

Abstract
Epilepsy is a chronic condition caused by an imbalance of normal excitatory and inhibitory forces in the brain. Antiepileptic drug therapy has been directed primarily toward reducing excitability through blockage of voltage-gated Na+ or Ca2+ channels, or increasing inhibition through enhancement of γ-aminobutyric acid currents. Prior to clinical studies, putative antiepileptic drugs are screened in animals, usually rodents. Maximal electrical shock, pentylenetetrazol, and kindling are typically used as non-mechanistic screens for antiseizure properties and the rotorod test for assessing acute toxicity. While antiseizure drug screening has been successful in bringing drugs to the market and improving our understanding of the pathophysiology of seizures, it should be emphasized that the vast majority of drug screening occurs in mature male rodents and involves models of seizures, not epilepsy. Effective drugs in acute seizures may not be effective in chronic models of epilepsy. Seizure type, clinical and electroencephalographic phenotype, syndrome, and etiology are often quite different in children with epilepsy than adults. Despite these age-related unique features, drugs used in children are generally the same as used in adults. As awareness of the unique features of seizures during development increases, it is anticipated that more drug screening in the immature animal will occur.
Keywords: seizures, epilepsy, mechanisms, convulsions, therapy
Introduction
All antiepileptic drugs (AEDs) are rigorously studied in animals, particularly rodents, before they are given to patients. Understanding how drugs are screened in animals is useful to the clinician, since the screening process is valuable in predicting the type of seizure in which the drug would be efficacious, as well as determining the mechanism of the drug's antiseizure action. Indeed, the dramatic discovery of the antiseizure properties of phenytoin was identified by Merritt and Putnam in 1938 using the electroshock-induced seizure model [1].
Animal modeling is used to enhance knowledge in a number of important areas. Modeling provides insight into fundamental neuronal mechanisms in epilepsy. Indeed, much of our understanding of seizure mechanisms comes from animal studies. Animal models are used to design new therapies with evaluations of both efficacy and toxicity of putative AEDs. Long-term consequences of seizures on learning, memory, and behavior can be studied using animal models. Finally, animal models in epilepsy can be used to determine new diagnostic techniques such as seizure prediction and detection.
Unfortunately, modeling in the developing animal has lagged far behind the adult animal. While animal models have been used to understand the unique features of seizures and epileptogenesis in the developing brain, there has been limited work in finding new therapeutic agents for seizures beginning in childhood. While AEDs that work well in the adult for partial and generalized seizures typically are effective in the developing animal, there are examples of AEDs that are more or less effective in the developing brain than in the adult brain. In addition, a major issue in pediatric AED development is the lack of efficacious and safe treatment for the epileptic syndromes. Conditions such as West syndrome, Lennox-Gastaut Syndrome, Dravet syndrome, Doose syndrome, Continuous Spike-Wave of Sleep, and Landau-Kleffner have few effective treatment options. As will be discussed, the lack of animal models has contributed to the delay in developing novel therapies.
In this review, basic pathophysiological mechanisms of seizures will be briefly reviewed which will lay the groundwork for a description of AED effects in animal models. As will be shown, a number of drug targets based on pathophysiological mechanisms of seizures have been successfully exploited with AEDs. Animal models will then be discussed, with a particular emphasis on models used in developing animals.
Mechanisms of Seizures
Epileptic seizures are characterized by hyperexcitability of central nervous system neurons. When a sufficient number of neurons synchronously depolarize and generate action potentials, a seizure begins. The location of the onset of the initial event and the propagation pattern of the discharge determine the behavioral changes that occur. Seizures are classified into two broad categories: (a) partial seizures (seizures beginning in a limited location in the brain) and (b) generalized seizures (seizures that are bilaterally symmetrical and without focal onset) [2]. The pathophysiological mechanisms of partial seizures and generalized seizures are different, although the common endpoint, such as a generalized tonic-clonic seizure, can be quite similar. Each seizure category will be briefly described.
Partial Seizures
The hallmark of partial seizures in the epileptic focus is the paroxysmal depolarization shift which is a large and sustained depolarization of neurons [3,4]. During a paroxysmal depolarization shift, the cell membrane near the soma undergoes a high voltage (10 to 15 mV) and prolonged (100 to 200 msec) depolarization. This long depolarization has the effect of generating a train of action potentials that are transmitted along the axon of the neuron. The paroxysmal depolarization shift can be generated by enhancement of Na+ or Ca2+ currents through voltage-gated channels or increased cation flow through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or N-methyl-D-aspartate (NMDA) ligand-gated channels. The paroxysmal depolarization shift is followed by a large hyperpolarization which serves to limit the duration of interictal paroxysms. This hyperpolarization is generated by currents through ionic channels, including γ-aminobutyric acid (GABA) and Ca2+-activated K+ channels. The paroxysmal depolarization shift corresponds to an interictal spike on the EEG and the subsequent hyperpolarization would correspond to the slow wave following the spike [4]. During a seizure, the epileptic neurons undergo a prolonged depolarization without a following repolarization. This prolonged paroxysmal depolarization shift results in continuous bursts of action potentials which propagate to other neurons. This recruitment of other neurons results in an electroencephalographic and clinical seizure.
While the paroxysmal depolarization shift has been useful in increasing our understanding of cellular physiology in epilepsy, the paroxysmal depolarization shift is seen in only a limited number of models. However, in virtually all animal models of epilepsy, there is a type of cellular “burst” discharge. Presumably, it is this enhanced cell excitability, or decreased inhibition, and synchrony that are responsible for both the “epileptic” EEG pattern and behavioral change. While it is clear that seizures begin at the cellular level, epilepsy is a disorder of a network of neurons that synchronously discharge together. The clinical phenotype of the seizure is dependent upon where the seizure initiates and propagates.
Figure 1 and 2 depict the primary mechanisms of antiepileptic drug action at excitatory and inhibitory synapses. In general, most antiepileptic drugs exert their action by attenuating excitatory currents, typically inward cationic currents. Drugs such as phenytoin and carbamazepine, felbamate, lamotrigine, topiramate, oxcarbazepine, valproate, and zonisamide have been demonstrated to attenuate voltage-gated Na+ channels in a use-dependent fashion [5]. Levetiracetam, lamotrigine, phenobarbital, felbamate, and topiramate target high-voltage Ca2+ channels [5]. Felbamate has a dual action, blocking NMDA channels and enhancing GABAergic effect [6]. Topiramate has been shown to attenuate non-NMDA (AMPA) and kainate currents [7]. Gabapentin and pregabalin bind to the α2γ subunit of the voltage-gated Ca2+ channel and modulate Ca2+ currents [8]. Recently, another target of AEDs, the H-channel, has been characterized. The H-channel (hyperpolarization induced, cyclic nucleotide gated), has a high permeability to K+ and tends to stabilize the membrane potential toward the resting potential against both hyperpolarizing and depolarizing inputs. AEDs, such as lamotrigine [9] and gabapentin [10] target the H-channels by increasing the hyperpolarization-activated cation current.
Figure 1.
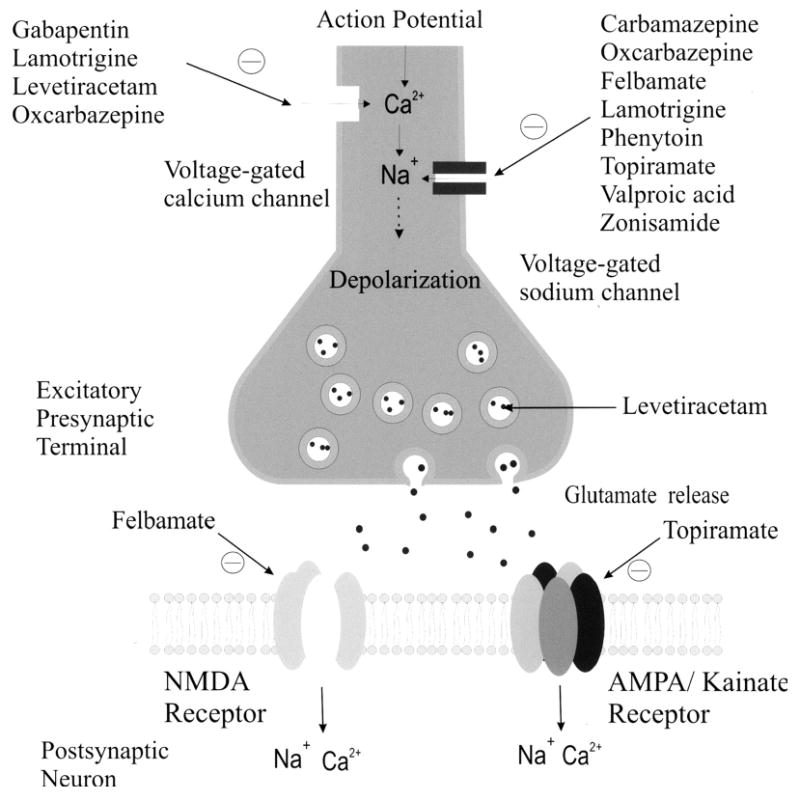
Excitatory pre- and postsynaptic neurons. AEDs may modify Na+ or Ca2+ channels or work at the postsynaptic NMDA or non-NMDA receptor.
