

The crystal structure of the choline-binding domain of Spr1274 (residues 44–129) has been solved at 2.38 Å resolution with three molecules in the asymmetric unit.
Keywords: Spr1274, Streptococcus pneumoniae, choline-binding domains
Abstract
Spr1274 is a putative choline-binding protein that is bound to the cell wall of Streptococcus pneumoniae through noncovalent interactions with the choline moieties of teichoic and lipoteichoic acids. Its function is still unknown. The crystal structure of the choline-binding domain of Spr1274 (residues 44–129) was solved at 2.38 Å resolution with three molecules in the asymmetric unit. It may provide a structural basis for functional analysis of choline-binding proteins.
1. Introduction
Choline is an important component of the eukaryotic cell membrane and is also found in a few prokaryotes (Garcia et al., 1998 ▶). It is not an essential nutrient for most bacteria. However, Streptococcus pneumoniae, the leading pathogen of acute respiratory infections, has an absolute nutritional requirement for choline (Garcia et al., 1998 ▶), which is covalently linked to teichoic acids and lipoteichoic acids on the cell surface (Tomasz, 1967 ▶). The presence of choline in the bacterial cell wall had been considered to be a unique characteristic of S. pneumoniae. However, it has recently also been found in other bacteria, for example S. oralis (Horne & Tomasz, 1993 ▶), S. mitis (Gillespie et al., 1996 ▶), Clostridium beijerinckii (Sanchez-Beato & Garcia, 1996 ▶), C. acetobutylicum (Sanchez-Beato et al., 1995 ▶) and others. Nevertheless, unlike S. pneumoniae, these bacteria do not need choline for growth.
In S. pneumoniae, the choline moieties can be specifically recognized by a series of surface proteins, the so-called choline-binding proteins (Gosink et al., 2000 ▶), which include cell murein hydrolases (LytA, LytB, LytC etc.; Lopez & Garcia, 2004 ▶) and other virulence factors involved in cellular adhesion and colonization (Rosenow et al., 1997 ▶). All choline-binding proteins are typical modular proteins with one functional domain and one or two choline-binding domains (CBDs) responsible for the noncovalent anchoring of choline-binding proteins to the choline moieties on the cell surface. The CBD is also referred to the choline-binding module (Martinez-Buey et al., 2007 ▶) and consists of several repeats of ∼20 residues called choline-binding repeats (ChBRs). The CBD, which was first found in S. pneumoniae (Garcia et al., 1998 ▶), has been identified in various proteins from a series of organisms, including CspA from C. beijerinckii, toxins A and B from C. difficile, glucan-binding protein from S. mutans, glycosyltransferases from both S. mutans and S. downei (Gosink et al., 2000 ▶) and Cpl-1 from pneumococcal bacteriophage (Hermoso et al., 2003 ▶).
The structure of CBD was first presented in the crystal structure of S. pneumoniae C-LytA (Fernandez-Tornero et al., 2001 ▶). To date, several structures of choline-binding proteins, including those of Cpl-1 (Hermoso et al., 2003 ▶), CbpF (Molina et al., 2009 ▶) and CbpE (Hermoso et al., 2005 ▶), have been solved. Their CBDs share a similar overall structure consisting of a peculiar β-solenoid. The major difference comes from their variable lengths; they are made up of different numbers of ChBRs (ten ChBRs in CbpE, eight in CbpF and six in C-LytA and Cpl-1).
The protein Spr1274 is a putative choline-binding protein encoded by S. pneumoniae R6. Sequence analysis shows that it contains a domain of unknown function at the N-terminus (residues 1–39) and a CBD consisting of three canonical ChBRs at the C-terminus (residues 51–129) (Fig. 1 ▶ a). We failed to obtain a crystal of the full-length protein. However, we solved the crystal structure of Spr1274-CBD (residues 44–129) at 2.38 Å resolution. Three molecules were present in the asymmetric unit, forming a pseudo-trimer. Our results may provide a structural basis for functional analysis of choline-binding proteins.
Figure 1.

(a) Sequence alignment of Spr1274 and the CBD of CbpF. The alignment was performed using ClustalX (Thompson et al., 1994 ▶) and ESPript (Gouet et al., 1999 ▶). The secondary-structure elements of Spr1274 are identified. The constituent domains of Spr1274 are displayed at the top of the alignment, with the functional domain in blue and the ChBRs in cyan or yellow (ChBR4 is not a canonical repeat). Residues involved in choline binding are indicated below with filled triangles (ChBS1, green; ChBS2, blue; ChBS3, yellow). (b) The overall structure of Spr1274-CBD (chain B). Secondary-structure elements are labelled (β-strands, turns and loops). The choline molecules (yellow) and residues (cyan) involved in choline binding are labelled and shown in stick representation.
