

Chlorite dismutase crystals belonged to space group P212121 and diffracted to 3.0 Å resolution at a third-generation synchrotron X-ray source.
Keywords: chlorite dismutase, b-type hemoproteins
Abstract
Chlorite dismutase from Dechloromonas aromatica RCB, a novel b-type hemoprotein that catalyzes O—O bond formation, has been crystallized. Synchrotron X-ray diffraction data have been collected to 3.0 Å resolution. The crystals belonged to space group P212121, with unit-cell parameters a = 122.7, b = 202.9, c = 247.1 Å.
1. Introduction
Oxygen-independent respiration provides a valuable energy supply to organisms in low-oxygen or anaerobic environments. As such, several species of anaerobes have evolved metabolic pathways to utilize ambient thermodynamically robust oxidants in their environment. Soluble inorganic oxides such as NO3 −, SeO3 2− and dimethyl sulfoxide (DMSO) have been shown to act as competent electron sinks in lieu of O2 (Holm & Donahue, 1993 ▶; McEwan, 2002 ▶). Dechloromonas aromatica RCB (Coates et al., 2001 ▶) is one organism which uses the high-potential oxidants perchlorate (ClO4 −) and chlorate (ClO3 −) as respiratory electron acceptors (Danielsson et al., 2003 ▶; Coates & Achenbach, 2004 ▶).
Oxochlorides, which include ClO4 −, ClO3 −, chlorite (ClO2 −) and hypochlorite (ClO−), are deleterious compounds that predominantly emanate from anthropogenic sources (Urbansky & Schock, 1999 ▶). Chlorite has recently been named as a top ten pollutant by the Environmental Protection Agency (2002 ▶). The health risks associated with chronic chlorite exposure include anemia, together with nervous system problems, in infants and young children (Michael, 1981 ▶; Kanitz et al., 1996 ▶). It is also chemically inert and highly soluble, leading to the build-up of appreciable concentrations within affected environments (Environmental Protection Agency, 2002 ▶). As various chemical and physical methods to remove chlorite and the other oxochlorides have proven ineffective, bioremediation may provide an attractive means of removal.
D. aromatica RCB is capable of catabolizing ClO4 − (or ClO3 −) to innocuous Cl− and O2 (Coates et al., 1999 ▶). This process involves two enzymes. The first is the molybdopterin-dependent enzyme perchlorate reductase (Pcr), which catalyzes the stepwise two-electron reductions of ClO4 − to ClO3 − and of ClO3 − to ClO2 − (Kengen et al., 1999 ▶; Bender et al., 2005 ▶). The ClO2 − is then decomposed into Cl− and O2 by the action of chlorite dismutase (Cld; van Ginkel et al., 1996 ▶; Stenklo et al., 2001 ▶; Xu & Logan, 2003 ▶).
Chlorite dismutase is a b-type hemoprotein containing a single heme per monomer (Bender et al., 2002 ▶; Thorell et al., 2002 ▶; Hagedoorn et al., 2002 ▶). Previously published crystal structures and native mass spectrometry have shown the Cld multimer to be pentameric (Ebihara et al., 2005 ▶; De Geus et al., 2009 ▶). Chemically, Cld-catalyzed decomposition is a remarkable process. The molecular oxygen product requires the formation of an O—O bond. This is the only example of O—O bond formation known in biology outside of photosystem II. Mechanistically, the reaction is of great interest in synthetic chemistry, particularly in developing models for artificial photosynthesis. Both photosynthetic and small-molecule-catalyzed O—O bond-forming reactions have been proposed to occur via high-valent metal–oxo intermediates (Betley et al., 2008 ▶). The presence of such intermediates in the Cld-catalyzed reaction was recently confirmed (Lee et al., 2008 ▶).
Structural insight into the active-site construction of Cld will be crucial to understanding O—O bond formation at the b-type heme. In the case of oxygen activation, the structures of several enzymatic b-type hemoproteins, including peroxidases (Berglund et al., 2002 ▶; Bonagura et al., 2003 ▶), catalases (Murshudov et al., 2002 ▶) and cytochromes P450 (Schlichting et al., 2000 ▶), have shown the strategic placement of distal and proximal active-site residues are key to this process. The environment of the Cld heme moiety that enables this hemoprotein to uniquely catalyze O—O bond formation has been partially defined by the crystal structure of Azospira oryza Cld (De Geus et al., 2009 ▶). Unfortunately, a portion of the structure close to the heme is disordered and as such a complete definition of the heme environment is lacking. The crystal structure of D. aromatica Cld will help to resolve the active-site structure and generate a more complete understanding of Cld catalysis.
