

A preliminary crystallographic analysis at 2.3 Å resolution of protein MJ1225 from M. jannaschii, a putative archaeal homolog of γ-AMPK, is described.
Keywords: MJ1225, γ-AMPK, CBS domains, archaea, Methanocaldococcus jannaschii
Abstract
In mammals, AMP-activated protein kinase (AMPK) is a heterotrimeric protein composed of a catalytic serine/threonine kinase subunit (α) and two regulatory subunits (β and γ). The γ subunit senses the intracellular energy status by competitively binding AMP and ATP and is thought to be responsible for allosteric regulation of the whole complex. This work describes the purification and preliminary crystallographic analysis of protein MJ1225 from Methanocaldococcus jannaschii, an archaeal homologue of γ-AMPK. The purified protein was crystallized using the hanging-drop vapour-diffusion method. Diffraction data for MJ1225 were collected to 2.3 Å resolution using synchrotron radiation. The crystals belonged to space group H32, with unit-cell parameters a = b = 108.95, c = 148.08 Å, α = β = 90.00, γ = 120.00°. Preliminary analysis of the X-ray data indicated that there was one molecule per asymmetric unit.
1. Introduction
Cystathionine β-synthase (CBS) domains are 60-residue-long motifs that were originally discovered in the enzyme cystathionine β-synthase (Bateman, 1997 ▶). Although their function is unknown, their importance is underlined by the range of hereditary diseases in humans that have been associated with mutations in their sequence (Zhang et al., 1999 ▶; Konrad et al., 2000 ▶; Cleiren et al., 2001 ▶; Blair et al., 2001 ▶; Bowne et al., 2002 ▶; Pusch et al., 2002 ▶; Haug et al., 2003 ▶). Crystal structures of the CBS domains of many proteins are now available (Meyer et al., 2007 ▶; Markovic & Dutzler, 2007 ▶; Day et al., 2007 ▶; Amodeo et al., 2007 ▶; Jin et al., 2007 ▶; Rudolph et al., 2007 ▶; Townley & Shapiro, 2007 ▶). CBS domains usually occur in tandem pairs, forming a so-called CBS pair or Bateman module (Bateman, 1997 ▶), although some proteins such as 5′-AMP-activated protein kinase (AMPK) and its homologues have revealed tetra-repeat units (Ignoul & Eggermont, 2005 ▶).
Adenosine 5′-monophosphate (AMP) activated protein kinase (AMPK) plays a key role in regulation of metabolism and acts as a central regulator of energy homeostasis in eukaryotic cells, such that an increase in the cellular AMP:ATP ratio results in the activation of AMPK, which then acts to inhibit anabolic pathways and activate catabolic pathways (Day et al., 2007 ▶; Zhang et al., 2009 ▶; Hardie, 2003 ▶). In mammals, AMPK is a heterotrimeric protein that is composed of a catalytic serine/threonine kinase subunit (α) and two regulatory subunits (β and γ). The γ AMPK subunit (γ-AMPK) senses the intracellular energy status by competitively binding AMP and ATP and is thought to be responsible for allosteric regulation of the whole complex (Cheung et al., 2000 ▶). Structurally, its three isoforms share the presence of four conserved CBS domains (two Bateman modules; Amodeo et al., 2007 ▶; Townley & Shapiro, 2007 ▶) which are connected by short peptide chains. This feature always results in a head-to-head orientation of the two Bateman domains and is also observed in other eukaryotes. On the other hand, the recent characterization of the ligand-binding sites of human γ-AMPK (Day et al., 2007 ▶; Jin et al., 2007 ▶; Amodeo et al., 2007 ▶; Townley & Shapiro, 2007 ▶; Xiao et al., 2007 ▶) has led some authors to propose a recognition motif for the ribose-phosphate moiety of AMP (Day et al., 2007 ▶). This motif (G-h-x-S/T-x-S/T-D) involves residues located in the third strand of the β-sheet and the first turn of the following helix of each CBS domain (Day et al., 2007 ▶; Proudfoot et al., 2008 ▶; Fig. 1 ▶). In general, it has been observed that those binding sites that lack the conserved aspartate that hydrogen bonds to the hydroxyls of the ribose ring of the nucleotide are not expected to bind phosphate-containing adenosine derivatives (Xiao et al., 2007 ▶). Nevertheless, substitution of such aspartates by lysine, arginine or bulky hydrophobic residues does not avoid AMP, ADP or ATP binding in proteins such as the chloride channel CLC-5 (Meyer et al., 2007 ▶), protein ATU1752 (PDB code 3fhm; A. U. Singer, G. Brown, M. Proudfoot, X. Xu, A. Dong, H. Cui, A. M. Edwards, A. Joachimiak, A. Savchenko & A. F. Yakunin, unpublished work), AMPK (Amodeo et al., 2007 ▶) and its yeast homologue SNF1 (Jin et al., 2007 ▶; Fig. 1 ▶). Thus, a wider spectrum of binding motifs seems to be involved in binding of phosphate-containing adenosine derivatives both in eukaryotes and in prokaryotes. In summary, the γ-AMPK regulatory subunit shows three basic structural requirements in all organisms for which it has been described: (i) it is a stand-alone protein that is not fused to other domains within the same polypeptide chain, (ii) it contains two contiguous Bateman domains (four CBS subunits) which are oriented in a head-to-head fashion and (iii) it binds AMP/ADP or ATP in two or three of its four potential ligand-binding sites. However, although several proteins fulfil these structural requirements and can be inferred from database searches, no prokaryotic homologue of γ-AMPK has been described to date and none of them has been structurally characterized. Based on the large number of stand-alone CBS-domain proteins observed in these genomes, some authors have recently pointed out that it is possible that nonhomologous kinase domains are regulated by stand-alone CBS-domain proteins in prokaryotes, forming energy-sensing kinase complexes that serve physiological roles analogous to those of AMPK (King et al., 2008 ▶). Alternatively, stand-alone CBS-domain proteins may interact directly with metabolic enzymes without making use of a central intermediary kinase (King et al., 2008 ▶).
Figure 1.

Sequence alignment of residues located in the last β-strand of the β-sheet and the following α-helix of each CBS domain in γ-AMPKs and structural homologues. The UniProt code for each protein fragment and the corresponding CBS domain in which it is located head each line. Known PDB entries are indicated in parentheses on the left together with the alternative potential ligand-binding motifs for AMP/ADP or ATP. The sequences have been grouped into five different blocks (separated by horizontal lines) corresponding to five putative ligand-binding motifs, where x is any amino acid and h is a hydrophobic amino acid. Those ligands for which a binding site has been experimentally determined are indicated in parentheses on the right; ϕ indicates that no adenosine binding has been observed (PDB codes 2qrc and 2v92). Alignment was carried out using the three-dimensional structure of human AMPK as reference. Residues involved in interactions with the phosphate moiety of AMP and included in the AMP-binding motif (G-h-x-S/T-x-S/T-D; Day et al., 2007 ▶) are marked with a red asterisk. Although it is not present in AMPKs, the first motif (G-h-x-S/T-x-K/R-D) has been observed to be involved in ADP/ATP binding in the chloride channel CLC-5 (PDB code 2j9l). MJ1225 sequences are marked for clarity.
A search of the Pfam database (http://pfam.sanger.ac.uk/) revealed 15 proteins in the Methanocaldococcus jannaschii genome that contained recognizable CBS domains (Martínez-Cruz et al., 2009 ▶). Remarkably, 11 of these proteins were stand-alone CBS-domain proteins and two of them, MJ1404 and MJ1225 (UniProtKB/Swiss-Prot Q58799 and Q58622, respectively), contained a tetra repeat of CBS subunits (Martínez-Cruz et al., 2009 ▶). Furthermore, a detailed comparison of the nucleotide-binding motif (Day et al., 2007 ▶) and the sequences of other CBS-domain proteins (Fig. 1 ▶) showed that at least three of the four putative ligand-binding sites found in MJ1225 might potentially bind AMP or related molecules, whereas other species such as proteins APE0234 from Aeropyrum pernix or PH1780 from Pyrococcus horikoshii lacked these structural features. All these data prompted us to initiate structural studies on protein MJ1225 as a potential archaeal homologue of γ-AMPK. The ORF of gene mj1225 codes for a polypeptide chain of 280 amino acids with a molecular weight of 31 719 Da. Its sequence consists of a tetra repeat of CBS domains (CBS1, residues 10–67; CBS2, residues 90–146; CBS3, residues 154–209; CBS4, residues 229–280) (http://smart.embl-heidelberg.de/). In this work, we present the cloning, expression, purification and preliminary crystallographic analysis of the full-length protein MJ1225 as a first step towards its three-dimensional and functional characterization.