2. Materials and methods
2.1. Protein expression and purification
The open reading frame (ORF) encoding Spr1274/AE008498 from S. pneumoniae strain R6 was first cloned into plasmid pLIM01 (Attali et al., 2008 ▶). The coding sequence of Spr1274-CBD (residues 44–129) was then amplified by PCR using the plasmid pLIM01 as the template. The product was cloned into a pET28a-derived expression vector between NotI and NdeI restriction sites with an N-terminal 6×His tag. The sequence of the construct was confirmed by DNA sequencing. Escherichia coli BL21 (DE3) cells (Novagen) was transformed with the plasmid pET28a-Spr1274-CBD and cultured at 310 K in LB medium containing 30 µg ml−1 kanamycin. Expression of the protein was induced for 4 h at 310 K by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.2 mM when the OD600 reached 0.6. Cells were harvested by centrifugation at 4000g for 10 min and resuspended in binding buffer (200 mM NaCl, 20 mM Tris–HCl pH 8.0, 100 mM choline chloride). After three cycles of freeze–thawing followed by sonication on ice, the lysate was clarified by centrifugation at 29 300g for 25 min at 277 K. The protein was purified using an Ni-affinity column (Qiagen) followed by size-exclusion chromatography (Amersham Biosciences). The purity of the protein was checked by SDS–PAGE. After exchange into storage buffer (50 mM NaCl, 20 mM Tris–HCl pH 8.0, 10 mM choline chloride, 14 mM β-mercaptoethanol), the protein was concentrated to 15 mg ml−1 using an Amicon Ultra 5 kDa cutoff concentrator (Millipore).
2.2. Crystallization, X-ray data collection, structure determination and refinement
The initial crystallization conditions for Spr1274-CBD were obtained from Crystal Screens I and II (Hampton Research) using the sitting-drop vapour-diffusion method at 291 K. Each drop contained 1 µl reservoir solution and 1 µl protein sample and was equilibrated against 100 µl reservoir solution. After optimization using the hanging-drop vapour-diffusion method, crystals appeared within one week at 291 K in a drop comprising a mixture of 4 µl protein solution (15 mg ml−1) and 2 µl reservoir solution containing 1.75 M ammonium sulfate, 0.1 M MES pH 6.5, 10% dioxane and reached maximal dimensions of 200 × 200 × 200 µm.
The Spr1274-CBD crystal was flash-frozen in liquid nitrogen using a cryoprotectant consisting of the reservoir solution supplemented with 20%(v/v) glycerol. Diffraction data were collected from a crystal cooled to 100 K in a liquid-nitrogen stream using a Rigaku MM007 X-ray generator (λ = 1.5418 Å) and a MAR Research 345 image-plate detector at the School of Life Sciences, University of Science and Technology of China (USTC, Hefei, China). The data were processed with MOSFLM (Leslie, 2006 ▶) and scaled using SCALA from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The crystal belonged to space group H3 with three molecules in the asymmetric unit. The structure was determined by the molecular-replacement method using the 1.67 Å resolution crystal structure of the choline-binding domain of CbpF (Molina et al., 2009 ▶; PDB code 2v05; 57.5% sequence homology) as the initial search model with the program MOLREP (Vagin & Isupov, 2001 ▶) in CCP4i (Potterton et al., 2003 ▶). The data set was severely anisotropic, with diffraction to 2.38 Å resolution along the a* and b* directions but only to 2.80 Å resolution along the c* direction. For this reason, the data were truncated along c* at 2.80 Å resolution and scaled with the diffraction anisotropy server (http://www.doe-mbi.ucla.edu/~sawaya/anisoscale/). After refinement with REFMAC5 (Murshudov et al., 1997 ▶; Potterton et al., 2003 ▶), the structure was finally refined to 2.38 Å resolution with an R factor of 22.3% and R free of 26.5%. PROCHECK (Potterton et al., 2003 ▶) indicated that 98.1% of the residues are in the most favoured regions and the rest are in allowed regions. Data-collection and refinement statistics are listed in Table 1 ▶. The final coordinates and structure factors have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb) under accession code 3hia.
Table 1. Crystal parameters, data collection and structure refinement.
