

Leucurolysin-a, a nonhaemorrhagic metalloproteinase from B. leucurus snake venom, has been crystallized in a free form and in a complexed form.
Keywords: Bothrops leucurus, Viperidae family, nonhaemorrhagic, snake-venom metalloproteinases
Abstract
Leucurolysin-a (leuc-a) is a class P-I snake-venom metalloproteinase isolated from the venom of the South American snake Bothrops leucurus (white-tailed jararaca). The mature protein is composed of 202 amino-acid residues in a single polypeptide chain. It contains a blocked N-terminus and is not glycosylated. In vitro studies revealed that leuc-a dissolves clots made either from purified fibrinogen or from whole blood. Unlike some other venom fibrinolytic metalloproteinases, leuc-a has no haemorrhagic activity. Leuc-a was sequenced and was crystallized using the hanging-drop vapour-diffusion technique. Crystals were obtained using PEG 6000 or PEG 1500. Diffraction data to 1.80 and 1.60 Å resolution were collected from two crystals (free enzyme and the endogenous ligand–protein complex, respectively). They both belonged to space group P212121, with very similar unit-cell parameters (a = 44.0, b = 56.2, c = 76.3 Å for the free-enzyme crystal).
1. Introduction
Snake venoms are an excellent source of pharmacologically active molecules (Mebs & Bailey, 1998 ▶). The snake venom gland synthesizes and stores for several weeks an extraordinary mixture of serine proteases, phospholipases A2, spreading factors and metalloproteinases (Wagstaff et al., 2008 ▶). A multifactorial process controls this lethal mixture to avoid damage to the gland and readily disengages when toxic potential is needed (Wagstaff et al., 2008 ▶). Physiochemical factors such as pH (Odell et al., 1998 ▶), the secretion of zymogens and the expression of low-affinity endogenous peptides of venom enzymes seem to be involved in the glandular storage-control mechanism (Wagstaff et al., 2008 ▶).
Among the active proteins in the venom, metalloproteinases play an important role during haemorrhage and coagulopathic pathologies following envenomation by most pit vipers and true vipers (Viperidae family; Bjarnason & Fox, 1994 ▶; Estevão-Costa et al., 2000 ▶). These snake-venom metalloproteinases (SVMPs) are synthesized in the venom gland as large multidomain proteins that include a proenzyme domain (Hite et al., 1994 ▶). SVMPs have been classified into four classes (P-I to P-IV; Kini & Evans, 1992 ▶; Bjarnason & Fox, 1994 ▶) on the basis of the number of additional domains that follow the metalloproteinase domain (Bello et al., 2006 ▶). Class P-I includes the small SVMPs that contain only the metalloproteinase domain (molecular weight of ∼23 kDa). Enzymes in class P-II contain an additional disintegrin-like domain following the metalloproteinase domain. SVMPs in class P-III contain the metalloproteinase domain, the disintegrin-like domain and a cysteine-rich domain. Finally, the SVMPs in class P-IV differ from those in class P-III by the presence of an additional lectin-like domain.
The ability of SVMPs to induce haemorrhage is associated with their capacity to hydrolyze proteins present in the basal membrane of the extracellular matrix, a structural framework surrounding the microvascular endothelial cells (Ohsaka & Lee, 1979 ▶; Bjarnason & Fox, 1994 ▶). The members of class P-I present a great variation in haemorrhagic activity (from high activity to none) despite their high amino-acid sequence similarity (Bjarnason & Fox, 1994 ▶; Tsai et al., 2000 ▶). It has been suggested that subtle differences in surface residues might be responsible for the variations in haemorrhagic activity (Sanchez & Swenson, 2007 ▶). Gong et al. (1998 ▶) and Watanabe et al. (2003 ▶) observed a high degree of structural identity in most regions of class P-I SVMPs, with the exception of a coil region (residues 153–176). This region, which surrounds the catalytic site, might be involved in the interaction with different substrates (Watanabe et al., 2003 ▶). Other hypotheses that only take the amino-acid sequence of P-I SVMPs into account have also been suggested (Tsai et al., 2000 ▶; Gasmi et al., 2000 ▶).