Figure 2.
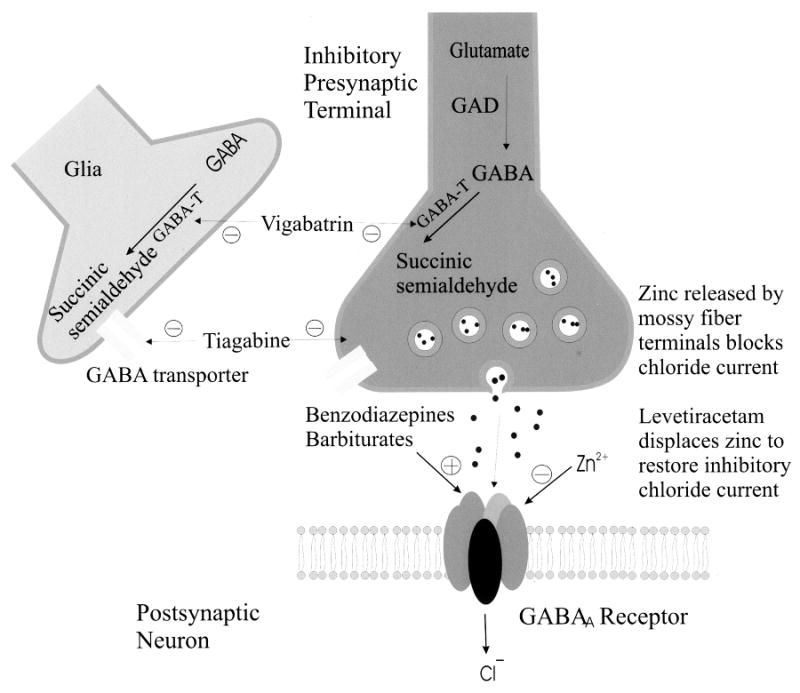
Inhibitory pre- and postsynaptic neurons. AEDs can enhance GABAA effect by increasing the frequency or duration of the channel opening. Levetiracetam can displace Zn2+, which reduces Cl- currents, from the GABAA . Vigabatrin inhibits GABA-transaminase thereby increasing intracellular GABA. Tiagabine inhibits the uptake of GABA from the synaptic cleft which increases extracellular GABA.
The mechanism of action of levetiracetam is not entirely clear. However, the drug blocks N-type Ca2+ channels and reverses the inhibition by the negative allosteric modulators zinc and beta-carbolines on neuronal GABA- and glycine-gated currents [11]. In addition, the drug binds to a synaptic protein (SV2) [12]. How binding to a synaptic protein results in reduced excitability (or enhanced inhibition) is not clear.
A large number of AEDs exert their effects through enhanced inhibition by augmenting GABA. This is accomplished by the AED acting directly at the GABAA site allosterically influencing the Cl− current (barbiturates, benzodiazepines, and felbamate), by antagonizing neuronal and glial reuptake of GABA (tiagabine), or by interfering with the metabolic breakdown of GABA (vigabatrin).
It is well recognized that the immature brain is more prone to seizures than the mature brain [13]. The underlying mechanisms responsible for this increased excitability during this period of life are not completely understood but are clearly age-dependent. In general, immature neurons and networks tend to oscillate and this inherent property is one that will facilitate the generation of pathological and pathogenic oscillations [14]. This is due to the higher input resistance of immature neurons that facilitate the generation of action potentials and enhance excitability. In addition, during the early postnatal period, at a time when the immature brain is highly susceptible to seizures [15,16], GABA exerts paradoxical excitatory action in all animal species and brain structures including primate in utero suggesting that this rule has been preserved throughout evolution [17]. Immature neurons are enriched with a high intracellular concentration of chloride leading to an efflux of chloride instead of an influx when GABA receptors are activated [18,19]. The lack of efficient GABAergic inhibition enhances excitability and may facilitate synchronicity [15,20]. The delayed maturation of postsynaptic G protein mediated GABAB inhibition also contributes to increased brain excitability [21]. The prolonged NMDA-mediated excitatory postsynaptic currents (EPSCs) in immature versus adult neurons will also promote the generation of network driven events [22].
The depolarizing effect of GABA in the immature brain may explain the limited efficacy of GABAergic drugs such as phenobarbital in newborns [23,24]. The unique physiology of excitatory and inhibitory neurotransmission in the developing brain is yet another powerful reason why AED screening should be performed in developing rats.
Absence Seizures
The basic underlying mechanism in generalized absence epilepsies involves thalamocortical circuitry and the generation of abnormal oscillatory rhythms in this neuronal network [25]. The neuronal circuit responsible for the generation of the oscillatory thalamocortical burst-firing observed during absence seizures. The circuit involved in thalamocortical burst-firing includes cortical pyramidal neurons, thalamic relay neurons, and the thalamic reticular nucleus (TRN). The principal synaptic connections of the thalamocortical circuit include glutamatergic fibers between neocortical pyramidal cells and the TRN; GABAergic connections between cells of the TRN which activate GABAA receptors; and GABAergic fibers from TRN neurons which activate GABAA and GABAB receptors on thalamic relay neurons (Fig. 3). The TRN is in a position to profoundly influence the flow of information between the thalamus and cerebral cortex. The TRN cells have rhythmic burst firing (oscillatory firing) during periods of sleep and continuous single spike firing (tonic firing) during wakefulness.
Figure 3.
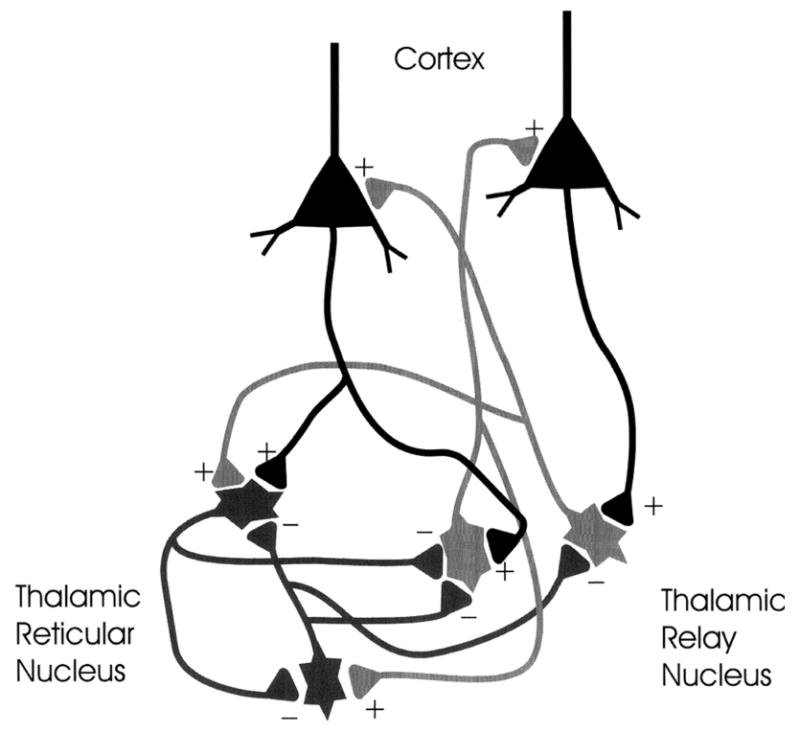
Circuitry of absence seizures. The cortex, thalamic reticular nucleus, and thalamic relay cells are the primary cell types involved in absence seizures. Enhanced inhibition through primary GABAB receptors in the thalamic relay serve to de-inactivate T-type Ca2+ channels and allow burst firing which result in absence seizures. The output of the reticular thalamic nucleus is inhibitory and serves to hyperpolarize the thalamic relay nucleus neurons.
