

Abstract
The combination of bone morphogenetic protein 7 (BMP7) and neurotrophins (e.g. brain-derived neurotrophic factor, BDNF) protects septal neurons during hypoglycemic stress. We investigated the signaling mechanisms underlying this synergistic protection. BMP7 (5 nM) increased phosphorylation and nuclear translocation of BMP-responsive Smads 1/5/8 within 30 min in cultures of rat embryonic septal neurons. BDNF (100 ng/ml) enhanced the BMP7-induced increase in phospho-Smad levels in both nucleus and cytoplasm; this effect was more pronounced after a hypoglycemic stress. BDNF increased both Akt and Erk phosphorylation, but pharmacological blockade of these kinase pathways (with wortmannin and U0126, respectively) did not reduce the Smad phosphorylation produced by the BMP7+BDNF combination. Inhibitors of casein kinase II (CK2) activity reduced the (BMP7 + BDNF)-induced Smad phosphorylation, and this trophic factor combination increased CK2 activity in hypoglycemic cultures. These findings suggest that BDNF can increase BMP-dependent Smad phosphorylation via a mechanism requiring CK2.
Keywords: basal forebrain, bone morphogenetic protein 7 (BMP7), brain-derived neurotrophic growth factor (BDNF), Smad 1/5/8, casein kinase II (CK2), hypoglycemia
Introduction
A combination of BMPs (6 and/or 7) and neurotrophins (nerve growth factor (NGF) and BDNF) protect septal neurons from both a hypoglycemic stress and a stress produced by inhibition of phosphatases 1A and 2A by okadaic acid (Nonner et al., 2001 & 2004). This combination was much more effective than either BMPs or neurotrophins alone. BMPs and neurotrophins also exert cooperative trophic effects in other neuronal populations (Bengtsson et al., 1998; Zhang et al., 1998; Farkas et al., 1999; Gratacòs et al., 2001; Althini et al., 2004). The goal of this study was to determine the mechanisms underlying the complementary effects of BMPs and neurotrophins in the protection of cultured embryonic septal neurons during hypoglycemia. Transient hypoglycemia in the early postnatal period is a frequent complication in infants born to diabetic mothers, and can produce brain damage (Burns et al., 2008; Tan et al., 2008)
Neurotrophins signal via TrkA, TrkB, TrkC and p75NTR receptors, and all of these receptors are expressed in cultures of basal forebrain neurons (Springer et al., 1987; Vazquez and Ebendal, 1991; Merlio et al., 1992). In these cultures, the neurons expressing TrkA and p75NTR are almost all cholinergic (Sobreviela et al., 1994); TrkB and TrkC receptors are more widespread (Merlio et al., 1992; Cheng & Mattson, 1994). The intracellular pathways that mediate the survival-promoting effects of neurotrophins include phosphatidylinositol 3-kinase (PI3K) in NGF-dependent PC12 cells, sympathetic neurons and neocortical neurons (Yao et al., 1995; Crowder et al., 1998; Mazzoni et al., 1999; Cheng et al., 2003; reviewed in Kaplan & Miller, 2000), mitogen-activated protein kinase (MAPK) in sympathetic ganglion neurons (Mazzoni et al., 1999; Atwal et al., 2000) and calcium-calmodulin in PC12 cells and neocortical neurons (Silva et al., 2000; Egea et al., 2001; Cheng et al., 2003).