2. Experimental
2.1. Sequence analysis
Sequence alignments of Cld homologs with D. aromatica RCB were conducted using the program ClustalW2 (Larkin et al., 2007 ▶).
2.2. Crystallization
Initial crystallization trials were carried out at 293 K using Hampton Research Crystal Screens 1 and 2. Recombinant protein was expressed and purified as previously described (Streit & DuBois, 2008 ▶). The recombinant protein was buffer-exchanged with an Amicon Ultra Centrifugal Device (Millipore) into 20 mM HEPES pH 7.0 and concentrated to 15 mg ml−1 as determined by standard Bradford analysis. Crystals were grown by hanging-drop vapor diffusion in VDX Plates (Hampton Research) using equal amounts (1.5 µl) of protein solution and reservoir solution. After one week, multiple small crystals were obtained from Hampton Research Crystal Screen 1 condition No. 46 (18% PEG 8000, 0.2 M calcium acetate, 0.1 M sodium cacodylate pH 6.5). This condition was optimized by a fine screen around the PEG 8000 concentration and by using MES buffer pH 6.5 in place of sodium cacodylate. Cocrystallizations of Cld with the substrate analogues sodium nitrite and sodium cyanide were also set up using these conditions.
2.3. Characterization
2.3.1. UV–visible spectrophotometry
Recombinant Cld was diluted accordingly in 20 mM HEPES pH 7.0 for spectral studies. A 40-fold excess of either sodium nitrite or potassium cyanide was added to 10 µM Cld for comparison to the unliganded Cld. Spectra were measured using a Cary 50 spectrophotometer (Varian).
2.3.2. Circular dichroism
Recombinant Cld was diluted to 100 µM with 20 mM HEPES pH 7.0 for circular-dichroism (CD) analysis. Spectra were measured using a Jasco J-710 spectrapolarimeter (Jasco). The resultant spectra were analyzed with the program suite DICHROWEB (Whitmore & Wallace, 2004 ▶, 2008 ▶).
2.4. X-ray data collection
Data collection was carried out on the 19ID-D SBC-CAT beamline at the Advanced Photon Source, Argonne, Illinois, USA. Cryogenic temperatures (100 K) were maintained during data collection. For data collection at 100 K, the crystal was first soaked for 10 min in a cryoprotectant solution containing 8 µl reservoir buffer and 2 µl 100% PEG 600. The crystal was then mounted in a nylon loop and flash-frozen in liquid nitrogen. Diffraction data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶). Further diffraction data analysis was conducted with the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
3.1. Sequence alignments
Structural genomics centers have determined the X-ray crystal structures of three Cld homologs from Thermus thermophilius (PDB code 1vdh; Ebihara et al., 2005 ▶), Bacillus stearothermophilus (PDB code 1t0t; M. Gilski, D. Borek, Y. Chen, F. Collart, A. Joachimiak & Z. Otwinowski, unpublished work) and Thermoplasma acidophilum (PDB code 3dtz; C. Chang, X. Xu, A. Savchenko, A. Edwards & A. Joachimiak, unpublished work). All three structures are presumed to be hemoproteins in their active state, but none contain heme as crystallized. None of these organisms are perchlorate respirers and reconstitution of the T. thermophilius homolog with heme did not lead to chlorite dismutase activity (Ebihara et al., 2005 ▶). As such, these distant homologs (their sequence identity to D. aromatica Cld is 14.5–19.0%) may have a different unidentified function. The low sequence identity with Cld was problematic in attempts to solve the D. aromatica RCB crystal structure by molecular replacement using these structures. Recently, the X-ray crystal structure of a known Cld from A. oryzae was determined (PDB code 2vxh; De Geus et al., 2009 ▶). The structure contains the intact heme moiety but crystallizes as a nonphysiological hexamer and with thiocyanate bound distally to the heme iron. As A. oryzae Cld is 97% identical in sequence to D. aromatica RCB Cld, it may be a better candidate for structure solution by molecular replacement.