The cellular events that underlie the ability of TRN neurons to shift between an oscillatory and tonic firing mode are the low threshold (T) Ca2+ spikes which are present in thalamocortical and TRN neurons. These T-type Ca2+ channels are involved in burst firing excitation and are associated with the change from oscillatory to burst firing in thalamocortical cells. Mild depolarization of these neurons is sufficient to activate these channels and to allow the influx of extracellular Ca2+ [26]. Further depolarization produced by Ca2+ inflow will exceed the threshold for firing a burst of action potentials. After T-channels are activated, they become inactivated rather quickly, hence the name transient. De-inactivation of T-channels requires a relatively lengthy hyperpolarization. GABAB receptor mediated hyperpolarization is a primary factor in the de-inactivation of T-channels.
The abnormal oscillatory rhythms in absence seizures could be caused by abnormalities of the T-type Ca2+ channels or enhanced GABAB function. In some animal models of absence seizures T-type Ca2+ channels activation in the TRN differs significantly from control animals [25,27]. These aberrant T-channels likely are the basis for absence seizures. In other models, there has been an increase in GABAB receptors in thalamic and neocortical neuronal populations compared to controls [28]. As would be predicted from the thalamocortical circuits involved in absence seizures, in animal models of absences, GABAB agonists produce an increase in seizure frequency while GABAB antagonists reduce seizure frequency [29,30]. Other neurotransmitter systems, i.e. serotonergic, noradrenergic, and cholinergic, can influence the thalamocortical circuits and therefore influence absence seizure frequency. However, the GABA system appears to be the critical system in the pathogenesis of absence seizures.
Recurrent collateral GABAergic fibers from the TRN neurons activate GABAA receptors on adjacent TRN neurons. Activating GABAA receptors in the TRN therefore results in an inhibition of inhibitory output to the thalamic relay neurons. Because of the decreased GABAB activation, there would be a reduced likelihood that Ca2+ de-inactivation would occur. This would result in decreased oscillatory firing. However, direct GABAA and GABAB activation of thalamic relay neurons would be expected to have detrimental effects, increasing hyperpolarization and therefore increasing the likelihood of de-inactivation of the T-channels.
As would be expected from these animal findings are the clinical observations that three drugs efficacious in the treatment of absence seizures, such as valproate, ethosuximide, and trimethadione, suppress T-currents. In addition, there is some clinical evidence that vigabatrin, which increases endogenous GABA levels and thereby increases the activation of GABAB receptors in the thalamic relay neurons, worsens absence seizures in patients. However, clonazepam, which preferentially activates GABA levels in the TRN can be a highly effective anti-absence drug [31].
Table 1 provides a summary of the major targets of the AEDs. It should be noted that multiple mechanisms of action are likely for each AED.
Table 1.
Mechanism of Antiepileptic Drugs (INaF = fast sodium currents; INaP = persistent sodium currents; HVA = high voltage activated). Modified from Rowaski and Loscher 5, with permission.
| Sodium Channel | Calcium Channels | H-Channels | GABA System | Glutamate Receptors | Other | |
|---|---|---|---|---|---|---|
| Benzodiazepines | GABAAR | |||||
| Carbamazepine | INaF | |||||
| Ethosuximide | T-type | |||||
| Felbamate | INaF | HVA | GABAAR | NMDA | ||
| Gabapentin | HVA | ↑ H-channel activated cation current | ||||
| Pregabalin | HVA | |||||
| Phenobarbital | GABAAR | |||||
| Phenytoin | INaF INaP | |||||
| Lamotrigine | INaF | HVA | ↑ H-channel activated cation current | |||
| Levetiracetam | HVA | Binds to synaptic vesicles | ||||
| Oxcarbazepine | INaF | |||||
| Tiagabine | GABA transporter | |||||
| Topiramate | INaF INaP | HVA | GABAAR | KA/AMPA | ||
| Valproate | INaF INaP | T-type | ||||
| Vigabatrin | GABA transaminase inhibitor | |||||
| Zonisamide | INaF |
Animal Screening
While in the past most AEDs were discovered fortuitously, more recently, AEDs have been designed to target one of the receptors or neurotransmitter involved in the genesis of seizures described above. Once a putative AED is identified, it is initially studied to determine whether it will be effective in generalized or partial seizures [32].
The maximal electroshock (MES) test, the subcutaneous pentylenetetrazol (scPTZ) test, and the electrical kindling model are the most commonly used models to evaluate potential new AEDs [33-37]. In the MES test an electric current of fixed intensity and duration is applied via ear-clips or corneal electrodes resulting in tonic extension of the hindlimbs. In the commonly used corneal model, a 60 Hz alternating current (mice, 50 mA; rats 150 mA) is delivered for 0.2 seconds through corneal electrodes. The animals receive a corneal anesthetic prior to the shock. Abolition of the hind limb tonic extensor component is taken as the end point for this test. Tonic extension is considered abolished if the hind limbs are not fully extended at 180 degrees with the plane of the body. Absence of this component suggests that the putative AED has the ability to prevent the spread of seizure discharge through neural tissue [32].
In the scPTZ test, a convulsive dose of PTZ is administered and the animal is observed for clonic seizures. The test drug is evaluated for its ability to suppress the clonic seizure. In both the MES and scPTZ, various doses of the test substance are evaluated.
The MES and scPTZ tests have markedly different pharmacological profiles [33]. The MES test identifies agents with activity against generalized tonic-clonic seizures while the scPTZ identifies compounds that are effective against absences and myoclonic seizures [38]. The MES model has served to identify AEDs that are functionally similar to phenytoin. AEDs that have a mechanism of action at the voltage-gated sodium channel are effective in this model [35]. The scPTZ model has proven to be a good predictor of clinical efficacy against generalized spike-wave epilepsies of the absence type. Compounds that suppress Ca2+ flux across T-type channels, such as ethosuximide or valproate, as well as compounds that enhance GABA receptor-mediated Cl− currents, such as the benzodiazepines are effective in this model. However, there are a number of drugs that block PTZ-induced seizures but are not effective against absence seizures, demonstrating one of the limitations of the model.
The MES and scPTZ tests have proven their value over decades of use. However, it has been demonstrated that the tests are not sufficient to detect effective AEDs. For example, levetiracetam is not effective in either the MES or scPTZ test but is highly effective in the kindling model [32,39,40]. Levetiracetam has been found to be highly effective in the 6-Hz seizure model. In this model, mice receive 32 mA, 6 Hz, stimulations delivered through corneal electrodes to induce a complex partial seizure. Typically, the seizure is characterized by a minimal clonic phase that is followed by stereotyped automatic behaviors [32]. Animals not displaying this behavior are considered protected.
As shown in Table 2, there appears to be a reasonable correlation between results obtained from the common animal tests and the anti-seizure profiles of AEDs. MES-induced tonic extension seizures are blocked by AEDs such as carbamazepine and phenytoin, which are known to inhibit voltage-sensitive Na+ channels, and by some drugs that enhance GABA-mediated inhibition. scPTZ-induced clonic seizures are blocked by AEDs acting at the GABAA receptor (benzodiazepine, barbiturates, valproate) and by an agent that reduces T-type Ca2+currents (ethosuximide). With the exception of ethosuximide, following full kindling, evoked seizures are blocked or attenuated by most of the standard AEDs.
Table 2.
Antiseizure profile of AEDs (+ = protective; - = non-protective; MES = maximal electroshock; scMET = subcutaneous pentylenetetrazol; scPIC = subcutaneous picrotoxin; AGS = audiogenic seizure susceptible).
| MES | scPTZ | scBIC | scPIC | AGS | 6 Hz | |
|---|---|---|---|---|---|---|
| Carbamazepine | + | - | - | + | + | +/- |
| Clonazepam | - | + | + | + | + | + |
| Ethosuximide | - | + | + | + | - | + |
| Felbamate | + | + | + | + | + | |
| Gabapentin | + | + | - | - | + | |
| Pregabalin | + | + | - | - | + | |
| Lamotrigine | + | - | - | - | + | +/- |
| Levetiracetam | - | - | + | - | + | |
| Phenobarbital | + | + | + | + | + | |
| Phenytoin | + | - | - | - | + | +/- |
| Oxcarbazepine | + | - | - | + | + | +/- |
| Tiagabine | - | + | - | + | + | + |
| Topiramate | + | - | - | - | + | - |
| Valproate | + | + | - | + | + | + |
| Vigabatrin | - | - | +/- | + | ||
| Zonisamide | + | + | - | - | + |
While the MES, scPTZ and kindling models are the best known antiseizure screening tests and are typically the first studies done with a new compound, a variety of other tests are available to further characterize the antiseizure effects of the compound. Chemically induced clonic seizures can be induced in mice by subcutaneous picrotoxin and bicuculline. Bicuculline is a GABAA receptor blocker while picrotoxin blocks the chloride channel associated with the GABAA receptor. In screening of potential anti-seizure drugs, the dosage of picrotoxin and bicuculline produce convulsions in over 97% of non-treated animals. Drugs that block the clonic seizures usually have an effect at the GABAA receptor.