BMPs signal via type I and type II BMP receptors (BMPR); type I BMPRs include activin receptor-like kinases (Alks), and type II receptors include BMPR-II and activin type II receptors (ActR). BMP7 can bind to several type I receptors (Alk2, BMPR-IA = Alk3 and BMPR-IB = Alk6) and to several type II receptors (BMPR-II, ActRII and ActRIIB, Macias-Silva et al., 1998). Mouse basal forebrain expresses mRNA for several type I (BMPR-IA, IB, Alk1) and type II (ActR-II, IIB) BMPRs and protein for BMPR-IB and II at early developmental stages; some BMPRs are also expressed in the adult (Lopez-Coviella et al., 2006). In the classical BMP signaling pathway, BMPR-II binds the BMP ligand and then recruits and phosphorylates BMPR-I, which in turn phosphorylates intracellular Smads 1, 5 and 8. These Smads then translocate to the nucleus where they regulate transcription of target genes (Lein et al., 2002, Angley et al., 2003, Chalazonitis et al., 2004, Lopez-Coviella et al., 2006). Transcripts for Smads1 and 5 and the co-Smad, Smad4, are also present in basal forebrain at early developmental stages as well as in the adult (Lopez-Coviella et al., 2006)
In addition to examining the above-mentioned pathways involved in neurotrophin- and BMP-mediated signalling, we also tested for a role of CK2 in stress protection. CK2 is a highly conserved serine/threonine kinase with two catalytic (α and/or α’) and two regulatory β subunits. Knockout of either the β regulatory subunit (Buchou et al., 2003) or the α catalytic subunit (Lou et al., 2008) is embryonic lethal in mice and knockout of both catalytic subunits is lethal in yeast (Hanna et al., 1995). CK2 has stress-protective effects in non-neuronal cells (reviewed in Blanquet, 2000), where CK2 activity is upregulated in response to certain stressors (Sayed et al. 2000; Davis et al., 2002; Fan et al., 2008). Neurotrophins increase CK2 activity in hippocampus (Blanquet, 1998; Arevalo & Rodriguez-Tébar et al. 2006), and CK2 is hypothesized to be stress-protective in brain (reviewed in Blanquet, 2000). CK2 has several anti-apoptotic effects (e.g. Klumpp et al. 2004), and neuronal CK2 levels correlate with neuronal survival following transient ischemia (Hu & Wielock, 1996). A CK2 inhibitor, 4,5,6,7-tetrabromobenzotriazole (TBB), blocks BDNF + BMP7-mediated protection of septal neurons during hypoglycemia (Nonner et al., unpublished observations).
Work presented here demonstrates that BMP7 increases Smad phosphorylation in septal neurons, and that BDNF enhances BMP7-induced Smad phosphorylation and nuclear translocation, especially after a hypoglycemic stress. These effects are blocked by inhibitors of CK2 activity, but not by inhibitors of PI3K, Erk or Ca2+-dependent signaling. These findings suggest that CK2 activation contributes to the synergistic stress-protective effects of BMP7 and BDNF in septal neurons.
Materials and methods
Cell culture
The septal region of the basal forebrain was dissected from embryonic day 15 rat embryos following euthanasia of pregnant rats with 100% CO2. Dissociated cells were plated using techniques described in Nonner et al. (2004); the plating density of 600 cells/mm2 was chosen to reduce paracrine effects from release of trophic factors by the cells. A defined medium (N5, Kawamoto & Barrett, 1986) was supplemented with 1 mg/ml of a 55 kDa serum fraction (Kaufman & Barrett, 1983), containing selenoprotein-P (Yan & Barrett, 1998). In these cultures, >95% of the cells were neurons (Tedeschi et al., 1986). Cells were maintained at 37 °C in 95% O2/5% CO2. The experimental procedures described below were initiated 5–7 days after plating. Culture dishes were coated with poly-L-lysine.
Hypoglycemic stress
Hypoglycemia was produced by washing cultures three times in medium containing normal salts (in mM: 125 NaCl, 2.7 KCl, 1.5 MgCl2, 0.05 MgSO4, 2 CaCl2, 0.83 NaH2PO4, 24 NaHCO3, 2 HEPES), 1 mg/ml bovine serum albumin (BSA) and 50 µM glucose (1% of the normal glucose concentration). Unless otherwise noted, cells were maintained in this hypoglycemic medium for 6 h with trophic factors added during the last hour. Inhibitors of signaling pathways were added 1 h prior to trophic factor addition. Unstressed control cultures were washed an equal number of times, but maintained throughout in the original N5 growth medium, except that the serum fraction was replaced with 1 mg/ml BSA to avoid possible effects of serum components on Smad signaling pathways (David et al., 2008). A salt solution (rather than the complete N5 medium) was used during the hypoglycemic stress to avoid possible use of the amino acids in N5 as an energy source. Fig. 1 in Supporting Information presents immunocytochemical evidence that a 6 h exposure to the salt solution was not by itself sufficient to induce the stress-induced changes reported here. Nonetheless, it remains possible that exposure to the salt solution added to the stress of hypoglycemia. Trophic factor addition was delayed so that any changes in the signaling pathways induced by hypoglycemia would be more likely to be present than if trophic factors were added at stress onset. Fig. 8 of Nonner et al. (1996) shows that addition of BDNF 6 h after onset of a hypoglycemic stress conferred the same degree of protection as addition at stress onset.