Although a large collection of amino-acid sequences of SVMPs have been accumulated and eight class P-I SVMP structures have been deposited in the Protein Data Bank (Berman et al., 2000 ▶) [adamalysin-II from Crotalus adamanteus (PDB code 1iag; Gomis-Rüth et al., 1993 ▶), atrolysin-C from C. atrox (PDB code 1htd; Zhang et al., 1994 ▶), H2-proteinase from Trimeresurus flavoviridis (PDB code 1wni; Kumasaka et al., 1996 ▶), acutolysins A and C from Agkistrodon acutus (PDB codes 1bsw and 1qua; Gong et al., 1998 ▶; Zhu et al., 1999 ▶), TM-3 from T. mucrosquamatus (PDB code 1kuf; Huang et al., 2002 ▶), F2 from A. acutus (PDB code 1yp1; Lou et al., 2005 ▶) and BaP1 from Bothrops asper (PDB code 1nd1; Watanabe et al., 2003 ▶)], a full explanation of the structural basis of haemorrhagic activity remains elusive (Fox & Serrano, 2005 ▶).
Leucurolysin-a (leuc-a; EC 3.4.3.4), a class P-I SVMP isolated from B. leucurus (white-tailed jararaca) venom, is a 23 kDa nonglycosylated α-fibrinogenase. It degrades fibrin clots directly and does not cause haemorrhage when injected (up to 100 µg) subcutaneously in mice (Bello et al., 2006 ▶; Sanchez & Swenson, 2007 ▶).
In the present work, we describe the complete primary structure assignment of leuc-a. The protein was also crystallized and initial X-ray diffraction studies were performed on two crystal forms using synchrotron radiation. Surprisingly, inspection of the preliminary electron-density maps for one of the X-ray data sets suggested the presence of an endogenous ligand in the active site of the enzyme. A comparative analysis between leuc-a and other class P-I structures may help in the identification of the specific amino-acid residues which determine the haemorrhagic effect of these proteinases. Additionally, the correct identification of the ligand in the leuc-a structure might also improve our understanding of the control regulation of SVMPs stored in the venom gland and of the development of specific inhibitors to improve antivenom efficacy.
2. Determination of the primary structure
Approximately 95% of the primary structure of leuc-a has been reported previously (Bello et al., 2006 ▶). However, the identities of a few residues, mainly in the N-terminal region, were not assigned. Thus, for a complete primary-structure assignment the identities of the remaining amino-acid residues were confirmed by automated Edman degradation and MALDI–TOF-TOF MS/MS.
A protein sample (3 mg), obtained accordingly to Bello et al. (2006 ▶), was reduced and S-alkylated using 4-vinylpyridine as described previously (Wilson et al., 1989 ▶). The reduced and alkylated protein was dissolved in 200 µl 8 M urea solution and diluted to 2 ml with 0.1 M NH4HCO3 pH 8.1 before proteolysis with trypsin [2%(w/w) enzyme/protein, 4 h at 310 K], chymotrypsin [2%(w/w) enzyme/protein, 3 h at 310 K] and Glu-C protease from Streptococcus aureus V8 [2%(w/w) enzyme/protein, 24 h at 310 K] individually. The protein was also cleaved with cyanogen bromide in 70%(v/v) aqueous TFA (trifluoroacetic acid) for 24 h at 293 K in the dark. The resulting peptides were purified by reverse-phase HPLC on a Vydac C18 small-pore column (25 cm × 4.6 mm; 201SP54) using extended (3 h) linear gradients of 0–50% acetonitrile in 0.1% aqueous TFA. The purified peptides were sequenced using a Shimadzu PPSQ-21A protein sequencer.
MALDI–TOF-TOF MS/MS was applied to sequence one of the peptides obtained after chymotrypsin proteolysis that was resistant to automated Edman degradation. The peptide, with a signal at m/z 1631.94 and sequence <EQFSPRYIELVVVA, displays a pyroglutamate as the first residue (represented as <E). The presence of pyroglutamic acid residues blocking the N-terminus has been reported for a number of SVMPs (Watanabe et al., 2003 ▶).