3.2. Crystallization
The final optimized conditions for crystal growth were 16% PEG 8000, 0.2 M calcium acetate, 90 mM MES pH 6.5 at 293 K. Crystals suitable for X-ray analysis appeared after 2 d by hanging-drop vapour diffusion. Inclusion of the substrate analogues sodium nitrite (25.5 mM) or sodium cyanide (25.5 mM) facilitated the growth of larger crystals. The crystals were reddish with a rod-like morphology (Fig. 1 ▶).
Figure 1.

Cocrystals of Cld complexed with nitrite. Average crystal size is 200 × 50 × 50 µm.
3.3. Characterization
3.3.1. UV–visible spectrophotometry
As purified, Cld exhibits a red color owing to a 392 nm Soret absorption peak in the UV–visible absorbance spectrum of the ferric protein. Red shifts of the Soret λmax to 405 and 420 nm confirmed binding of the ligands nitrite and cyanide, respectively, to the chromophore (Fig. 2 ▶). Cld cocrystals with nitrite and cyanide exhibit a pinker hue arising from the red-shifting of the Soret absorption band.
Figure 2.

UV–visible absorbance spectra for unbound Cld (blue), nitrite-bound Cld (red) and cyanide-bound Cld (green).
3.3.2. Circular dichroism
Analysis of the Cld CD spectrum suggested a mix of α-helical (28%) and β-sheet (38%) content (Fig. 3 ▶). The high proportion of β-sheet content is unique amongst the enzymatic b-type hemoprotein family and is consistent with the crystal structures of the D. aromatica RCB Cld homologs. CD spectra were also collected for Cld with bound nitrite and cyanide, but these were identical to the spectrum of unbound Cld.
Figure 3.

Circular-dichroism spectrum of Cld.
3.4. Preliminary X-ray diffraction studies
The diffraction quality of the Cld crystals and Cld cocrystals varied greatly. Multiple Cld crystals and Cld–cyanide cocrystals were screened (∼30 each) with a range of cyroprotectants (e.g. PEG 400, glycerol, MPD), but none were found to be suitable for data collection. Unliganded Cld crystals diffracted weakly (8.0 Å in-house), while Cld–cyanide cocrystals had split diffraction spots and smearing suggesting multiple lattices. The diffraction quality of Cld–nitrite cocrystals was far superior and these were used for data collection. However, the Cld–nitrite cocrystals had to be extensively screened to find suitable candidates for X-ray data collection. Generally, only one out of every ten crystals could be used for data collection following handling and freezing, as crystalline fragility led to loss of diffraction quality. Handling appeared to be the primary problem, as crystals mounted in capillaries displayed the same loss of diffraction quality. Nitrite cocrystals of Cld diffracted to 5.0 Å resolution on an in-house Cu Kα rotating-anode X-ray source. Exposure of Cld crystals to a third-generation synchrotron X-ray source provided diffraction to 3.0 Å resolution. A crystal grown in 16% PEG 8000, 0.2 M calcium acetate, 25.5 mM sodium nitrite, 90 mM MES pH 6.5 was used for X-ray data collection. Data-collection statistics are provided in Table 1 ▶ and a diffraction image is shown in Fig. 4 ▶. The crystals belonged to the orthorhombic space group P212121, with unit-cell parameters a = 122.7, b = 202.9, c = 247.1 Å. Analysis of the Matthews coefficient (Matthews, 1968 ▶) suggested a total of three to four Cld pentamers per asymmetric unit, with a solvent content ranging from 66 to 50%, respectively. The data were of good quality, with an overall R merge of 8.9% and an estimated mosaicity of 0.32°. The average B factor derived from the Wilson plot was 49.2 Å2 (French & Wilson, 1978 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Peak | Inflection point | Native (remote) | |
|---|---|---|---|
| No. of crystals used in collection† | 6 | 3 | 1 |
| Wavelength (Å) | 1.7393 | 1.7416 | 0.9784 |
| Temperature (K) | 100 | 100 | 100 |
| Space group | P212121 | P212121 | P212121 |
| Unit-cell parameters | |||
| a (Å) | 122.99 | 122.98 | 122.7 |
| b (Å) | 201.65 | 201.65 | 202.9 |
| c (Å) | 246.89 | 247.00 | 247.1 |
| Resolution range (Å) | 50–3.76 (4.00–3.76) | 50–3.50 (3.70–3.50) | 50–3.00 (3.11–3.00) |
| No. of unique reflections | 62515 (6157) | 78393 (7728) | 123152 (12094) |
| Rmerge‡ (%) | 18.2 (37.4) | 18.3 (34.5) | 8.9 (33.0) |
| Completeness (%) | 99.9 (99.9) | 99.3 (94.7) | 99.7 (99.5) |
| Redundancy | 22.9 (22.9) | 15.0 (14.8) | 8.3 (8.4) |
| Mean I/σ(I) | 38.8 (24.05) | 27.0 (15.0) | 21.2 (6.0) |
| Experimental 〈ΔF〉〈F〉§ (%) | 1.42 | 1.05 | N/A |
| Calculated 〈ΔF〉〈F〉§ (%) | 2.21 | 1.14 | N/A |
The crystals used for data collection at the peak wavelength were grown at pH 6.5, whereas the crystals used for the inflection-point wavelength data collection were grown at pH 9.0.