The γ-hydroxybutyrate spike-and-wave model of absence seizures is used to evaluate AEDs that may have an effect on absence seizures [41]. This model has behavioral and EEG features similar to human absence seizures and shows pharmacologic specificity for anti-absence drugs such as ethosuximide and trimethadione. Moreover, the absence seizures induced by these agents are exacerbated by GABAergic agonists, a property unique to experimental absence seizures. The model is useful in studying mechanisms of pathogenesis of absence seizures as well as in screening for anti-absence seizure activity of potential antiepileptic drugs.
The kindling model is widely accepted as an excellent animal model for epileptogenesis whereby recurrent electrical or chemical stimulations induce a progressive increase in seizure intensity and duration. The model is useful in studying the mechanisms of epileptogenesis and is ideal for studying molecules that may interfere with the epileptogenic process [42-44]. In the fully kindled state the model is useful for identifying activity against partial seizures with secondary generalization [45]. To measure antiepileptogenic properties, the compound can be administered during kindling to determine if the rate of kindling or afterdischarge durations are altered, while anticonvulsive properties of drugs can be measured by giving the compound prior to a kindling stimulus in an animal previously fully kindled. Both techniques have been used extensively to test compounds. [40,46-50].
While the models mentioned here are the most common, as shown in Tables 3 and 4, there are a wide variety of models that have been studied. The models include a variety of chemoconvulsant agents, electrical stimulation, spontaneous mutations, transgenic and targeted gene knockout models of ion channels, transporters, and neurotransmission pathways, hyperthermia, hypoxia, trauma, intracerebral vascular occlusion, and radiation. Drug testing has been employed in some of the disorders listed.
Table 3.
Animal models of seizures – acquired.
| Electrical | Chemical Kindling |
| Maximal Electrical Shock | Pentylenetetrazol |
| Self-Sustaining Status Epilepticus | Beta Carboline |
| Kindling | Picrotoxin |
| Chemoconvulsant | Bicuculline |
| Pentylenetetrazol (high dose) | Cocaine |
| Bicuculline | Lidocaine |
| Picrotoxin | Hypoxia |
| Kainic Acid | Hypoxic-induced seizures |
| Pilocarpine | Focally injected convulsants |
| Flurothyl | Alumina gel |
| Chemo-non-convulsive | Tetanus toxin |
| γ-hydroxybutyrate | Cepahalosporins |
| Penicillin | Iron |
| THIP | Sodium penicillin |
| PTZ (low dose) | Trauma |
| AY-9944 | Fluid-percussion brain injury |
| MAM-AY (Double hit) | Stroke |
| Hyperthermia | Vascular occlusion |
| Experimental febrile seizures | Radiation |
| In utero radiation model |
Table 4.
Animal Models of Seizures – Genetic.
| Spontaneous Single-Locus Mouse Mutants | Spontaneous Seizures and Developmental Disruption |
| Spike-wave | Flathead rat |
| Tottering | Tish rat |
| Lethargic | Otxl-/- mouse |
| Ducky | P35-/- mouse |
| Stargazer | Models of Absence Inbred |
| SWE | Genetic Absence Epilepsy Rats from Strasbourg (GAERS) |
| Mocha2j | Wistar Albino Glaxo/Rijswijk (WAG/Rij) |
| Coloboma | Transgenic and targeted gene knockout models of |
| Convulsions | AKv1.1a – K+ channel transgene |
| Dilute lethal | Gabbr1 |
| Jimpy | Gabbr3 |
| Jittery | Gad2 |
| Megencephaly | Glra1 |
| Quaking | Hcn2 |
| Staggerer | Htr2c |
| Torpid | Kcna1 |
| Varitint waddler | Kcnc2 |
| Wabbler-lethal | Kcnq2 |
| Weaver | Scn1b |
| Writher | Scn2a |
| Convulsions evoked by sensory stimuli | Slc9a1 |
| Frings | Slc12a5 |
| Lurcher |
Toxicity of AEDs
Early testing of AEDs also includes some measures of acute toxicity. Determining the toxicity of putative AEDs is as important as determining efficacy. Screening tests usually assess motor function initially. Cognitive testing is rarely done early in the course of AED testing due to costs and time required. Commonly used tests for acute toxicity screening include the following:
Rotorod Test
This is an easily performed test where the mouse or rat is placed on a rotating rod. The measure is how long the rodent can remain on the rod. Rats that have toxicity will fall off the rod sooner than non-toxic or control rats.
Positional Sense Test
In this model the hind limbs of the rodent are slowly lowered over the edge of a table [32]. Normally the rats will quickly lift its leg back to a normal position. Neurological deficit is indicated by the inability of the animal to correct rapidly such as an abnormal position of the limb.
Gait and Posture Test
Signs of neurological damage include an irregular, ataxic gait, or abnormal body posture. A number of standardized tests are available to quantify these deficits [51,52].
Antiseizure quantification is done by calculating the median effective dose that prevents seizures in 50% of the animals (ED50) in the MES, scPTZ, or 6 Hz kindling test and the median toxic dose (TD50) that causes impairment in the rotorod test. The protective index is the ratio of the toxic dose to the effective dose (TD50/ED50). Numbers below 1 would indicate that drug has too much toxicity to be considered further. Ideally, AEDs should have high ratios with the dose causing toxicity much higher than the dose that prevents seizures.
Developmental Animal Models
It is well recognized that seizure type, clinical and electroencephalographic phenotype, syndrome, and etiology are often quite different in children than adults. The mechanisms underlying seizures in the developing brain can be different than in the adult. Therapies that may be effective in treating seizures in the adult brain may not be appropriate in the developing brain. Likewise, AEDs may have age-related toxicity. The likelihood of lamotrigine-induced rash [53], gabapentin-induced behavioral abnormalities [54,55], phenobarbital-induced hyperactivity [56], and valproate-induced hepatotoxicity [57,58] are all more likely to occur in children, than adults. Despite these age-related unique features of AEDs, the majority of AEDs used in children are the same as used in adults.
There have been relatively few studies evaluating the effects of AEDs across ages. In most cases, the AEDs used in adult models have been as effective in immature rats as adult rats. For example, gabapentin suppresses motor seizures induced by scPTZ during both early postnatal development as well as in adult rats [59]. Likewise, clobazam was found to be generally effective across developmental ages in a model of cortical epileptic afterdischarges [60]. However, this is not always the case. Haugvicova et al. [61] found a marked effect of age in response to vigabatrin in scPTZ-treated rats, with vigabatrin showing a greater effect in younger rats than older rats. The lack of comparative studies of AED effect across ages severely limits our ability to determine if AEDs have age-dependent effects in regard to efficacy and adverse reactions.
In addition to a lack of studies of AEDs across ages, there has been a lack of drug development in the epileptic encephalopathies. AEDs are typically screened in mature rats that have acute seizures rather than epilepsy and the limited number of animal models for the severe epileptic encephalopathies in children has also been a major hindrance in developing new AEDs [62]. An example of this situation is the case of vigabatrin where it has been shown that the drug is quite successful in the treatment of infantile spasms [63]. This was not predicted by animal models. Likewise, adrenocorticotrophin hormone (ACTH) has little role in treating seizures in adults, but is quiet effective in the treatment of infantile spasms, an epilepsy syndrome confined to children [63,64].
Fortunately, there are a number of developmental animal models of seizures and epilepsy which mimic childhood seizure conditions. Since these models have considerable potential for screening new compounds they will be briefly reviewed here.
Hypoxia-Induced Seizures
It has been observed that rats exposed to hypoxia have a clear age-dependence in susceptibility to seizure activity. Most rats at postnatal (P) 10 will have seizures when exposed to hypoxia; whereas, younger and older rats rarely have hypoxic-induced seizures. While it is not possible to accurately equate ages in rats and humans, a P10 rat would be roughly equivalent to a full term infant or toddler [65]. In this model, rats are exposed to global hypoxia while in an airtight chamber. Nitrogen is infused into the chamber to create a graded hypoxia over 15 minutes. The animals develop tonic-clonic head and trunk movements approximately 3-7 minutes into the hypoxic period. These brief seizures occur repetitively and become progressively longer. The EEG shows rapid spike activity that starts as low amplitude activity and gradually increases in amplitude over 5-10 seconds.
