ATP-dependent reaction kinetics for the production of ADP by AKMM from 293 to 313 K. ATP was used as the variable substrate at concentrations of 0.1, 0.5, 1, 5, 10, 50, 100 and 500 µM at each temperature. The concentrations of AKMM (9 nM) and MgAMP (1400 µM) were kept constant. The kinetic constants determined from these experiments are summarized in Table 1 ▶. (b) AMP-dependent reaction kinetics for the production of ADP by AKMM from 293 to 313 K. AMP was used as the variable substrate at concentrations of 0.1, 0.5, 1, 5, 10, 50, 100 and 500 µM at each temperature. The concentrations of AKMM (9 nM) and MgATP (1400 µM) were kept constant. The kinetic constants determined from these experiments are summarized in Table 1 ▶. (c) Thermal denaturation midpoints (T m) of AKMM and the AKMM–AP5A complex from circular dichroism. The midpoint of the transition for unfolding of AKMM at 20 µM occurs at 321.1 K, while the midpoint of unfolding for the AKMM–AP5A complex is 340.6 K, 19.5 K higher than for the free protein. (d) Temperature profile for the activity of M. marinus and Bacillus AKs. The temperature–activity profile for AKMM has a much broader temperature range that is more similar to that of the mesophilic AK. The data for B. globisporus and B. subtilis AKs were taken from Bae & Phillips (2004 ▶).
Table 1. Kinetic parameters for M. marinus AK at various temperatures.
| KM (µM) | Vmax (µM min−1 per nM of protein) | |||
|---|---|---|---|---|
| Temperature (K) | ATP | AMP | ATP | AMP |
| 295 | 2.0 ± 0.3 | 21.4 ± 6.7 | 0.24 ± 0.01 | 2.2 ± 0.2 |
| 303 | 1.8 ± 0.2 | 24.0 ± 7.0 | 0.22 ± 0.01 | 2.6 ± 0.2 |
| 313 | 5.0 ± 0.3 | 21.6 ± 4.0 | 0.72 ± 0.01 | 3.4 ± 0.2 |
3.2. Thermal denaturation followed by circular dichroism (CD)
Thermal stability was assessed using CD by following the changes in molar ellipticity with increasing temperature at 220 nm. As shown in Fig. 1 ▶(c), the midpoint of the transition for AKMM unfolding occurs at 321.1 K. CD experiments at concentrations of 2, 20 and 40 µM showed no concentration-dependence, which is consistent with the expected monomeric solution state. The stability of the ligand-bound closed conformation was estimated in the presence of 100 µM AP5A. It has previously been shown that AKBG, AKBS and AKGS are greatly stabilized when bound to AP5A (Counago et al., 2008 ▶; Bae & Phillips, 2004 ▶). Likewise, the midpoint of unfolding for AKMM–AP5A is 19.5 K higher than that of the free protein.
3.3. Structural overview of AKMM
The overall structure and topology of AKMM closely resemble those of AKBG, AKBS and AKGS, consistent with the strong sequence identity (72.8, 70.5 and 71.9% respectively) observed across this family of proteins (Fig. 2 ▶). Like other AKs, AKMM is a member of the α/β class, with a central β-sheet region surrounded by several α-helices that produce the characteristic AMP-binding, LID and CORE domains. Primary sequence alignment of the two most closely related family members, AKMM and AKBG, shows that the C-terminal region (residues 184–216) is the most divergent. AKs from Gram-positive species including AKMM also contain a conserved-sequence zinc-binding motif (Cys-X 2-Cys-X 16-Cys-X 2-Cys/Asp; Fig. 3 ▶ a; Bae & Phillips, 2004 ▶; Gilles et al., 1994 ▶; Glaser et al., 1992 ▶).
Figure 2.

Primary sequence alignment of M. marinus AK and Bacillus AKs. The sequences were aligned using the program ClustalW and represented using the program BOXSHADE (Thompson et al., 1994 ▶). AKMM shares high amino-acid sequence identity with other Bacillus AKs, including those of the psychrophile B. globisporus (AKBG; 72.8%), the mesophile B. subtilis (AKBS; 70.5%) and the thermophile G. stearothermophilus (AKGS; 71.9%).
Figure 3.

(a) Structure of psychrophilic M. marinus AK. The bound Zn2+ ion is shown as a gray sphere in the LID domain of the enzyme. The bound AP5A molecule (green) spans the active site of the enzyme. The LID domain (red) is in the fully closed conformation. (b) Electron density for bound Zn2+ and surrounding ligands in AKMM. The coordination of Zn2+ (purple) to AKMM is clearly visible in the (2F o − F c) σA-weighted electron-density map contoured at 1.5σ. The bound Zn2+ is coordinated to Cys130, Cys133, Cys150 and Asp153 in a manner similar to that in other Gram-positive AKs.