BMP7 (combined with 5 nM BMP6 in Fig. 1) was applied at the optimal concentration for increasing ChAT activity (5 nM, Nonner et al., 2001). BMP7 induced the same degree of Smad phosphorylation whether alone or combined with BMP6. Neurotrophins were applied at 100 ng/ml, which produces maximal stimulation of ChAT activity. Some experiments used both NGF and BDNF, but in most cases, the neurotrophin was BDNF alone, since the majority of septal neurons express TrkB. Also, BDNF alone was as effective as when combined with NGF in upregulating Smad phosphorylation in the presence of BMP7.
Fig. 1. BMP6/7 and a BMP-neurotrophin combination increase nuclear and cytoplasmic P-Smad levels.

Septal cells at 5 days in vitro were treated for 1 h with BMP6/7 alone or in combination with BDNF+NGF, then stained with an anti-P-Smad 1/5/8 antibody and counterstained with DAPI. Average values of nuclear and cytoplasmic fluorescence were measured as detailed in Fig. 1 of Supporting Information. A) Log scale scatter plot of P-Smad fluorescence in the nucleus (y axis) and cytoplasm (x axis). Each point represents one neuron. Points situated above the 45° identity line represent cells in which nuclear fluorescence exceeded cytoplasmic fluorescence. B) Ratio of nuclear to cytoplasmic P-Smad fluorescence, a measure of nuclear translocation. * indicates significant difference from control or between the indicated trophic factor groups, assessed with one-way ANOVA followed by Newman-Keuls test (p<0.05, n≥22 cells per group, from two different experiments).
Immunocytochemistry and Western blots
Cells were fixed in 4% paraformaldehyde and blocked in phosphate-buffered saline (PBS) with 0.1% Triton containing 10% donkey serum. Primary antibodies (incubated at 4 °C overnight) were rabbit anti-phospho Smad1/5/8 (1:200, gift from Dr. C.H. Heldin, Uppsala University, Uppsala, Sweden; or 1:50, Cell Signaling Technology, Danvers, MA, USA; these antibodies recognize Smad sites that become phosphorylated in response to activation of BMP receptors); mouse anti-casein kinase IIα and goat anti-casein kinase IIα’ (both at 1:50, Santa Cruz Biotechnology, Santa Cruz, CA, USA); and mouse anti-BMP receptor II (1:75, R&D Systems, Minneapolis, MN, USA). Secondary antibodies, all from Molecular Probes (Eugene, OR, USA), were 488 donkey anti-mouse, 1:2000; 488 donkey anti-rabbit, 1:300; and 555 donkey anti-rabbit, 1:1000. Mounting medium with 4',6-diamidino-2-phenylindole (DAPI) was used for nuclear counterstain (Vector Labs, Burlingame, CA, USA).
Cells were imaged using a Hamamatsu-ER CCD camera (Bridgewater, NJ, USA) and a 60X oil-immersion objective, numerical aperture, 1.45 (Olympus, Center Valley, PA, USA). All images of control and experimental treatment groups were collected using identical exposure time, gain and magnification. Custom macros written in an image analysis program (V++, Digital Optics, Browns Bay, Auckland, New Zealand) were used to measure fluorescence of nuclear and cytoplasmic phospho-Smads 1/5/8 in cells counter-stained with DAPI (details in Supporting Information Fig. 2). In all experiments, controls without primary antibodies gave negligible staining.