2. Materials and methods
2.1. Cloning, expression and purification of MJ1225
The MJ1225 protein from M. jannaschii was amplified by PCR using genomic cDNA as a template, which was kindly provided by Sung-Hou Kim, University of Berkeley, California, USA. The primers used were 5′-CACCATGTTTGTGAGAGTCATGAAAATTG-3′ (forward) and 5′-TTAAGCAAAGTATTTTAAAACATCC-3′ (reverse). The amplified DNA was cloned into vector pET101/D-TOPO (Invitrogen). This plasmid was transformed into chemically competent Escherichia coli strain BL21Star (DE3) One Shot (Invitrogen; Studier & Moffatt, 1986 ▶; Grunberg-Manago, 1999 ▶). The cells were grown in Luria–Bertani medium containing 100 µg ml−1 ampicillin for 12 h at 310 K without using IPTG induction. The cells were harvested by centrifugation at 4000g for 15 min at 277 K. The cell pellet (about 30 g in weight) was resuspended in 90 ml lysis buffer (100 mM HEPES pH 7.0, 1 mM EDTA, 1 mM benzamidine, 0.1 mM PMSF) and lysed by sonication in a Labsonic P sonicator (Sartorius) for 10 × 12 s at 90% amplitude, keeping the cells on ice to prevent overheating. The cell debris was separated by ultracentrifugation at 120 000g in a 70 Ti rotor (Beckman) for 25 min at 277 K. Since the source organism of MJ1225 is a hyperthermophile, the first purification step consisted of a heat-shock, in which the clarified lysate from the previous centrifugation step was heated at 348 K for 30 min. The proteins precipitated by the heat-shock were removed by centrifugation at 4000g in an SX4250 rotor (Beckman) for 15 min at 277 K. The supernatant was then injected onto a 5 ml HiTrap Q column (GE Healthcare) at a flow rate of 1 ml min−1, washed with several column volumes of buffer A (50 mM HEPES pH 7.0, 1 mM EDTA, 1 mM β-mercaptoethanol) and eluted with a stepped gradient of buffer B (50 mM HEPES pH 7.0, 1 M NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol) over 30 min. Fractions containing the protein of interest were pooled and diluted with two volumes of buffer A before being injected onto a 1 ml HiTrap Blue column (GE Healthcare) at a flow rate of 1 ml min−1. The column was washed with buffer A as described previously and eluted with a 0–100% gradient of buffer B over 20 min. The fractions of interest were combined and concentrated using Vivaspin centrifugal concentrators (5000 molecular-weight cutoff) to a volume of approximately 0.5 ml and injected onto a Superdex75 HR 10/300 column (GE Healthcare). The protein was eluted at a flow rate of 0.5 ml min−1 with an isocratic gradient using buffer C (50 mM HEPES pH 7.0, 200 mM NaCl, 1 mM EDTA). Fractions containing pure MJ1225 were pooled and concentrated using Vivaspin concentrators (5000 molecular-weight cutoff) to a final concentration of 160 mg ml−1 for crystallization trials. The concentration of the protein was estimated by Bradford assay (Bradford, 1976 ▶). The identity of MJ1225 was confirmed by mass spectrometry.