Fig. 1 ▶ shows the complete amino-acid sequence of leuc-a, including some corrections to the previously described annotations, aligned with the class P-I haemorrhagic SVMP BaP1 from B. asper venom. The primary structures, which displayed 78% identity, were aligned using the program ClustalW (Higgins et al., 1996 ▶).
Figure 1.

Complete primary structure of leucurolysin-a from B. leucurus. The Leuc-a and BaP1 amino-acid sequences were aligned with ClustalW (Higgins et al., 1996 ▶). Cysteine amino-acid residues involved in disulfide bridges are highlighted. Bold letters indicate conserved amino-acid residues.
3. Crystallization
Lyophilized native leuc-a was dissolved in Milli-Q water to a final concentration of 10.0 mg ml−1. Crystallization trials were performed using the vapour-diffusion method at 291 K. Initial sitting-drop crystallization plates were automatically prepared using a HoneyBee 963 crystallization robot (Digilab Genomic Solutions, USA) at the Laboratório Nacional de Luz Síncrotron (Brazilian Synchrotron Light Laboratory). A total of 594 different crystallization conditions were tested. For each condition, the crystallization drop was formed of 0.3 µl protein solution and an equal volume of screening solution and set up for crystallization against 80 µl screening reservoir solution. Within two weeks, more than 30 conditions showed well formed crystals. A few of these conditions were selected and refinement of crystallization conditions was performed in hanging-drop crystallization plates to improve the crystal quality and size. The well diffracting crystals used in this work were obtained when a 1.0 µl protein drop mixed with an equal volume of reservoir solution was equilibrated over 100 mM MIB pH 5.0, 15–25%(w/v) PEG 6000 or 1500 and 100 M ammonium acetate solution (Fig. 2 ▶). All crystals were obtained using samples from the same purification batch.
Figure 2.

Typical crystal of leucurolysin-a from B. leucurus. Crystals were obtained using PEG 1500 or PEG 6000 as precipitant in MIB buffer pH 5.0. The crystal depicted in the picture is 0.1 mm in the largest dimension.
4. Data collection and processing
Single leuc-a crystals were harvested using nylon loops (Hampton Research) and transferred from the crystallization drop to 10 µl of a cryogenic solution containing the crystallization solution with 10%(v/v) ethylene glycol for a few seconds. Crystals were then flash-cooled to 100 K in a cold nitrogen stream and used for data collection. Diffraction data from two crystals (data sets 1 and 2) were collected with a rotation range of 1.0° per image and exposure times of 90 and 35 s, respectively. Data were acquired on a MAR CCD image-plate detector using the D03B-MX1 beamline at the Laboratório Nacional de Luz Síncrotron (Polikarpov et al., 1998 ▶). The crystal-to-detector distance was set to 75 mm and a total of 295 images were collected to a resolution of 1.80 Å for data set 1; the crystal-to-detector distance was set to 50 mm and a total of 240 images were collected to a resolution of 1.60 Å for data set 2. Data were indexed, integrated and scaled with the HKL-2000 package (Otwinowski & Minor, 1997 ▶). Diffraction data statistics for both data sets are shown in Table 1 ▶.
Table 1. Summary of diffraction data statistics of leucurolysin-a crystals.
Values in parentheses are for the highest resolution shells.
| Data set 1 | Data set 2 | |
|---|---|---|
| Wavelength (Å) | 1.425 | |
| Space group | P212121 | |
| Unit-cell parameters (Å) | a = 44.0, b = 56.2, c = 76.3 | a = 44.0, b = 58.6, c = 76.3 |
| Resolution range (Å) | 50.0–1.80 (1.84–1.80) | 50.0–1.60 (1.64–1.60) |
| No. of observations | 185231 (3294) | 139715 (7294) |
| No. of unique reflections | 17761 (914) | 26476 (1722) |
| Data completeness (%) | 98.1 (77.5) | 99.0 (99.0) |
| 〈I/σ(I)〉 | 38.7 (2.8) | 11.1 (3.1) |
| Redundancy | 10.4 (3.6) | 5.3 (4.2) |
| Rmerge† | 0.054 (0.365) | 0.136 (0.412) |
| Matthews value (Å3 Da−1) | 2.05 | 2.14 |
| Solvent content (%) | 40.1 | 42.7 |
R
merge = 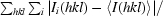
 , where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
Phasing was performed by molecular replacement using AMoRe (Navaza, 2001 ▶) from the CCP4 program suite v.6.1.1 (Collaborative Computational Project, Number 4, 1994 ▶). The model template used for molecular replacement was a full molecule of BaP1 (Watanabe et al., 2003 ▶). In each data set, a single leuc-a molecule in the asymmetric unit was obtained as a solution during molecular replacement. Initial rigid-body fitting performed by AMoRe yielded an R factor of 43.5% for both data sets.