Values in parentheses are for the highest resolution bin.
| Data processing | |
| Space group | H3 |
| Unit-cell parameters (, ) | a = b = 140.610, c = 46.050, = = 90, = 120 |
| Resolution range () | 43.072.38 (2.472.38) |
| Unique reflections | 12995 (1089) |
| Unique reflections after truncation | 11503 (406) |
| Completeness (%) | 94.7 (80.0) |
| Completeness after truncation (%) | 83.2 (51.4) |
| I/(I) | 21.72 (5.40) |
| R merge † (%) | 6.1 (20.9) |
| Redundancy | 3.7 |
| Refinement statistics | |
| Resolution range () | 20.002.38 (2.472.38) |
| R factor‡/R free § (%) | 22.3/26.5 (33.5/35.7) |
| No. of protein atoms | 1896 |
| No. of water atoms | 114 |
| R.m.s.d.¶ bond length () | 0.011 |
| R.m.s.d. bond angles () | 1.245 |
| Average of B factors (2) | 34.54 |
| Ramachandran plot†† | |
| Most favoured (%) | 98.1 |
| Additional allowed (%) | 1.9 |
| Outliers (%) | 0 |
| PDB code | 3hia |
R
merge = 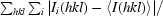
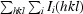 , where I
i(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the mean value for reflection hkl; summations are over all reflections.
, where I
i(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the mean value for reflection hkl; summations are over all reflections.
R factor = 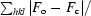
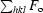 , where F
o and F
c are the observed and calculated structure-factor amplitudes, respectively.
, where F
o and F
c are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated with 5% of the data excluded from the refinement.
Root-mean-square deviation from ideal values.
Categories were defined by MolProbity.
3. Results and discussion
3.1. Overall structure
The final model contains three molecules in the asymmetric unit. All residues of chain B could be fitted well into the electron-density map, whereas the C-terminal regions of both chains A and C were disordered. Residues Asn114–Thr115 and residues Tyr119–Gly126 of chain C were incorporated into the final map with low occupancy.
The overall structure of Spr1274-CBD resembles those of other CBDs of known structure. Spr1274-CBD and the corresponding region of CbpF-CBD share a sequence identity of 57.5% and the average r.m.s.d. between their main-chain atoms is about 1.2 Å. Spr1274-CBD is formed by three canonical ChBRs (ChBR1, residues 52–71; ChBR2, residues 72–92; ChBR3, residues 93–112) and a C-terminal tail (residues 113–129) (Fig. 1 ▶ a). Each ChBR comprises a β-hairpin followed by a long connecting loop. The four consecutive hairpins are strictly arranged to form a left-handed superhelical fold with the shape of a triangular prism. Although it lacks the conserved motif (GWXK…WYY-ϕ…GXM…, where X represents any residue and ϕ represents a hydrophobic residue) of ChBRs, the C-terminus still follows the superhelix and thus is considered to be ChBR4 of Spr1274. A similar C-terminal tail has also been found in many other choline-binding proteins such as CbpE (Hermoso et al., 2005 ▶) and CbpF (Molina et al., 2009 ▶).
3.2. Choline-binding sites
Choline has been considered to be a determinant element for the quaternary structure of CBDs (Fernandez-Tornero et al., 2001 ▶). Similar to other choline-binding proteins of known structure, the choline-binding sites (ChBSs) are located on the three lateral faces of superhelix. Each ChBS is formed by the interface between consecutive hairpin pairs: ChBR1 and ChBR2 for ChBS1, ChBR2 and ChBR3 for ChBS2 and ChBR3 and ChBR4 for ChBS3 (Fig. 1 ▶). In the final map of Spr1274-CBD, with the exception of ChBS3 of chain A and B, the choline-binding sites of the three chains clearly present choline molecules. However, only the trimethylamine head of choline can be found in most ChBSs owing to degradation of choline chloride, with the exception of ChBS2 of chain A, which presents a full choline molecule. Three methyl groups of choline fill a hydrophobic pocket constituted by three conserved aromatic residues from the hairpins of the ChBSs (Trp53, Trp60, Tyr81 in ChBS1; Trp73, Trp80, Tyr102 in ChBS2; Trp94, Trp101, Tyr119 in ChBS3) plus a hydrophobic residue (ChBS1, Met89; ChBS2, Leu110; ChBS3, Trp127) from the following connecting loop (Fig. 1 ▶). The cation–π interaction between the positive charge of the quaternary amine of choline and the electron-rich systems of the aromatic rings enhances the binding by electrostatic interactions (Fernandez-Tornero et al., 2001 ▶). Because ChBS3 is the last choline-binding site, ChBR4 does not need to contain the two conserved tryptophans for the next ChBS. Thus, the lack of hydrophobic residues in ChBR4 does not affect its role in binding a choline molecule.