The seizures are blocked by systemic administration of the AMPA receptor antagonist 6-niytro-7-sulfamoylbenzo(f)quinoxaline-2,3-dione (NBQX). There is no antiseizure effect of the NMDA receptor antagonists MK-801, GABAA receptor agonists (lorazepam and phenobarbital) or the conventional AED phenytoin [66]. However, topiramate effectively suppresses the acute seizures in a dose-related manner [67,68].
Experimental Febrile Seizures
Prolonged febrile seizures have been modeled by hyperthermia [69-72]. In this model, rat pups at P10 are placed in a 3 liter jar and their core temperature increased using a regulated stream of heated air. Seizures are generated by maintaining hyperthermia (40-42 °C) for 30 minutes. This procedure typically results in seizures by 24 minutes. The seizures caused by the hyperthermia are limbic in semiology and involve the hippocampal formation [70]. Seizures consist of arrest of heat-induced hyperkinesis followed by facial automatisms, often followed by body flexion. These behavioral events correlate with electrographic hippocampal seizures [70,71,73].
The seizures induce transient neuronal injury but no cell death [71,74]. T2-weighted signal changes have been found in the hippocampus following the hyperthermic seizures [75]. The prolonged experimental febrile seizures cause profound and enduring alterations in the expression of several channel genes [73,76]. Following the febrile seizure, there is enhanced long-term hippocampal excitability [70] with spontaneous seizures and interictal epileptiform activity in a proportion of the rats [72]. The features of the model replicate the human condition quite well.
Hyperthermic seizures are prevented by phenobarbital, valproate, and the benzodiazepines but not by phenytoin [77,78]. While hyperthermic seizures mimic human febrile seizures in response to AEDs and long-term sequelae, it is important to note that the mechanisms responsible for seizures in a child with endogenous fever is likely to be different from seizures induced by raising temperatures through exogenous means. Nevertheless, this model has been shown to be highly valuable.
Recurrent seizures
The volatile agent flurothyl (bis-2, 2, 2-triflurothyl ether) is a potent and rapidly acting central nervous system stimulant that produces seizures within minutes of exposure [79]. Because of its reliability and reproducibility, it is frequently used to induce recurrent seizures in young animals. Serial administration of flurothyl models recurrent generalized tonic-clonic seizures [80].
The typical way that flurothyl is administered is through inhalation. Rats are placed in a plastic container that allows them to turn, rear, and move about and liquid flurothyl is administered via a pump at a constant rate onto filter paper suspended in the center of the container where it evaporates. When inducing recurrent seizures, rats are exposed to flurothyl until tonic extension of both the forelimbs and hind limbs is observed. The animals are then quickly removed from the chamber and allowed to recover in room air. Because the seizures end within 30 seconds of placing the animals in room air, the investigator has control of the duration and severity of the seizures. For example, by maintaining the animal in the testing chamber with continuous administration of flurothyl, status epilepticus can be elicited [81]. For the purpose of recurrent seizures, the rats are usually removed from the chamber once the tonic phase begins.
There are age-related differences in the clinical features of flurothyl-induced seizures [82]. Gatt and colleagues [83] administered flurothyl to rats at postnatal days 5, 10, 20, 30, and 70 and monitored both behavioral and EEG changes. In all age groups flurothyl inhalation initially resulted in agitation with increased exploring of the testing chamber. Swimming movements were prominent in P5 pups, followed by abrupt onset of tonus. Clonic seizures were not fully developed before P10. Behavioral features of the seizures did not change after P20 and consisted of myoclonic jerks followed by forelimb clonus, wild running, loss of posture and severe tonic posturing. Urinary and fecal incontinence and salivation were common.
Recurrent flurothyl seizures during the first two weeks of life result in no discernible cell loss [84-86]. However, when the animals are studied as adults, there is extensive synaptic reorganization of the axons and terminals of the dentate granule cells, the so-called mossy fibers [86,87]. The sprouting of mossy fibers occurs in both the molecular region of the dentate granule cells as well as the CA3 hippocampal subfield.
Recurrent flurothyl seizures during early development can alter the formation of new neurons. McCabe et al. [88] studied the extent of neurogenesis in the granule cell layer of the dentate gyrus over multiple time points following a series of 25 flurothyl-induced seizures administered between P0 and P4. Rats with neonatal seizures had a significant reduction in the number of the thymidine analog 5-bromo-2′-deoxyuridine-5′-monophosphate- (BrdU) labeled cells in the dentate gyrus and hilus compared to the control groups when the animals were sacrificed either 36 hours or two weeks after the BrdU injections. The reduction in BrdU-labeled cells continued for six days following the last seizure. BrdU-labeled cells largely co-localized with the neuronal marker neuron-specific nuclear protein and rarely co-localized with the glial cell marker glial fibrillary acidic protein (GFAP), providing evidence that a very large percentage of the newly formed cells were neurons. Immature rats subjected to a single seizure did not differ from controls in number of BrdU-labeled cells. In comparison, adult rats undergoing a series of 25 flurothyl-induced seizures had a significant increase in neurogenesis compared to controls. This study indicates that following recurrent seizures in the neonatal rat, there is a reduction in newly born granule cells. These seizure-induced decreases in neurogenesis is consistent with prior studies by Wasterlain and colleagues [89-91] who concluded that recurrent neonatal seizures, while not causing cell death, resulted in reduced cell number.
Following neonatal seizures there is impairment of visual spatial memory in the Morris water maze with rats subjected to recurrent flurothyl seizures during the neonatal period requiring longer times to find the platform than controls [84,86,87,92,93]. This impairment occurs when the rats are tested either during adolescence or when fully mature. Recurrent seizures are critical in causing the cognitive impairment. When only a single flurothyl seizure is induced at P6, no impairment in water maze performance occurs [93]. Animals subjected to recurrent flurothyl seizures between P15-20 also have been shown to have impairment of auditory discrimination [92].
Recurrent flurothyl seizures in immature rats do not lead to spontaneous recurrent seizures. However, animals subjected to multiple flurothyl-induced seizures demonstrate a kindling phenomenon with a decreased latency to forelimb clonus 84. In addition, recurrent flurothyl seizures are associated with a reduced seizure threshold when examined at an older age [86,94].
The basis for the reduced seizure threshold following recurrent flurothyl seizures has been addressed using in vitro recordings [95,96]. Rats that experience neonatal seizures have decreases in the amplitude of spontaneous inhibitory postsynaptic currents in CA3 [96]. In addition, intracellular recordings of CA1 and CA3 pyramidal neurons from neonatal flurothyl-induced seizures revealed impairment in spike frequency adaptation. In addition, the after-hyperpolarizing potentials following a spike train were markedly reduced when compared with controls. In contrast, no significant alterations in the firing properties of CA3 pyramidal neurons were found. It was concluded that neonatal seizures lead to persistent changes in intrinsic membrane properties of CA1 pyramidal neurons. These alterations are consistent with an increase in neuronal excitability and may contribute to the behavioral deficit and epileptogenic predisposition observed in rats that experienced repeated neonatal seizures. In addition, rats that experienced neonatal seizures have a reduction in the amplitude of spontaneous inhibitory postsynaptic currents [96].
While the model has been widely used to assess the effects of convulsive seizures during development, AEDs have not been extensively studied in this model. In unpublished work from our laboratory we found that gabapentin was effective against flurothyl-induced seizures across ages.
Absence seizures
Although there are a number of genetic and pharmacologically-induced models of absence seizures, most of the animals start having seizures when they are adults. One exception is the AY-9944 model. In this model, AY-9944 is given to rat pups. AY-9944 inhibits the reduction of 7-dehydrocholesterol to cholesterol [97-100]. For reasons that are not entirely clear, the rats begin having atypical absences during preadolescence [98]. Behaviorally, atypical absence seizures in the AY-9944 model are more complex than typical absence seizures with a gradual onset and offset of the seizures. In addition, the EEG is not time-locked with the ictal behavior. The absences are atypical in that they have a slower frequency of spike-wave than in animals with typical absences, involve hippocampal structures more than typical absences, and are associated with cognitive impairment [97]. Of significant interest is the finding that GABAB antagonists improve the cognitive deficits in these animals without having a discernible effect on the spike-wave activity [101].
The seizures in the model are blocked or attenuated by ethosuximide, benzodiazepines, and valproic acid and are worsened by phenytoin or carbamazepine [97].
Infantile Spasms
The development of effective therapies for infantile spasms has been significantly hindered by the lack of adequate animal models [62]. Fortunately, there is now more interest in developing animal models of infantile spasms [102].