Inspection of (F o − F c, ϕcalc) and (2F o − F c, ϕcalc) electron-density maps obtained from data set 2 after a few cycles of restrained refinement using REFMAC v.5.5 (Murshudov et al., 1997 ▶) allowed us to observe unidentified electron density in the catalytic site of the enzyme; refinement against data set 1 did not reveal such density. Further cycles of restrained refinement and water insertion using data set 2 lowered the R factor and R free to 17.6% and 23.2%, respectively. The contour of the unidentified electron density obtained after refinement, and data accumulated in this field, suggest the presence of an endogenous inhibitory peptide.
Several endogenous inhibitors of SVMPs have been isolated and characterized from the venoms of A. acutus (Lou et al., 2005 ▶), A. halys blomhoffii, C. adamanteus, B. jararaca, T. flavoviridis, T. mucrosquamatus and T. gramineus (Kato et al., 1966 ▶; Lo, 1972 ▶; Lou et al., 2005 ▶). Of the eight class P-I SVMP structures deposited in the Protein Data Bank (Berman et al., 2000 ▶), only in the structures of TM-3 and F2 was an endogenous inhibitor (peptide) observed in the active site of the enzyme (Huang et al., 2002 ▶; Lou et al., 2005 ▶). Moreover, very recently BaP1 was crystallized with a peptidomimetic inhibitor in the active site (Lingott et al., 2009 ▶).
Taken together, these facts suggest that data sets 1 and 2 correspond to a free-enzyme crystal and an endogenous ligand–protein complex crystal, respectively. Further model refinement and biochemical assays are being conducted to fully describe the enzyme, the endogenous ligand and their complex.
Acknowledgments
We acknowledge the contribution of Andréia N. Meza and Celisa C. Tonoli in technical assistance during crystallization trials performed at the Brazilian Synchrotron Light Laboratory. This work was supported by the Brazilian Synchrotron Light Laboratory (D03B-MX1-6335/6938), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, INCT Toxinas), Fundação de Desenvolvimento à Pesquisa do Estado de Minas Gerais (FAPEMIG, Rede Mineira de Estudos de Estrutura e Função de Biomoléculas, Rede-170/08) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). RNF is the recipient of a scholarship from FAPEMIG. AMCP and BR are the recipients of a fellowship and scholarship, respectively, from CNPq.