Western blots were performed as described in Supporting material 1
Assay for CK2 activity
Cells were washed twice in PBS and homogenized in a lysis buffer (in mM: 150 NaCl, 50 Tris-HCl, 25 β-glycerol phosphate, 10 NaF, 5 Na pyrophosphate, 2 EGTA, 1 thioglycolate, 0.1% Triton, pH 7.45). Inhibitor cocktails were as described above for Western blots; an anti-tyrosine phosphatase cocktail (Sigma-Aldrich) was also added. Protein concentrations were measured in the cleared lysates (Bradford assay) and equivalent amounts of protein (2.5 – 5 µg) were used for the kinase assay. Lysates (5 – 10 µl) were incubated for 12 min at 30 °C with a CK2 substrate peptide (RRRDDDSDDD, 200 µM, Upstate/Millipore, Temecula, CA, USA) and 1 – 10 µCi γ32P-ATP (Perkin-Elmer, Waltham, MA, USA). The assay was performed following the manufacturer’s protocol except that following trichloroacetic acid addition, samples were centrifuged (8000 rpm, 15 min) to remove phosphorylated cellular proteins. 25 µl of the supernatant was pipetted onto P81 phosphocellulose (Upstate), washed, placed in vials with scintillation fluid (ScintiSafe, Fisher Scientific, Pittsburgh, PA, USA) and counted with a scintillation counter. The assay was calibrated using recombinant CK2 (CKII, New England Biolabs, Ipswich, MA, USA). With this assay, 1 unit (or 2 ng) of CKII corresponds to 10−3 pmol phosphate transferred to the substrate peptide (200 µM) in 1 min at 30 °C in a reaction volume of 50 µl.
Reagents
Trophic factors: recombinant human BMP6 and BMP7 (Calbiochem, LaJolla, CA, USA); recombinant human NGF and BDNF (Alomone Labs, Jerusalem, Israel). Pharmacological inhibitors: PI3K/Akt pathway, wortmannin (100 nM, Calbiochem), 1L-6-Hydroxymethyl-chiro-inositol 2-(R)-2-O-methyl-3-O-octadecylcarbonate (=Akt inhibitor, 5 µM, Alexis, San Diego, CA); MAPK pathway: U0126 (10 µM, Sigma-Aldrich); calmodulin kinase II pathway, intracellular Ca2+ buffer 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA-AM, 25 µM, Sigma-Aldrich); inhibitor of CK2 as a tetramer or as free catalytic subunits, 4,5,6,7-tetrabromobenzotriazole (TBB, 20 µM) and 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB, 20 µM); inhibitor of CK2 as a tetramer, but not as free catalytic subunits, 4,5,6,7-tetrabromobenzimidazole (TBBz, 20 µM, all CK2 inhibitors from Sigma-Aldrich).
Statistical Analyses
Averages (from at least 3 experiments performed on different biological samples) are expressed as means ± SEM. Data were analyzed using GraphPad Prism software (LaJolla, CA, USA) with a one-way analysis of variance followed by Dunnett’s test for multiple comparisons to control or by Newman-Keuls test for pairwise comparison. Regression analysis with non-parametric correlation (Spearman test, GraphPad InStat, La Jolla, CA, USA) was used to test for changes in phospho-Smad levels and CK2 activity over time.
Results
Neurotrophin enhancement of BMP7 signaling is greater during hypoglycemia
Phosphorylation of Smads 1/5/8 by BMPR type I indicates activation of the BMP signaling pathway. Fig. 1 shows results of immunocytochemical experiments in which phosphorylated Smads 1/5/8 (P-Smad) were fluorescently labeled as described in Materials and Methods. Fig. 1A shows a scatter plot of a representative experiment comparing average P-Smad fluorescence in the cytoplasmic and nuclear compartments of individual neurons. Neurons exposed to BMP6/7 for 1 hr (open circles) showed more intense P-Smad staining than controls (open squares) in both compartments. P-Smad staining was further increased when neurotrophins (BDNF and NGF, filled circles) were also present. In most cells treated with BMPs ± neurotrophins, the average intensity of nuclear staining exceeded that of cytoplasmic staining. Fig. 1B summarizes results of these experiments, plotted as the average ratio of nuclear/cytoplasmic P-Smad staining, a measure of translocation. BMP6/7 increased this ratio, and the ratio was further increased (by 14%) when neurotrophins were also present. Additional immunocytochemical experiments confirmed the presence of BMPR type II in both cholinergic and non-cholinergic neurons (Fig. 3 in Supporting Information).
In Western blots of both nuclear and cytoplasmic subcellular fractions (Fig. 2A, B), P-Smad levels increased within 30 min following BMP7 addition, and this increase was sustained for at least 2 h (Fig. 2C, D). Sister cultures treated with both BMP7 and BDNF showed a greater increase in nuclear P-Smad at 30 and 60 min than cultures treated with BMP7 alone (Fig. 2C). Possible reasons for the apparent decrease in nuclear P-Smads after 90–120 min in BMP7 + BDNF are considered in the Discussion.