The crystal structure of AKMM was solved to a resolution of 2.0 Å and then refined to an R factor of 18.0% and an R free of 22.6% (Table 2 ▶). AKMM was crystallized with the inhibitor AP5A bound to the active site. Superimposition of the Cα atoms of AKMM and AKBG shows that the structures are very similar, with an overall r.m.s.d. of 0.58 Å. A comparison of temperature factors suggests that AKMM (2.0 Å resolution; average main-chain B factor of 16.4 Å2) is well ordered compared with AKBG (2.25 Å resolution; average main-chain B factor of 49.5 Å2) and AKBS (1.90 Å resolution; average main-chain B factor of 27.7 Å2), although the static disorder of the crystal also contributes to this. In order to analyze the flexibility of these enzymes and minimize the issues associated with crystal quality, data collection or refinement, the relative B factors were calculated by taking the mean B factor of every residue and dividing it by the mean B factor of the whole protein (Violot et al., 2005 ▶). In this analysis, about 55% of the residues in AKBG showed higher relative B factors than those in AKMM, whereas only 10% of the residues displayed a lower relative temperature factor. These results suggested an increase in the flexibility of AKBG, which in general correlates with the lower thermostability. In contrast, the relative B factors for AKBS and AKGS showed a significantly smaller number of residues with higher flexibility and a larger number with lower flexibility, consistent with their thermostability (46% higher and 25% lower for AKBS and 39% higher and 25% lower for AKGS). In addition, the average local structural entropy (LSE) values of AKBG, AKMM and AKBS were calculated for the CORE domains (Chan et al., 2004 ▶). AKMM has a lower average LSE value than that of AKBG, which correlates with the higher thermostability and highlights the importance of entropy in the stability of protein structures.
Table 2. Data-collection and refinement statistics for the M. marinus AK structure.
Values in parentheses are for the last shell.
| Data collection | |
| Wavelength (Å) | 1.5418 |
| Resolution (Å) | 33.42–2.00 (2.07–2.00) |
| Space group | C2 |
| Molecules per ASU | 1 |
| Unit-cell parameters (Å, °) | a = 92.6, b = 46.5, c = 62.7, α = γ = 90.0, β = 98.7 |
| Unique reflections | 17786 (1713) |
| Average redundancy | 2.81 (2.46) |
| Completeness (%) | 98.7 (96.9) |
| Rmerge† (%) | 2.9 (8.0) |
| Output 〈I/σ(I)〉 | 24.0 (9.4) |
| Refinement | |
| Rwork‡ (%) | 18.0 (21.2) |
| Rfree§ (%) | 22.6 (27.0) |
| R.m.s.d.¶ from ideality | |
| Bonds (Å) | 0.019 |
| Angles (°) | 1.9 |
| Average B factor (Å2) | 19.12 |
| Wilson plot B factor (Å2) | 7.9 |
| Ramachandran plot†† | |
| Most favored regions (%) | 96.7 |
| Additional allowed regions (%) | 3.3 |
| PDB code | 3fb4 |
R
merge = 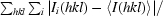
 , where I(hkl) is the measured intensity for reflections with indices hkl.
, where I(hkl) is the measured intensity for reflections with indices hkl.
R
work = 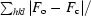
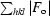 for all data with F
o > 2σ(F
o), excluding data used to calculate R
free.
for all data with F
o > 2σ(F
o), excluding data used to calculate R
free.
R
free =
for all data with F
o > 2σ(F
o) that were excluded from refinement.
Root-mean-square deviation.
Calculated using PROCHECK (Laskowski et al., 1996 ▶).
The LID domain of AKMM is fully closed, as observed in the structures of AKBS and AKBG. The bound metal modeled as a Zn2+ is clearly evident in the LID domain of our structure and is coordinated to Cys130, Cys133, Cys150 and Asp153 (Fig. 3 ▶ b). The carboxylate of Asp153 shows a bidentate coordination to the Zn2+. As observed in AKBG, one of the interactions from the O atom of the bidentate ligand Asp153 is longer than the other (Bae & Phillips, 2004 ▶; Alberts et al., 1998 ▶). The inhibitor AP5A is bound occupying both the ATP/ADP and AMP/ADP sites, as has been observed in the AKBG, AKBS and AKGS complexes.