2.2. Mass-spectrometric analysis
SDS–PAGE gel bands containing MJ1225 were subjected to in-gel tryptic digestion according to Shevchenko et al. (1996 ▶), with minor modifications. The gel piece was swollen in a digestion buffer containing 50 mM NH4HCO3 and 12.5 ng µl−1 trypsin (Roche Diagnostics) in an ice bath. After 30 min the supernatant was removed and discarded, 20 µl 50 mM NH4HCO3 was added to the gel piece and the digestion was allowed to proceed at 310 K overnight. Prior to MS analysis, the sample was acidified by adding 5 µl 0.5% TFA. 0.5 µl digested sample was directly spotted onto the MALDI target and then mixed with 0.5 µl α-cyano-4-hydroxycinnamic acid (CHCA) matrix solution [20 µg µl−1 in acetonitrile/0.1% TFA, 70:30(v:v)]. Peptide mass fingerprinting was performed on a Bruker Autoflex III mass spectrometer (Bruker Daltonics, Bremen, Germany). Positively charged ions were analyzed in reflector mode using delayed extraction. The spectra were obtained by randomly scanning the sample surface. About 600–800 spectra were averaged to improve the signal-to-noise ratio. Spectra were externally calibrated, resulting in a mass accuracy of <50 p.p.m. when external calibration was performed and typically of <20 p.p.m. in the case of internal calibration. Protein identification was performed by searching a nonredundant protein database (NCBI) using the Mascot search engine (http://matrixscience.com). The following parameters were used for database searches: missed cleavages, 1; allowed modifications, carbamidomethylation of cysteine (complete) and oxidation of methionine (partial). Ultimately, MJ1225 expressed in E. coli was wild type and did not contain any mutations in the amino-acid sequence according to the M. jannaschii genome-sequence database.
2.3. Crystallization of MJ1225
Suitable protein concentrations were determined using the Pre-Crystallization Screen (PCT; Hampton Research). Initial crystallization screens were set up using the hanging-drop vapour-diffusion technique in 24-well VDX plates (Hampton Research). Trials were carried out with a variety of commercial screens from Hampton Research (Crystal Screens 1 and 2, PEG/Ion Screen and SaltRX). Drops consisted of 0.5 µl protein solution at 160 mg ml−1 (in 100 mM HEPES pH 7.0, 1 mM β-mercaptoethanol, 1 mM EDTA) and 0.5 µl reservoir solution and were equilibrated over a reservoir volume of 500 µl at a constant temperature of 291 K. Initial experiments yielded square plates that grew using 2 M ammonium sulfate, 2%(v/v) PEG 400, 100 mM HEPES pH 7.5. After extensive optimization, the best diffraction-quality crystals grew in 2.4 M ammonium sulfate, 2.8% PEG 400, 100 mM HEPES pH 7.5 in the presence of detergents (114 mM FOS-choline 8 or 11 mM FOS-choline 10). The crystals appeared in 2 d and grew to maximum dimensions of about 0.5 × 0.5 × 0.4 mm within 5–6 d (Fig. 2 ▶).
Figure 2.

Crystals of MJ1225 grown by vapour diffusion at 293 K in 2.4 M ammonium sulfate, 2.8% PEG 400, 100 mM HEPES pH 7.5 with 11 mM FOS-choline 10 as an additive.
2.4. Preliminary crystallographic analysis
Prior to data collection, the crystals were transferred to crystallization buffer containing 30% glycerol as a cryoprotectant for a few seconds before being flash-cooled by directly immersing them into liquid nitrogen at 93 K. Crystals were mounted for X-ray data collection using either CryoLoops (Hampton Research) or MicroMounts loops (MiTeGen). Data sets were collected in-house using a CCD detector mounted on a Microstar-H rotating-anode X-ray generator (Bruker) operated at 60 kV and 100 mA with Helios optics and a copper target (Cu Kα; λ = 1.542 Å) and on beamline ID14.1 at the ESRF synchrotron (Grenoble, France). Diffraction data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). Preliminary analysis of the data sets was performed using the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). Native crystals of MJ1225 diffracted to 2.3 Å resolution (Fig. 3 ▶) and belonged to space group H32, with unit-cell parameters a = b = 108.95, c = 148.08 Å, α = β = 90, γ = 120°. The presence of one or two molecules within the asymmetric unit gave Matthews coefficients of 2.66 and 1.33 Å3 Da−1, respectively (Matthews, 1968 ▶), and solvent contents of 53.78% and 7.57%. After careful analysis of the self-rotation function, we estimated that one molecule per asymmetric unit was the most probable value. Taking into account the fact that MJ1225 crystallizes in space group H32, native data sets were tested for twinning. Intensity statistics, including the cumulative-intensity plot and the moments of normalized intensities (Stanley, 1972 ▶; Dauter, 2003 ▶), were obtained using the Twinning Server (http://www.doe-mbi.ucla.edu/Services/Twinning; Yeates, 1997 ▶). No twinning was detected. The data-collection statistics are summarized in Table 1 ▶.