The clinical phenotype of infantile spasms can be modeled by the administration of NMDA to rat pups [103-105]. These NMDA-induced seizures resembled clinical spasms with onset of spasm-like activity in immature rats, occurrence of spasms in clusters, and an EEG which resembled hypsarhythmia. In addition, rats with NMDA-induced flexion seizures have impaired cognition when studied in adulthood [106]. However, the pharmacological response of the spasm-like behavior differed from children with infantile spasms in that the rats did not respond to glucocorticoids [103].
Recently, the model was altered by Velisek and colleagues [105], who reasoned that since infantile spasms may be secondary to disorders of the hypothalamic-pituitary-adrenal system [107], alteration of the adrenal brain axis may predispose the immature brain to spasms. Since prenatal administration of a glucocorticoid can impair brain hypothalamic-pituitary-adrenal function [108], the authors administered betamethasone to pregnant rats. They then gave the offspring NMDA at P15. The rats developed spasm-like behavior with associated high-amplitude discharges followed by an attenuation of the EEG or a high-amplitude discharge followed by a rhythmic afterdischarge. Importantly, ACTH significantly increased the latency to onset of the spasms. While the model does not totally mimic the human condition [109], the model has considerable potential for screening AEDs.
Conclusions
Animal models have taught us much about the pathophysiology of seizures and have been critical in getting a large number of safe and effective AEDs to the market. While children have benefited enormously from these drugs, screening of putative AEDs rarely includes immature animals. Early screening of putative AEDs in developing animals may allow investigators to find drugs that have an age-specific effect. Current AED screening is useful in evaluating drugs that work in partial and generalized seizures but do not identify drugs that work in specific pediatric syndromes such as the epileptic encephalopathies. Unfortunately, including developing animals in early stages of drug testing will increase costs and time without a guarantee that novel compounds that work in children will be identified. Fortunately, investigators have now developed animal models of hypoxia-induced seizures, febrile seizures, infantile spasms, and recurrent generalized tonic-clonic seizures and it is anticipated that these models will spur the study of new therapies.
Acknowledgments
Supported by the Western Massachusetts Epilepsy Awareness Fund, Friends of Shannon McDermott, and the NINDS (Grants: NS27984 and NS44295).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Merritt HH, Putnam TJ. A new series of anticonvulsant drugs tested by experiments on animals. Arch Neurol Psych. 1938;39:1003–1015. [Google Scholar]
- 2.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:249–260. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto H, Ajmone-Marsan C. Cortical cellular phenomena in experimental epilepsy: ictal manifestations. Exp Neurol. 1964;25:305–326. doi: 10.1016/0014-4886(64)90026-3. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto H, Ajmone-Marsan C. Cortical cellular phenomena in experimental epilepsy: interictal manifestations. Exp Neurol. 1964;80:286–304. doi: 10.1016/0014-4886(64)90025-1. [DOI] [PubMed] [Google Scholar]
- 5.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 6.Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: Opposing effects on N-methyl-D-aspartate and GABBAA receptors. Ann Neurol. 1994;35:229–234. doi: 10.1002/ana.410350216. [DOI] [PubMed] [Google Scholar]
- 7.Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41 Suppl 1:S3–S9. [PubMed] [Google Scholar]
- 8.Dooley DJ, Donovan CM, Meder WP, Whetzel SZ. Preferential action of gabapentin and pregabalin at P/Q-type voltage-sensitive calcium channels: inhibition of K+-evoked [3H]-norepinephrine release from rat neocortical slices. Synapse. 2002;45:171–190. doi: 10.1002/syn.10094. [DOI] [PubMed] [Google Scholar]
- 9.Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- 10.Surges R, Freiman TM, Feuerstein TJ. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia. 2003;44:150–156. doi: 10.1046/j.1528-1157.2003.36802.x. [DOI] [PubMed] [Google Scholar]
- 11.Rigo JM, Hans G, Nguyen L, Rocher V, Belachew S, Malgrange B, et al. The antiepileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glyucine-gated currents. Br J Pharmacol. 2002;136:659–672. doi: 10.1038/sj.bjp.0704766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055–1063. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Ari Y, Holmes GL. The multiple facets of gamma-aminobutyric acid dysfunction in epilepsy. Curr Opin Neurol. 2005;18:141–145. doi: 10.1097/01.wco.0000162855.75391.6a. [DOI] [PubMed] [Google Scholar]
- 15.Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590–600. doi: 10.1111/j.0953-816x.2003.03152.x. [DOI] [PubMed] [Google Scholar]
- 16.Jensen FE, Baram TZ. Developmental seizures induced by common early-life insults: short- and long-term effects on seizure susceptibility. Ment Retard Dev Disabil Res Rev. 2000;6:253–257. doi: 10.1002/1098-2779(2000)6:4<253::AID-MRDD4>3.0.CO;2-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khazipov R, Esclapez M, Caillard O, Bernard C, Khalilov I, Tyzio R, et al. Early development of neuronal activity in the primate hippocampus in utero. J Neurosci. 2001;21:9770–9781. doi: 10.1523/JNEUROSCI.21-24-09770.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 20.Dzhala VI, Staley KJ. Transition from interictal to ictal activity in limbic networks in vitro. J Neurosci. 2003;23:7873–7880. doi: 10.1523/JNEUROSCI.23-21-07873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nurse S, Lacaille JC. Late maturation of GABA(B) synaptic transmission in area CA1 of the rat hippocampus. Neuropharmacology. 1999;38:1733–1742. doi: 10.1016/s0028-3908(99)00122-7. [DOI] [PubMed] [Google Scholar]
- 22.Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit composition shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- 24.Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 25.Holmes GL. Models for generalized seizures. Suppl Clin Neurophysiol. 2004;57:415–424. doi: 10.1016/s1567-424x(09)70379-4. [DOI] [PubMed] [Google Scholar]
- 26.Crunelli V, Leresche N. A role for GABAB receptors in excitation and inhibition of thalamocortical cells. TINS. 1991;14:16–21. doi: 10.1016/0166-2236(91)90178-w. [DOI] [PubMed] [Google Scholar]
- 27.Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249–5257. doi: 10.1523/JNEUROSCI.5546-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin FH, Cao Z, Hosford DA. Increased number of GABAB receptors in the lethargic (lh/lh) mouse model of absence epilepsy. Brain Res. 1993;608:101–106. doi: 10.1016/0006-8993(93)90779-m. [DOI] [PubMed] [Google Scholar]
- 29.Snead OC., III Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;37:146–157. doi: 10.1002/ana.410370204. [DOI] [PubMed] [Google Scholar]
- 30.Hosford DA, Clark S, Cao Z, Wilson WA, Lin FH, Morrisett RA, et al. The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science. 1992;257:398–401. doi: 10.1126/science.1321503. [DOI] [PubMed] [Google Scholar]
- 31.Huguenard JR, Prince DA. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. J Neurophysiol. 1994;71:2576–2581. doi: 10.1152/jn.1994.71.6.2576. [DOI] [PubMed] [Google Scholar]
- 32.White HS, Woodhead JH, Wilcox KS, Stables JP, Kupferberg HJ, Wolf HH. Discovery and preclinical development of antiepileptic drugs. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. Fifth. Philadephia: Lippincott Williams & Williams; 2002. pp. 36–48. [Google Scholar]
- 33.White HS. Clinical significance of animal seizure models and mechanism of action studies of potential antiepileptic drugs. Epilepsia. 1997;38(Suppl 1):S9–S17. doi: 10.1111/j.1528-1157.1997.tb04523.x. [DOI] [PubMed] [Google Scholar]