4. Discussion
The AK family of proteins has emerged as a prototype for the study of protein folding, adaptive protein evolution, dynamics and design (Counago et al., 2006 ▶, 2008 ▶; Henzler-Wildman et al., 2007 ▶; Whitford et al., 2007 ▶, 2008 ▶; Bae et al., 2008 ▶; Lu & Wang, 2008 ▶; Arora & Brooks, 2007 ▶; Bae & Phillips, 2004 ▶, 2006 ▶; Wolf-Watz et al., 2004 ▶; Criswell et al., 2003 ▶; Nguyen et al., 2008 ▶). AKs have been isolated from a range of extremophilic and temperate organisms and serve as a living record of adaptation, providing insights into protein evolution. In order to expand our understanding of extremophilic adaptation, we have determined the structure and elucidated the enzymatic properties of an AK from the psychrophile M. marinus. Surprisingly, the enzymatic properties of AKMM are quite close to those of mesophiles such as B. subtilis and this suggests that M. marinus experiences temperature ranges in which excellent enzyme function over a broad temperature range (293–313 K) has been retained for the success of the organism.
We compared the activity profile and structure of AKMM with those of B. globisporus and B. subtilis. M. marinus is a closely related psychrophilic subspecies of B. globisporus and therefore we expected AKMM to have quite comparable properties and structure to those of AKBG. Although no detailed kinetics are available for the psychrophilic AKBG, Bae & Phillips (2004 ▶) showed that the total activity of AKBG was maximal at 308 K and decreased by ∼30% from 308 to 313 K. AKMM shows no loss in activity over the same range (Fig. 1 ▶ d). In addition, the denaturation temperature of AKMM (321.1 K) is quite close to that of AKBS (320.7 K) (Bae & Phillips, 2004 ▶; our data). The relative magnitude of the K M for ATP (2–5 µM) is about tenfold lower than that for AMP (21–23 µM), while those of AKBS are nearly equal over a comparable temperature range (∼10 and 11–26 µM, respectively; Counago et al., 2008 ▶). The K M for AMP of AKMM displayed less temperature-dependence than that of AKBS, although in the case of AKBS only a modest threefold change is observed over a comparable temperature range (293–313 K; Counago et al., 2008 ▶).
Our findings suggest that AKMM is a broad-range adenylate kinase that in many respects is more consistent with a mesophilic enzyme than might have been expected from previous work on the related psychrophilic B. globisporus AKBG. M. marinus grows over a slightly broader temperature range (274–302 K) than B. globisporus (271–296 K) and suggests that the organism may require an AK with a broader functional range in order to deal with a more challenging environment. Previous work has shown that even transient loss of AK function in cells leads to an irreversible loss of viability (Counago & Shamoo, 2005 ▶) and therefore AK function may be well buffered for the entire range of temperatures an organism might experience in nature.
Supplementary Material
PDB reference: adenylate kinase, 3fb4, r3fb4sf
Acknowledgments
This work was supported by grants from the National Science Foundation (0641792) and The Robert A. Welch Foundation (C-1584). The Rice University Crystallographic Core Facility is supported by a Kresge Science Initiative endowment grant.
References
- Alberts, I. L., Nadassy, K. & Wodak, S. J. (1998). Protein Sci.7, 1700–1716. [DOI] [PMC free article] [PubMed]
- Arora, K. & Brooks, C. L. (2007). Proc. Natl Acad. Sci. USA, 104, 18496–18501. [DOI] [PMC free article] [PubMed]
- Atkinson, D. E. (1968). Biochemistry, 7, 4030–4034. [DOI] [PubMed]
- Bae, E., Bannen, R. M. & Phillips, G. N. Jr (2008). Proc. Natl Acad. Sci. USA, 105, 9594–9597. [DOI] [PMC free article] [PubMed]
- Bae, E. & Phillips, G. N. Jr (2004). J. Biol. Chem.279, 28202–28208. [DOI] [PubMed]
- Bae, E. & Phillips, G. N. Jr (2006). Proc. Natl Acad. Sci. USA, 103, 2132–2137. [DOI] [PMC free article] [PubMed]
- Berry, M. B. & Phillips, G. N. Jr (1998). Proteins, 32, 276–288. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chan, C. H., Liang, H. K., Hsiao, N. W., Ko, M. T., Lyu, P. C. & Hwang, J. K. (2004). Proteins, 57, 684–691. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Counago, R., Chen, S. & Shamoo, Y. (2006). Mol. Cell, 22, 441–449. [DOI] [PubMed]
- Counago, R. & Shamoo, Y. (2005). Extremophiles, 9, 135–144. [DOI] [PubMed]
- Counago, R., Wilson, C. J., Pena, M. I., Wittung-Stafshede, P. & Shamoo, Y. (2008). Protein Eng. Des. Sel.21, 19–27. [DOI] [PubMed]
- Criswell, A. R., Bae, E., Stec, B., Konisky, J. & Phillips, G. N. Jr (2003). J. Mol. Biol.330, 1087–1099. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, California, USA.