3.3. Interactions between monomers in an asymmetric unit
Three chains in one asymmetric unit form an interesting noncanonical pseudo-trimer (Fig. 2 ▶ a). Chains A and C are antiparallel and interact with the two termini of chain B. No common threefold axis can be found in the pseudo-trimer, but two twofold axes are present between chains A and B and between chains A and C (Fig. 2 ▶ b and 2 ▶ c). Chains A and B interact with each other through their ChBR1–ChBR2 regions, including loop0, loop1 and β3, which bury an interface of ∼611.6 Å2. The overall shape is like the letter V, with an angle of ∼60° between the two superhelices; their interaction mainly involves hydrophobic interactions, including residues Trp60, Phe62, Thr65, Ala70, Trp73, Val74, Val76, Asn77 and Trp80. Three hydrogen bonds can be found between Gln75 and Gln75′, and between Asn64 and Asn77′. Chains A and C are antiparallel, with an interface of ∼385.5 Å2, and their interaction is again mainly sustained by hydrophobic interactions, including residues Asn86, Gly87, Gln96, Val97, Gly99 and Tyr102. A pair of hydrogen bonds can be found between Gly98 (located in Turn3) of chain A and Ser85′′ (located in loop2) of chain C. The C-terminal region of chain B also interacts with the N-terminal region of chain C, with an area of ∼336.6 Å2 but without a twofold axis.
Figure 2.

(a) Overall structure of the three chains in an asymmetric unit. (b) Interactions between chains A (red) and B (blue). (c) Interactions between chains A (red) and C (yellow). The secondary structures involved in interaction are labelled and the residues involved in hydrogen bonds are labelled and shown in stick representation.
Although there are three molecules forming a ‘trimer’ in an asymmetric unit, other data suggest that Spr1274 and Spr1274-CBD are mainly present as monomers and dimers in solution (data not shown). Further experiments are requried in order to clarify the interactions between the monomers present in the crystal structure and their relation to function.
Supplementary Material
PDB reference: choline-binding domain of Spr1274, 3hia, r3hiasf
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Program 30870488) and the Ministry of Science and Technology of China (Project 2009CB918800), and ANR Jeunes Chercheurs 2005 (ANR-05-JCJC-0049-01) grant to AMDG and by the FPG EUR-INTAFAR LSHM-CT-2004-512138 project.
References
- Attali, C., Frolet, C., Durmort, C., Offant, J., Vernet, T. & Di Guilmi, A. M. (2008). Infect. Immun. 76, 466–476. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Fernandez-Tornero, C., Lopez, R., Garcia, E., Gimenez-Gallego, G. & Romero, A. (2001). Nature Struct. Biol. 8, 1020–1024. [DOI] [PubMed]
- Garcia, J. L., Sanchez-Beato, A. R., Medrano, F. J. & Lopez, R. (1998). Microb. Drug Resist. 4, 25–36. [DOI] [PubMed]
- Gillespie, S. H., Ainscough, S., Dickens, A. & Lewin, J. (1996). J. Med. Microbiol. 44, 35–40. [DOI] [PubMed]
- Gosink, K. K., Mann, E. R., Guglielmo, C., Tuomanen, E. I. & Masure, H. R. (2000). Infect. Immun. 68, 5690–5695. [DOI] [PMC free article] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Hermoso, J. A., Lagartera, L., Gonzalez, A., Stelter, M., Garcia, P., Martinez-Ripoll, M., Garcia, J. L. & Menendez, M. (2005). Nature Struct. Mol. Biol. 12, 533–538. [DOI] [PubMed]
- Hermoso, J. A., Monterroso, B., Albert, A., Galan, B., Ahrazem, O., Garcia, P., Martinez-Ripoll, M., Garcia, J. L. & Menendez, M. (2003). Structure, 11, 1239–1249. [DOI] [PubMed]
- Horne, D. S. & Tomasz, A. (1993). J. Bacteriol. 175, 1717–1722. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Lopez, R. & Garcia, E. (2004). FEMS Microbiol. Rev. 28, 553–580. [DOI] [PubMed]
- Martinez-Buey, R. M., Monterroso, B., Menendez, M., Diakun, G., Chacon, P., Hermoso, J. A. & Diaz, J. F. (2007). J. Mol. Biol. 365, 411–424. [DOI] [PubMed]
- Molina, R., Gonzalez, A., Stelter, M., Perez-Dorado, I., Kahn, R., Morales, M., Campuzano, S., Campillo, N. E., Mobashery, S., Garcia, J. L., Garcia, P. & Hermoso, J. A. (2009). EMBO Rep. 10, 246–251. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Rosenow, C., Ryan, P., Weiser, J. N., Johnson, S., Fontan, P., Ortqvist, A. & Masure, H. R. (1997). Mol. Microbiol. 25, 819–829. [DOI] [PubMed]
- Sanchez-Beato, A. R. & Garcia, J. L. (1996). Gene, 180, 13–21. [DOI] [PubMed]
- Sanchez-Beato, A. R., Ronda, C. & Garcia, J. L. (1995). J. Bacteriol. 177, 1098–1103. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Tomasz, A. (1967). Science, 157, 694–697. [DOI] [PubMed]
- Vagin, A. A. & Isupov, M. N. (2001). Acta Cryst. D57, 1451–1456. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: choline-binding domain of Spr1274, 3hia, r3hiasf