Fig. 2. Time course of Smad 1/5/8 phosphorylation following addition of BMP7 ± BDNF.
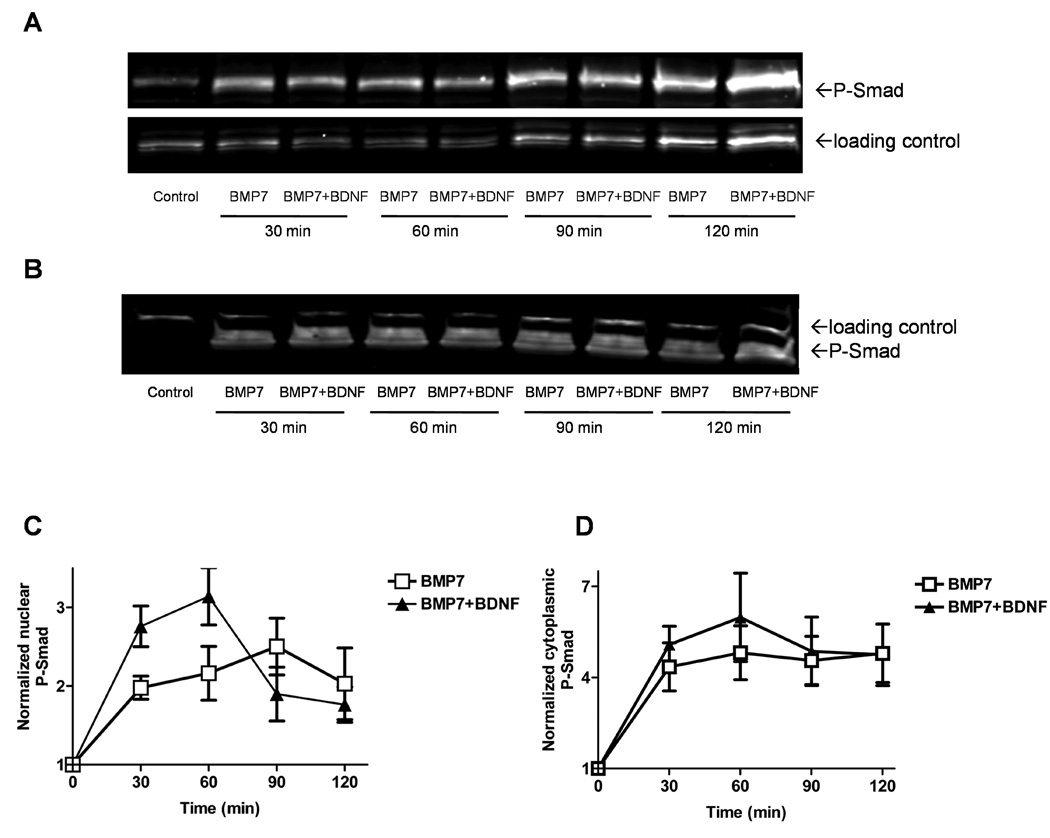
Western blots of nuclear (A) and cytoplasmic (B) fractions from lysates of cells grown for 5 to 7 days in culture after the indicated incubation periods with BMP7 or BMP7 plus BDNF. Loading controls for the cytoplasmic and nuclear compartments were poly (ADP-ribose) polymerase and TATA box binding protein, respectively. C, D) Quantification of nuclear (C) and cytoplasmic (D) blots like those shown in A and B. Averaged band intensities for P-Smad were normalized to their respective loading controls and this ratio was normalized to that of controls not exposed to trophic factors (plotted at 0 min). Regression analysis with Spearman non-parametric correlation showed that BMP7+BDNF increased nuclear P-Smad more than BMP7 alone during the first 60 min of incubation (data from ≥ 4 different experiments, p<0.01).
Fig. 3 shows that a 6 h exposure to hypoglycemia alone increased P-Smad fluorescence in both the nucleus (73% increase, compare Fig. 3A & B) and cytoplasm (70% increase, Fig. 3C & D), compared to non-stressed controls. BMP7 increased both nuclear and cytoplasmic P-Smad fluorescence by a greater percentage in hypoglycemic cultures than in non-stressed cultures. The enhancement of P-Smad fluorescence produced by the combination of BMP7 and neurotrophin was also greater under hypoglycemic conditions (40% greater in nucleus, 37% greater in cytoplasm, compared to BMP7 alone; see also Fig. 6). BDNF alone had no significant effect on P-Smad fluorescence in either control or hypoglycemic conditions (Fig. 3, see also Fig. 5, Fig. 6). These results suggest that hypoglycemia enhances P-Smad signaling even in the absence of added trophic factors and enhances the stimulatory effects of both BMP7 and the BMP7+ BDNF combination on P-Smad signaling.