- Gilles, A. M., Glaser, P., Perrier, V., Meier, A., Longin, R., Sebald, M., Maignan, L., Pistotnik, E. & Bârzu, O. (1994). J. Bacteriol.176, 520–523. [DOI] [PMC free article] [PubMed]
- Girons, I. S., Gilles, A. M., Margarita, D., Michelson, S., Monnot, M., Fermandjian, S., Danchin, A. & Barzu, O. (1987). J. Biol. Chem.262, 622–629. [PubMed]
- Glaser, P., Presecan, E., Muriel, D., Witold, K., Surewicz, W. K., Mantsch, H. H., Barzu, O. & Gilles, A. N. (1992). Biochemistry, 31, 3038–3043. [DOI] [PubMed]
- Henzler-Wildman, K. A., Lei, M., Thai, V., Kerns, J. S., Karplus, M. & Kern, D. (2007). Nature (London), 450, 913–916. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- John, D. M. & Weeks, K. M. (2000). Protein Sci.9, 1416–1419. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., Rullmann, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. (1996). J. Biomol. NMR, 8, 477–486. [DOI] [PubMed]
- Lu, Q. & Wang, J. (2008). J. Am. Chem. Soc.130, 4772–4783. [DOI] [PubMed]
- Muller, C. W. & Schulz, G. E. (1998). J. Mol. Biol.202, 909–912. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nguyen, P. Q., Liu, S., Thompson, J. C. & Silberg, J. J. (2008). Protein Eng. Des. Sel.21, 303–310. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Potterton, E., McNicholas, S., Krissinel, E., Cowtan, K. & Noble, M. (2002). Acta Cryst. D58, 1955–1957. [DOI] [PubMed]
- Potterton, L., McNicholas, S., Krissinel, E., Gruber, J., Cowtan, K., Emsley, P., Murshudov, G. N., Cohen, S., Perrakis, A. & Noble, M. (2004). Acta Cryst. D60, 2288–2294. [DOI] [PubMed]
- Rüger, H. J., Fritze, D. & Sproer, C. (2000). Int. J. Syst. Evol. Microbiol.50, 1305–1313. [DOI] [PubMed]
- Rüger, H. J. & Richter, G. (1979). Int. J. Syst. Bacteriol.29, 196–203.
- Sheng, X. R., Li, X. & Pan, X. M. (1999). J. Biol. Chem.274, 22238–22242. [DOI] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gilbson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Vieille, C., Krishnamurthy, H., Hyun, H. H., Savchenko, A., Yan, H. & Zeikus, J. G. (2003). Biochem. J.372, 577–585. [DOI] [PMC free article] [PubMed]
- Violot, S., Aghajari, N., Czjzek, M., Feller, G., Sonan, G. K., Gouet, P., Gerday, C., Haser, R. & Receveur-Brechot, V. (2005). J. Mol. Biol.348, 1211–1224. [DOI] [PubMed]
- Vonrhein, C., Bonisch, H., Schafer, G. & Schulz, G. E. (1998). J. Mol. Biol.282, 167–179. [DOI] [PubMed]
- Whitford, P. C., Gosavi, S. & Onuchic, J. N. (2008). J. Biol. Chem.283, 2042–2048. [DOI] [PubMed]
- Whitford, P. C., Miyashita, O., Levy, Y. & Onuchic, J. N. (2007). J. Mol. Biol.366, 1661–1671. [DOI] [PMC free article] [PubMed]
- Wolf-Watz, M., Thai, V., Henzler-Wildman, K. A., Hadjipavlou, G., Eisenmesser, E. Z. & Kern, D. (2004). Nature Struct. Mol. Biol.11, 945–949. [DOI] [PubMed]
- Yoon, J. H., Weiss, N., Lee, K. C., Lee, I. S., Kang, K. H. & Park, Y. H. (2001). Int. J. Syst. Evol. Microbiol.51, 2087–2093. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: adenylate kinase, 3fb4, r3fb4sf
PDB reference: adenylate kinase, 3fb4, r3fb4sf