R
merge = 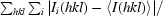
 , where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple measurements.
, where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple measurements.
Phasing power per one Fe per monomer. For significant anomalous signal 〈ΔF〉〈F〉 > 0.8.
Figure 4.

Section of an X-ray diffraction image from a Cld–nitrite cocrystal. The red-colored arcs show resolution (from left to right, the arcs show 3.0, 4.0 and 8.0 Å resolution).
Multiple anomalous dispersion (MAD) experiments were conducted on Cld–nitrite cocrystals. In conjunction with the high-energy remote (native) data, the synchrotron X-ray wavelength was tuned for data collection at the peak and inflection point of the Fe K edge determined by a fluorescence scan of the crystals (Table 1 ▶). As Cld–nitrite cocrystals incurred radiation damage towards the end of data collection, multiple crystals were merged to produce highly redundant and complete data sets. Overall, the quality of the merged Fe peak and inflection data was fair, with an R merge of 18.2 and 18.3%, respectively. The presence of significant anomalous signal, based on comparing the amplitudes of Bijvoet-related reflections, from both the Fe peak and inflection data sets was confirmed with SHELXC (Sheldrick, 2008 ▶). Attempts to solve the structure via Fe-MAD phasing and molecular replacement are currently under way.
Acknowledgments
This research was supported by the National Institutes of Health (R01 GM-66569 to CMW; R03 ES-14390 to JLD). BS was supported by an Environmental Protection Agency STAR fellowship (FP-91690601-0). Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute and we thank Can Ergenekan for his support. X-ray data were collected at the Kahlert Structural Biology Laboratory (KSBL) at The University of Minnesota and beamline 19-ID-D, SBC-CAT at the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, Illinois, USA. Argonne is operated by UChicago Argonne LLC for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The KSBL is supported by a Minnesota Partnership for Biotechnology and Medical Genomics grant SPAP-05-0013-P-FY06. We thank Ed Hoeffner for KSBL support and Steve Ginell and the staff at Sector 19, APS for their support.