Fig. 3. Hypoglycemia increases the enhancement of nuclear and cytoplasmic P-Smad levels produced by both BMP7 alone and a BMP7 + BDNF combination.
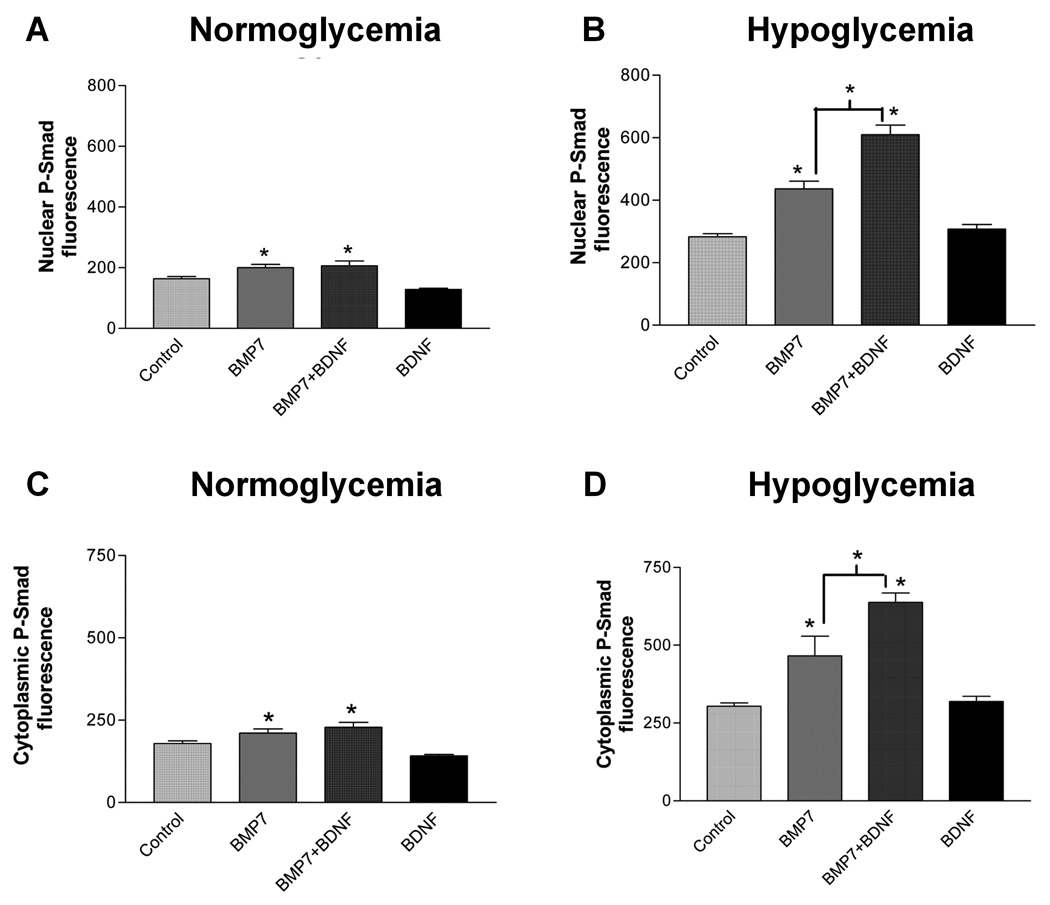
Cultures grown for 5 days in vitro were subjected to hypoglycemic stress (B, D) or to serum-free medium with normal glucose (A, C). During the last hour, BMP7 alone, BDNF alone, or BMP7 + BDNF were added, and nuclear (A, B) and cytoplasmic (C, D) P-Smad fluorescence were measured, as in Fig. 1. * indicates significant increase from control or indicated group with p<0.01, using one-way ANOVA followed by Newman-Keuls test; each group represents measurements of P-Smad fluorescence in at least 25 cells.