- 34.White HS, Johnson M, Wolf HH, Kupferberg HJ. The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Neurol Sci. 1995;16:73–77. doi: 10.1007/BF02229077. [DOI] [PubMed] [Google Scholar]
- 35.Rho JM, Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40:1471–1483. doi: 10.1111/j.1528-1157.1999.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 36.Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia. 1978;19:409–428. doi: 10.1111/j.1528-1157.1978.tb04507.x. [DOI] [PubMed] [Google Scholar]
- 37.Krall RL, Penry JK, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: I. History and a program for progress. Epilepsia. 1978;19:393–408. doi: 10.1111/j.1528-1157.1978.tb04506.x. [DOI] [PubMed] [Google Scholar]
- 38.Kupferberg HJ, Schmutz M. Screening of new compounds and the role of the pharmaceutical industry. In: Engel J Jr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: 1997. pp. 1417–1434. [Google Scholar]
- 39.Klitgaard H, Matagne A, Gobert J, Wulfert E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol. 1998;353:191–206. doi: 10.1016/s0014-2999(98)00410-5. [DOI] [PubMed] [Google Scholar]
- 40.Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284:474–479. [PubMed] [Google Scholar]
- 41.Snead OC., III Pharmacological models of generalized absence seizures in rodents. J Neural Transm Suppl. 1992;35:7–19. doi: 10.1007/978-3-7091-9206-1_2. [DOI] [PubMed] [Google Scholar]
- 42.Stables JP, Bertram E, Dudek FE, Holmes G, Mathern G, Pitkanen A, et al. Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia. 2003;44:1472–1478. doi: 10.1111/j.0013-9580.2003.32803.x. [DOI] [PubMed] [Google Scholar]
- 43.McNamara JO. Kindling model of epilepsy. Adv Neurol. 1986;44:303–318. [PubMed] [Google Scholar]
- 44.McNamara JO, Byrne MC, Dasheiff RM, Fitz JG. The kindling model of epilepsy: a review. Prog Neurobiol. 1980;15:139–159. doi: 10.1016/0301-0082(80)90006-4. [DOI] [PubMed] [Google Scholar]
- 45.McNamara JO. Development of new pharmacological agents for epilepsy: lessons from the kindling model. Epilepsia. 1989;30 1:S13–S18. doi: 10.1111/j.1528-1157.1989.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 46.Stratton SC, Large CH, Cox B, Davies G, Hagan RM. Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model. Epilepsy Res. 2003;53:95–106. doi: 10.1016/s0920-1211(02)00254-1. [DOI] [PubMed] [Google Scholar]
- 47.Hashimoto Y, Araki H, Futagami K, Kawasaki H, Gomita Y. Effects of valproate, phenytoin, and zonisamide on clonic and tonic seizures induced by acute and repeated exposure of mice to flurothyl. Physiol Behav. 2003;78:465–469. doi: 10.1016/s0031-9384(03)00013-1. [DOI] [PubMed] [Google Scholar]
- 48.Loscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50:105–123. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- 49.Namba T, Morimoto K, Sato K, Yamada N, Kuroda S. Antiepileptogenic and anticonvulsant effects of NBQX, a selective AMPA receptor antagonist, in the rat kindling model of epilepsy. Brain Res. 1993;638:36–44. doi: 10.1016/0006-8993(94)90630-0. [DOI] [PubMed] [Google Scholar]
- 50.Holmes GL, Weber DA. The effect of progesterone on kindling: a developmental study. Brain Res. 1984;318:45–53. doi: 10.1016/0165-3806(84)90061-0. [DOI] [PubMed] [Google Scholar]
- 51.Pellis SM, Pellis VC. Posture. In: Whishaw IQ, Kolb B, editors. The Behavior of the Laboratory Rat A Handbook with Tests. Oxford: Oxford University Press; 2005. pp. 121–128. [Google Scholar]
- 52.Muir G. Locomotion. In: Whishaw IQ, Kolb B, editors. The Behavior of the Laboratory Rat A Handbook with Tests. Oxford: Oxford University Press; 2005. pp. 150–161. [Google Scholar]
- 53.Hirsch LJ, Weintraub DB, Buchsbaum R, Spencer HT, Straka T, Hager M, et al. Predictors of Lamotrigine-associated rash. Epilepsia. 2006;47:318–322. doi: 10.1111/j.1528-1167.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 54.Wolf SM, Shinnar S, Kang H, Gil KB, Moshé SL. Gabapentin toxicity in children manifesting as behavioral changes. Epilepsia. 1995;36:1203–1205. doi: 10.1111/j.1528-1157.1995.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 55.Khurana DS, Riviello J, Helmers S, Holmes G, Anderson J, Mikati MA. Efficacy of gabapentin therapy in children with refractory partial seizures. J Pediatr. 1996;128:829–833. doi: 10.1016/s0022-3476(96)70336-0. [DOI] [PubMed] [Google Scholar]
- 56.Schubert R. Attention deficit disorder and epilepsy. Pediatr Neurol. 2005;32:1–10. doi: 10.1016/j.pediatrneurol.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 57.Dreifuss FE, Santilli N, Langer DH, Sweeney KP, Moline KA, Menander KB. Valproic acid hepatic fatalities: a retrospective review. Neurology. 1987;37:379–385. doi: 10.1212/wnl.37.3.379. [DOI] [PubMed] [Google Scholar]
- 58.Dreifuss FE, Langer DH, Moline KA, Maxwell JE. Valproic acid hepatic fatalities: US experience since 1984. Neurology. 1989;39:201–207. doi: 10.1212/wnl.39.2.201. [DOI] [PubMed] [Google Scholar]
- 59.Mares P, Haugvicova R. Anticonvulsant action of gabapentin during postnatal development in rats. Epilepsia. 1997;38:893–896. doi: 10.1111/j.1528-1157.1997.tb01254.x. [DOI] [PubMed] [Google Scholar]
- 60.Slamberova R, Mares P, Vorlicek J. Clobazam exerts an anticonvulsant action in immature rats. Physiol Res. 1998;47:301–305. [PubMed] [Google Scholar]
- 61.Haugvicova R, Kubova H, Mares P. Does vigabatrin possess an anticonvulsant action against pentylenetetrazol-induced seizures in developing rats? Physiol Res. 2002;51:363–370. [PubMed] [Google Scholar]
- 62.Stafstrom CE, Holmes GL. Infantile spasms: criteria for an animal model. Int Rev Neurobiol. 2002;49:391–411. doi: 10.1016/s0074-7742(02)49023-x. [DOI] [PubMed] [Google Scholar]
- 63.Mackay MT, Weiss SK, ms-Webber T, Ashwal S, Stephens D, Ballaban-Gill K, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62:1668–1681. doi: 10.1212/01.wnl.0000127773.72699.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Snead OCI, Benton JW, Myers GL. ACTH and prednisone in childhood seizure disorders. Neurology. 1983;33:966–970. doi: 10.1212/wnl.33.8.966. [DOI] [PubMed] [Google Scholar]
- 65.Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jensen FE, Blume H, Alvarado S, Firkusny I, Geary C. NBQX blocks acute and late epileptogenic effects of perinatal hypoxia. Epilepsia. 1995;36:966–972. doi: 10.1111/j.1528-1157.1995.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 67.Koh S, Tibayan FD, Simpson JN, Jensen FE. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569–575. doi: 10.1111/j.0013-9580.2004.69103.x. [DOI] [PubMed] [Google Scholar]
- 68.Koh S, Jensen FE. Topiramate blocks perinatal hypoxia-induced seizures in rat pups. Ann Neurol. 2001;50:366–372. doi: 10.1002/ana.1122. [DOI] [PubMed] [Google Scholar]
- 69.Chen K, Baram TZ, Soltesz I. Febrile seizures in the immature rat model modify neuronal excitability long-term. Nature Medicine. 1999;5:888–894. doi: 10.1038/11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dube C, Chen K, Eghbal-Ahmadi M, Brunson K, Soltesz I, Baram TZ. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann Neurol. 2000;47:336–344. [PMC free article] [PubMed] [Google Scholar]
- 71.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dube C, Richichi C, Bender RA, Chung G, Litt B, Baram TZ. Temporal lobe epilepsy after experimental prolonged febrile seizures: prospective analysis. Brain. 2006;129:911–922. doi: 10.1093/brain/awl018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bender RA, Dubé C, Gonzalez-Vega R, Mina EW, Baram TZ. Mossy fiber plasticity and enhanced hippocampal excitability, without hippocampal cell loss or altered neurogenesis, in an animal model of prolonged febrile seizures. Hippocampus. 2004;13:399–412. doi: 10.1002/hipo.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dube C, Yu H, Nalcioglu O, Baram TZ. Serial MRI after experimental febrile seizures: altered T2 signal without neuronal death. Ann Neurol. 2004;56:709–714. doi: 10.1002/ana.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brewster AL, Bernard JA, Gall CM, Baram TZ. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol Dis. 2005;19:200–207. doi: 10.1016/j.nbd.2004.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olson JE, Scher MS, Holtzman D. Effects of anticonvulsants on hyperthermia-induced seizures in the rat pup. Epilepsia. 1984;25:96–99. doi: 10.1111/j.1528-1157.1984.tb04161.x. [DOI] [PubMed] [Google Scholar]