Figure 3.

Representative X-ray diffraction image from native MJ1225. The crystal was exposed for 2 s over a 1° oscillation range. The edge of the detector corresponds to a resolution of 1.80 Å.
Table 1. Data-processing statistics for native MJ1225.
Values in parentheses are for the outer resolution shell.
| Beamline | ID14-1 |
| Wavelength (Å) | 0.934 |
| Total No. of reflections | 336885 |
| No. of unique reflections | 15295 |
| Resolution range (Å) | 50–2.3 |
| Space group | H32 |
| Unit-cell parameters (Å) | |
| a = b | 108.95 |
| c | 148.08 |
| Completeness (%) | 99.2 (98.9) |
| Redundancy | 11.9 (8.9) |
| Rmerge† (%) | 8.9 (27.9) |
| Mean I/σ(I) | 26.5 (7.5) |
| Wilson B value (Å2) | 46.5 |
R
merge = 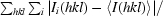
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Acknowledgments
We thank Professor Sung-Hou Kim from the University of California at Berkeley for providing us with the genomic DNA from M. jannaschii and Dr José Miguel Mancheño from Instituto ‘Rocasolano’, CSIC, Madrid and the staff of ESRF beamline ID14-1 for support during synchrotron data collection. We also thank Dr Felix Elortza from the Proteomics Service at CIC bioGUNE for mass-spectrometric analysis and Beatriz González Callejas for maintenance of the in-house X-ray equipment. This research was supported by program grants from the Basque Government (ETORTEK IE05-147, IE07-202), Diputación Foral de Bizkaia (Exp. 7/13/08/2006/11 and 7/13/08/2005/14), the Spanish Ministry of Education (SAF2005-00855) and SICI CONSOLIDER Project (CSD2008-00005) as well as from a postdoctoral fellowship from CIC bioGUNE.
References
- Amodeo, G. A., Rudolph, M. J. & Tong, L. (2007). Nature (London), 449, 492–495. [DOI] [PubMed]
- Bateman, A. (1997). Trends Biochem. Sci.22, 12–13. [DOI] [PubMed]
- Blair, E., Redwood, C., Ashrafian, H., Oliveira, M., Broxholme, J., Kerr, B., Salmon, A., Ostman-Smith, I. & Watkins, H. (2001). Hum. Mol. Genet.10, 1215–1220. [DOI] [PubMed]
- Bowne, S. J., Sullivan, L. S., Blanton, S. H., Cepko, C. L., Blackshaw, S., Birch, D. G., Hughbanks-Wheaton, D., Heckenlively, J. R. & Daiger, S. P. (2002). Hum. Mol. Genet.11, 559–568. [DOI] [PMC free article] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Cleiren, E., Bénichou, O., Van Hul, E., Gram, J., Bollerslev, J., Singer, F. R., Beaverson, K., Aledo, A., Whyte, M. P., Yoneyama, T., deVernejoul, M. C. & Van Hul, W. (2001). Hum. Mol. Genet.10, 2861–2867. [DOI] [PubMed]
- Cheung, P. C., Salt, I. P., Davies, S. P., Hardie, D. G. & Carling, D. (2000). Biochem. J.346, 659–669. [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dauter, Z. (2003). Acta Cryst. D59, 2004–2016. [DOI] [PubMed]