References
- Bender, K. S., O’Connor, S. M., Chakraborty, R., Coates, J. D. & Achenbach, L. A. (2002). Appl. Environ. Microbiol.68, 4820–4826. [DOI] [PMC free article] [PubMed]
- Bender, K. S., Shang, C., Chakraborty, R., Belchik, S. M., Coates, J. D. & Achenbach, L. A. (2005). J. Bacteriol.187, 5090–5096. [DOI] [PMC free article] [PubMed]
- Berglund, G. I., Carlsson, G. H., Smith, A. T., Szoke, H., Henriksen, A. & Hajdu, J. (2002). Nature (London), 417, 463–468. [DOI] [PubMed]
- Betley, T. A., Wu, Q., Van Voorhis, T. & Nocera, D. G. (2008). Inorg. Chem.47, 1849–1861. [DOI] [PubMed]
- Bonagura, C. A., Bhaskar, B., Shimizu, H., Li, H., Sundaramoorthy, M., McRee, D. E., Goodin, D. B. & Poulos, T. L. (2003). Biochemistry, 42, 5600–5608. [DOI] [PubMed]
- Coates, J. D. & Achenbach, L. A. (2004). Nature Rev. Microbiol.2, 569–580. [DOI] [PubMed]
- Coates, J. D., Chakraborty, R., Lack, J. G., O’Connor, S. M., Cole, K. A., Bender, K. S. & Achenbach, L. A. (2001). Nature (London), 411, 1039–1043. [DOI] [PubMed]
- Coates, J. D., Michaelidou, U., Bruce, R. A., O’Connor, S. M., Crespi, J. N. & Achenbach, L. A. (1999). Appl. Environ. Microbiol.65, 5234–5241. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Danielsson, H., Stenklo, T. K., Karlsson, J. & Nilsson, T. (2003). Appl. Environ. Microbiol.69, 5585–5592. [DOI] [PMC free article] [PubMed]
- De Geus, D. C., Thomassen, E. A., Hagedoorn, P. L., Pannu, N. S., van Duijn, E. & Abrahams, J. P. (2009). J. Mol. Biol.387, 192–206. [DOI] [PubMed]
- Ebihara, A., Okamoto, A., Kousumi, Y., Yamamoto, H., Masui, R., Ueyama, N., Yokoyama, S. & Kuramitsu, S. (2005). J. Struct. Funct. Genomics, 6, 21–32. [DOI] [PubMed]
- Environmental Protection Agency (2002). Perchlorate Environmental Contamination: Toxicological Review and Risk Characterization, http://cfpub.epa.gov/ncea/CFM/recordisplay.cfm?deid=24002.
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Ginkel, C. G. van, Rikken, G. B., Kroon, A. G. & Kengen, S. W. (1996). Arch. Microbiol.166, 321–326. [DOI] [PubMed]
- Hagedoorn, P. L., De Geus, D. C. & Hagen, W. R. (2002). Eur. J. Biochem.269, 4905–4911. [DOI] [PubMed]
- Holm, R. H. & Donahue, J. P. (1993). Polyhedron, 12, 571–589.
- Kanitz, S., Franco, Y., Patrone, V., Caltabellotta, M., Raffo, E., Riggi, C., Timitilli, D. & Ravera, G. (1996). Environ. Health Perspect.104, 516–520. [DOI] [PMC free article] [PubMed]
- Kengen, S. W., Rikken, G. B., Hagen, W. R., van Ginkel, C. G. & Stams, A. J. (1999). J. Bacteriol.181, 6706–6711. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Lee, A. Q., Streit, B. R., Zdilla, M., Abu-Omar, M. A. & DuBois, J. L. (2008). Proc. Natl Acad. Sci. USA, 105, 15654–15659. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- McEwan, A. G., Ridge, J. P., McDevitt, C. A. & Hugenholtz, P. (2002). Geomicrobiol. J.19, 3–21.
- Michael, G. E. (1981). Arch. Environ. Health, 36, 20–27. [DOI] [PubMed]
- Murshudov, G. N., Grebenko, A. I., Brannigan, J. A., Antson, A. A., Barynin, V. V., Dodson, G. G., Dauter, Z., Wilson, K. S. & Melik-Adamyan, W. R. (2002). Acta Cryst. D58, 1972–1982. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Schlichting, I., Berendzen, J., Chu, K., Stock, A. M., Maves, S. A., Benson, D. E., Sweet, R. M., Ringe, D., Petsko, G. A. & Sligar, S. G. (2000). Science, 287, 1615–1622. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Stenklo, K., Thorell, H. D., Bergius, H., Aasa, R. & Nilsson, T. (2001). J. Biol. Inorg. Chem.6, 601–607. [DOI] [PubMed]
- Streit, B. R. & DuBois, J. L. (2008). Biochemistry, 47, 5271–5280. [DOI] [PMC free article] [PubMed]
- Thorell, H. D., Karlsson, J., Portelius, E. & Nilsson, T. (2002). Biochim. Biophys. Acta, 1577, 445–451. [DOI] [PubMed]
- Urbansky, E. T. & Schock, M. R. (1999). J. Environ. Manage.56, 79–95.
- Whitmore, L. & Wallace, B. A. (2004). Nucleic Acids Res.32, W668–W673. [DOI] [PMC free article] [PubMed]
- Whitmore, L. & Wallace, B. A. (2008). Biopolymers, 89, 392–400. [DOI] [PubMed]
- Xu, J. & Logan, B. E. (2003). J. Microbiol. Methods, 54, 239–247. [DOI] [PubMed]