- 78.Dubé CM, Baram TZ. Complex febrile seizures - an experimental model in immature rodents. In: Pitkänen A, Schwartzkroin PA, Moshé S, editors. Models of Seizures and Epilepsy. Burlington, Massachusetts: Elsevier; 2006. pp. 333–340. [Google Scholar]
- 79.Truitt EB, Ebersberger EM, Ling ASC. Measurement of brain excitability by use of hexaflurodiethyl ether (Indoklon) J Pharmacol Exp Ther. 1960;129:445–453. [Google Scholar]
- 80.Moshé SL, Velísek L, Holmes GL. Developmental aspects of experimental generalized seizures induced by pentylenetetrazol, bicuculline and flurothyl. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic Generalized Epilepsies. London: John Libbey; 1994. pp. 51–64. [Google Scholar]
- 81.Sperber EF, Haas KZ, Romero MT, Stanton PK. Flurothyl status epilepticus in developing rats: behavioral, electrographic, histological and electrophysiological studies. Develop Brain Res. 1999;116:59–68. doi: 10.1016/s0165-3806(99)00075-9. [DOI] [PubMed] [Google Scholar]
- 82.Sperber EF, Moshé SL. Age-related differences in seizure susceptibility to flurothyl. Dev Brain Res. 1988;39:295–297. doi: 10.1016/0165-3806(88)90033-8. [DOI] [PubMed] [Google Scholar]
- 83.Gatt G, Veliskova J, Liu Z, Moshé SL, Holmes GL. Ontogeny of flurothyl-induced seizures: A behavioral and EEG electroencephalographic analysis. Epilepsia. 1993;34(Suppl 6):63. [Google Scholar]
- 84.Liu Z, Yang Y, Silveira DC, Sarkisian MR, Tandon P, Huang LT, et al. Consequences of recurrent seizures during early brain development. Neuroscience. 1999;92:1443–1454. doi: 10.1016/s0306-4522(99)00064-0. [DOI] [PubMed] [Google Scholar]
- 85.Riviello P, de Rogalski Landrot I, Holmes GL. Lack of cell loss following recurrent neonatal seizures. Brain Res Dev Brain Res. 2002;135:101–104. doi: 10.1016/s0165-3806(02)00302-4. [DOI] [PubMed] [Google Scholar]
- 86.Holmes GL, Gairsa JL, Chevassus-Au-Louis N, Ben-Ari Y. Consequences of neonatal seizures in the rat: morphological and behavioral effects. Ann Neurol. 1998;44:845–857. doi: 10.1002/ana.410440602. [DOI] [PubMed] [Google Scholar]
- 87.Huang L, Cilio MR, Silveira DC, McCabe BK, Sogawa Y, Stafstrom CE, et al. Long-term effects of neonatal seizures: a behavioral, electrophysiological, and histological study. Brain Res Dev Brain Res. 1999;118:99–107. doi: 10.1016/s0165-3806(99)00135-2. [DOI] [PubMed] [Google Scholar]
- 88.McCabe BK, Silveira DC, Cilio MR, Cha BH, Liu Z, Sogawa Y, et al. Reduced neurogenesis after neonatal seizures. J Neurosci. 2001;21:2094–2103. doi: 10.1523/JNEUROSCI.21-06-02094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wasterlain CG. Effects of neonatal status epilepticus on rat brain development. Neurology. 1976;26:975–986. doi: 10.1212/wnl.26.10.975. [DOI] [PubMed] [Google Scholar]
- 90.Wasterlain CG. Neonatal seizures and brain growth. Neuropaediatrie. 1978;9:213–228. doi: 10.1055/s-0028-1091482. [DOI] [PubMed] [Google Scholar]
- 91.Wasterlain CG, Plum F. Vulnerability of developing rat brain to electroconvulsive seizures. Archives of Neurology. 1973;29:38–45. doi: 10.1001/archneur.1973.00490250056006. [DOI] [PubMed] [Google Scholar]
- 92.Neill J, Liu Z, Sarkisian M, Tandon P, Yang Y, Stafstrom CE, et al. Recurrent seizures in immature rats: effect on auditory and visual discrimination. Dev Brain Res. 1996;95:283–292. doi: 10.1016/0165-3806(96)00099-5. [DOI] [PubMed] [Google Scholar]
- 93.Bo T, Jiang Y, Cao H, Wang J, Wu X. Long-term effects of seizures in neonatal rats on spatial learning ability and N-methyl-d-aspartate receptor expression in the brain. Brain Res Dev Brain Res. 2004;152:137–142. doi: 10.1016/j.devbrainres.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 94.Sogawa Y, Monokoshi M, Silveira DC, Cha BH, Cilio MR, McCabe BK, et al. Timing of cognitive deficits following neonatal seizures: relationship to histological changes in the hippocampus. Brain Res Dev Brain Res. 2001;131:73–83. doi: 10.1016/s0165-3806(01)00265-6. [DOI] [PubMed] [Google Scholar]
- 95.Villeneuve N, Ben-Ari Y, Holmes GL, Gaiarsa JL. Neonatal seizures induced persistent changes in intrinsic properties of CA1 rat hippocampal cells. Ann Neurol. 2000;47:729–738. [PubMed] [Google Scholar]
- 96.Isaeva E, Isaev D, Khazipov R, Holmes GL. Selective impairment of GABAergic synaptic transmission in the flurothyl model of neonatal seizures. Eur J Neurosci. 2006;23:1559–1566. doi: 10.1111/j.1460-9568.2006.04693.x. [DOI] [PubMed] [Google Scholar]
- 97.Cortez MA, McKerlie C, Snead OC., III A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology. 2001;56:341–349. doi: 10.1212/wnl.56.3.341. [DOI] [PubMed] [Google Scholar]
- 98.Persad V, Cortez MA, Snead OC., III A chronic model of atypical absence seizures: studies of developmental and gender sensitivity. Epilepsy Res. 2002;48:11–119. doi: 10.1016/s0920-1211(01)00319-9. [DOI] [PubMed] [Google Scholar]
- 99.Chan KF, Jia Z, Murphy PA, Burnham WM, Cortez MA, Snead OC., III Learning and memory impairment in rats with chronic atypical absence seizures. Exp Neurol. 2004;190:328–336. doi: 10.1016/j.expneurol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 100.Stewart LS, Bercovici E, Shukla R, Serbanescu I, Persad V, Mistry N, et al. Daily rhythms of seizure activity and behavior in a model of atypical absence epilepsy. Epilepsy Behav. 2006;9:564–572. doi: 10.1016/j.yebeh.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 101.Chan KF, Burnham WM, Jia Z, Cortez MA, Snead OC., III GABAB receptor antagonism abolishes the learning impairments in rats with chronic atypical absence seizures. Eur J Pharmacol. 2006;541:64–72. doi: 10.1016/j.ejphar.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 102.Stafstrom CE, Moshe SL, Swann JW, Nehlig A, Jacobs MP, Schwartzkroin PA. Models of pediatric epilepsies: strategies and opportunities. Epilepsia. 2006;47:1407–1414. doi: 10.1111/j.1528-1167.2006.00674_1.x. [DOI] [PubMed] [Google Scholar]
- 103.Kabova R, Liptakova S, Slamberova R, Pometlova M, Velisek L. Age-specific N-methyl-D-aspartate-induced seizures: perspectives for the West syndrome model. Epilepsia. 1999;40:1357–1369. doi: 10.1111/j.1528-1157.1999.tb02006.x. [DOI] [PubMed] [Google Scholar]
- 104.Mares P, Velisek L. N-methyl-D-aspartate (NMDA)-induced seizures in developing rats. Brain Res Dev Brain Res. 1992;65:185–189. doi: 10.1016/0165-3806(92)90178-y. [DOI] [PubMed] [Google Scholar]
- 105.Velisek L, Jehle K, Asche S, Veliskova J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61:109–119. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- 106.Stafstrom CE, Sasaki-Adams DM. NMDA-induced seizures in developing rats cause long-term learning impairment and increased seizure susceptibility. Epilepsy Res. 2003;53:129–137. doi: 10.1016/s0920-1211(02)00258-9. [DOI] [PubMed] [Google Scholar]
- 107.Baram TZ. Pathophysiology of massive infantile spasms: perspective on the putative role of brain adrenal axis. Ann Neurol. 1993;33:231–236. doi: 10.1002/ana.410330302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Welberg LA, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience. 2001;104:71–79. doi: 10.1016/s0306-4522(01)00065-3. [DOI] [PubMed] [Google Scholar]
- 109.Baram TZ. Models for infantile spasms: an arduous journey to the Holy Grail. Ann Neurol. 2007;61:89–91. doi: 10.1002/ana.21075. [DOI] [PMC free article] [PubMed] [Google Scholar]