- Day, P., Sharff, A., Parra, L., Cleasby, A., Williams, M., Hörer, S., Nar, H., Redemann, N., Tickle, I. & Yon, J. (2007). Acta Cryst. D63, 587–596. [DOI] [PubMed]
- Grunberg-Manago, M. (1999). Annu. Rev. Genet.33, 193–227. [DOI] [PubMed]
- Hardie, D. G. (2003). Endocrinology, 144, 5179–5183. [DOI] [PubMed]
- Haug, K. et al. (2003). Nature Genet.33, 527–532. [DOI] [PubMed]
- Ignoul, S. & Eggermont, J. (2005). Am. J. Physiol. Cell Physiol.289, C1369–C1378. [DOI] [PubMed]
- Jin, X., Townley, R. & Shapiro, L. (2007). Structure, 15, 1285–1295. [DOI] [PubMed]
- King, N. P., Lee, T. M., Sawaya, M. R., Cascio, D. & Yeates, T. O. (2008). J. Mol. Biol.380, 181–192. [DOI] [PMC free article] [PubMed]
- Konrad, M., Vollmer, M., Lemmink, H. H., van den Heuvel, L. P., Jeck, N., Vargas-Poussou, R., Lakings, A., Ruf, R., Deschênes, G., Antignac, C., Guay-Woodford, L., Knoers, N. V., Seyberth, H. W., Feldmann, D. & Hildebrandt, F. (2000). J. Am. Soc. Nephrol.11, 1449–1459. [DOI] [PubMed]
- Markovic, S. & Dutzler, R. (2007). Structure, 15, 715–725. [DOI] [PubMed]
- Martínez-Cruz, L. A., Encinar, J. A., Kortazar, D., Prieto, J., Gómez, J., Fernández-Millán, P., Lucas, M., Arribas, E. A., Fernández, J. A., Martínez-Chantar, M. L., Mato, J. M. & Neira, J. L. (2009). Biochemistry, 48, 2760–2776. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Meyer, S., Savaresi, S., Forster, I. C. & Dutzler, R. (2007). Nature Struct. Mol. Biol.14, 60–67. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Proudfoot, M., Sanders, S. A., Singer, A., Zhang, R., Brown, G., Binkowski, A., Xu, L., Lukin, J. A., Murzin, A. G., Joachimiak, A., Arrowsmith, C. H., Edwards, A. M., Savchenko, A. V. & Yakunin, A. F. (2008). J. Mol. Biol.375, 301–315. [DOI] [PMC free article] [PubMed]
- Pusch, M. (2002). Hum. Mutat.19, 423–434. [DOI] [PubMed]
- Rudolph, M. J., Amodeo, G. A., Iram, S. H., Hong, S. P., Pirino, G., Carlson, M. & Tong, L. (2007). Structure, 15, 65–74. [DOI] [PubMed]
- Townley, R. & Shapiro, L. (2007). Science, 315, 1726–1729. [DOI] [PubMed]
- Shevchenko, A., Wilm, M., Vorm, O. & Mann, M. (1996). Anal. Chem.68, 850–858. [DOI] [PubMed]
- Stanley, E. (1972). J. Appl. Cryst.5, 191–194.
- Studier, F. W. & Moffatt, B. A. (1986). J. Mol. Biol.189, 113–130. [DOI] [PubMed]
- Xiao, B., Heath, R., Saiu, P., Leiper, F. C., Leone, P., Jing, C., Walker, P. A., Haire, L., Eccleston, J. F., Davis, C. T., Martin, S. R., Carling, D. & Gamblin, S. J. (2007). Nature (London), 449, 496–500. [DOI] [PubMed]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–358. [PubMed]
- Zhang, R., Evans, G., Rotella, F. J., Westbrook, E. M., Beno, D., Huberman, E., Joachimiak, A. & Collart, F. R. (1999). Biochemistry, 38, 4691–4700. [DOI] [PubMed]
- Zhang, B. B., Zhou, G. & Li, C. (2009). Cell Metab.9, 407–416. [DOI] [PubMed]