References
- Bello, C. A., Hermogenes, A. L. N., Magalhães, A., Veiga, S. S., Gremski, L. H., Richardson, M. & Sanchez, E. F. (2006). Biochimie, 88, 189–200. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res.28, 235–242. [DOI] [PMC free article] [PubMed]
- Bjarnason, J. B. & Fox, J. W. (1994). Pharmacol. Ther.62, 325–372. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Estevão-Costa, M. I., Diniz, C. R., Magalhães, A., Markland, F. S. & Sanchez, E. F. (2000). Thromb. Res.99, 363–376. [DOI] [PubMed]
- Fox, J. W. & Serrano, S. M. T. (2005). Toxicon, 45, 969–985. [DOI] [PubMed]
- Gasmi, A., Srairi, N., Karoui, H. & El Ayeb, M. (2000). Biochim. Biophys. Acta, 1481, 209–212. [DOI] [PubMed]
- Gomis-Rüth, F. X., Kress, L. F. & Bode, W. (1993). EMBO J.12, 4151–4157. [DOI] [PMC free article] [PubMed]
- Gong, W., Zhu, X., Liu, S., Teng, M. & Niu, L. (1998). J. Mol. Biol.283, 657–668. [DOI] [PubMed]
- Higgins, D. G., Thompson, J. D. & Gibson, T. J. (1996). Methods Enzymol.266, 383–402. [DOI] [PubMed]
- Hite, L. A., Jia, L. G., Bjarnason, J. B. & Fox, J. W. (1994). Arch. Biochem. Biophys.308, 182–191. [DOI] [PubMed]
- Huang, K.-F., Chiou, S.-H., Ko, T.-P. & Wang, A. H.-J. (2002). Eur. J. Biochem.269, 3047–3056. [DOI] [PubMed]
- Kato, H., Iwanaga, S. & Suzuki, T. (1966). Experientia, 22, 49–50. [DOI] [PubMed]
- Kini, R. M. & Evans, H. J. (1992). Toxicon, 30, 265–293. [DOI] [PubMed]
- Kumasaka, T., Yamamoto, M., Moriyama, H., Tanaka, N., Sato, M., Katsube, Y., Yamakawa, Y., Omori-Satoh, T., Iwanaga, S. & Ueki, T. (1996). J. Biochem.119, 49–57. [DOI] [PubMed]
- Lingott, T., Schleberger, C., Gutierrez, J. M. & Merfort, I. (2009). Biochemistry, 48, 6166–6174. [DOI] [PubMed]
- Lo, T. P. (1972). J. Chin. Biochem. Soc.1, 39–46.
- Lou, Z., Hou, J., Liang, X., Chen, J., Qiu, P., Liu, Y., Li, M., Rao, Z. & Yan, G. (2005). J. Struct. Biol.152, 195–203. [DOI] [PubMed]
- Mebs, D. & Bailey, G. S. (1998). Enzymes from Snake Venoms, pp. 1–10. Colchester: Alaken Inc.
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed]
- Odell, G. V., Ferry, E. C., Vick, L. M., Fenton, A. W., Decker, L. S., Cowell, R. L., Ownby, C. L. & Gutierrez, J. M. (1998). Toxicon, 36, 1801–1806. [DOI] [PubMed]
- Ohsaka, A. & Lee, C. Y. (1979). Snake Venoms, pp. 480–546. Berlin: Springer-Verlag.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Polikarpov, I., Perles, L. A., de Oliveira, R. T., Oliva, G., Castellano, E. E., Garratt, R. C. & Craievich, A. (1998). J. Synchrotron Rad.5, 72–76. [DOI] [PubMed]
- Sanchez, E. F. & Swenson, S. (2007). Curr. Pharm. Anal.3, 147–157.
- Tsai, I.-H., Wang, Y.-M., Chiang, T.-Y., Chen, Y.-L. & Huang, R.-J. (2000). Eur. J. Biochem.267, 1359–1367. [DOI] [PubMed]
- Wagstaff, S. C., Favreau, P., Cheneval, O., Laing, G. D., Wilkinson, M. C., Miller, R. L., Stocklin, R. & Harrison, R. A. (2008). Biochem. Biophys. Res. Commun.365, 650–656. [DOI] [PubMed]
- Watanabe, L., Shannon, J. D., Valente, R. H., Rucavado, A., Alape-Giron, A., Kamiguti, A. S., Theakston, R. D. G., Fox, J. W., Gutierrez, J. M. & Arni, R. K. (2003). Protein Sci.12, 2273–2281. [DOI] [PMC free article] [PubMed]
- Wilson, K. J., Yuan, P. M., Findlay, J. B. C. & Geisow, M. J. (1989). Protein Sequencing: A Practical Approach, pp. 3–41. Oxford: IRL Press.
- Zhang, D. C., Botos, I., Gomis-Rüth, F. X., Doll, R., Blood, C., Njoroge, F. G., Fox, J. W., Bode, W. & Meyer, E. F. (1994). Proc. Natl Acad. Sci. USA, 91, 8447–8451. [DOI] [PMC free article] [PubMed]
- Zhu, X., Teng, M. & Niu, L. (1999). Acta Cryst. D55, 1834–1841. [DOI] [PubMed]