Fig. 6. A CK2 inhibitor reduces the (BMP7 + BDNF)-induced increase in P-Smad under both normoglycemic (A) and hypoglycemic (B) conditions.
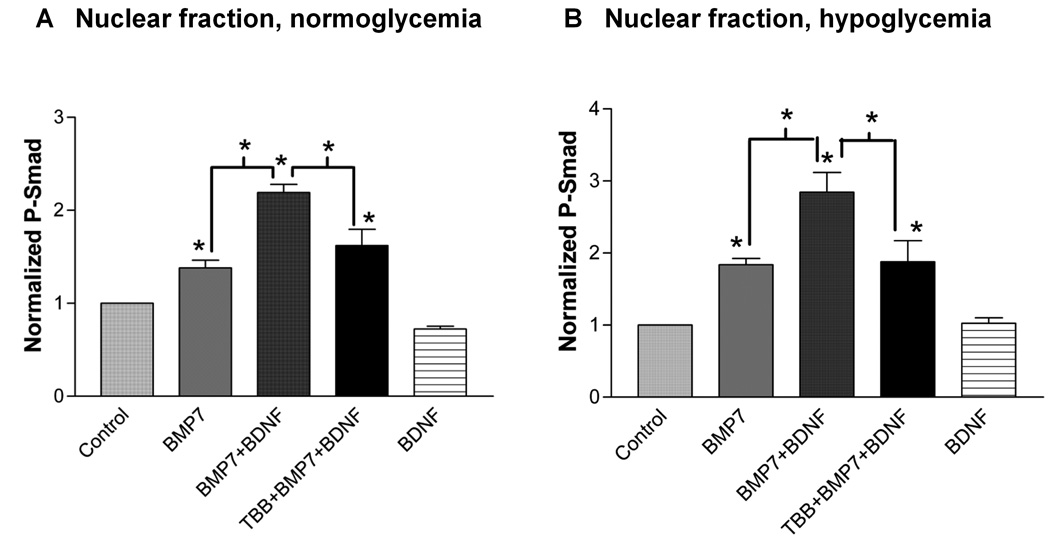
Nuclear fractions prepared from lysates were exposed to the indicated trophic factor(s) for 1 h, with some cultures also exposed to TBB for 1 h prior to and during trophic factor exposure. P-Smad fluorescence from Western blots was normalized to both non-treated control and loading control (TBP). * indicates significant difference from control or indicated group, one-way ANOVA followed by Newman-Keuls test (p<0.05, n=4 experiments).
Fig. 5. The increase in nuclear Smad fluorescence produced by (BMP7 + BDNF) is not reduced by inhibitors of PI3K/Akt, Mek/Erk, or Ca2+-dependent signaling (A), but is reduced by a CK2 inhibitor (B).

Cells under hypoglycemic conditions were pre-treated with pharmacological inhibitors for 1 h prior to and during a 1 h incubation with the indicated trophic factors. Nuclear P-Smad fluorescence was measured immunocytochemically, as described for Fig. 1. Wortmannin (Wort, 100 nM), Akt inhibitor (5 µM), U0126 (10 µM), BAPTA-AM (25 µM), TBB (20 µM). * indicates significant difference from control or indicated group, one-way ANOVA followed by Newman-Keuls test (p<0.001, n=6 to 54 cells).
BDNF enhancement of BMP7-induced Smad phosphorylation is independent of PI3K/Akt and Mek/Erk pathways, but requires CK2
Supporting Fig. 4 demonstrates increased levels of phosphorylated Akt (P-Akt) and p42-44 Erk (P-Erk) on Western blots within 15 min after addition of BDNF. The PI3K inhibitor wortmannin (100 nM) selectively blocked the increase in P-Akt and the Mek1/2 inhibitor U0126 (10 µM) selectively blocked the increase in P-Erk. Fig. 4A–C shows Western blot evidence that BMP7 did not increase Akt or Erk phosphorylation, and that the combination of BMP7 with BDNF did not increase Akt or Erk phosphorylation above the levels achieved by BDNF alone. Fig. 4 also shows that the BDNF-induced increase in Akt and Erk phosphorylation was not blocked by TBB (20 µM), an inhibitor of CK2 (Sarno et al., 2005).
Fig. 4. BDNF-induced increases in phosphorylated Akt and Erk are not modified by BMP7 or a CK2 inhibitor, TBB.
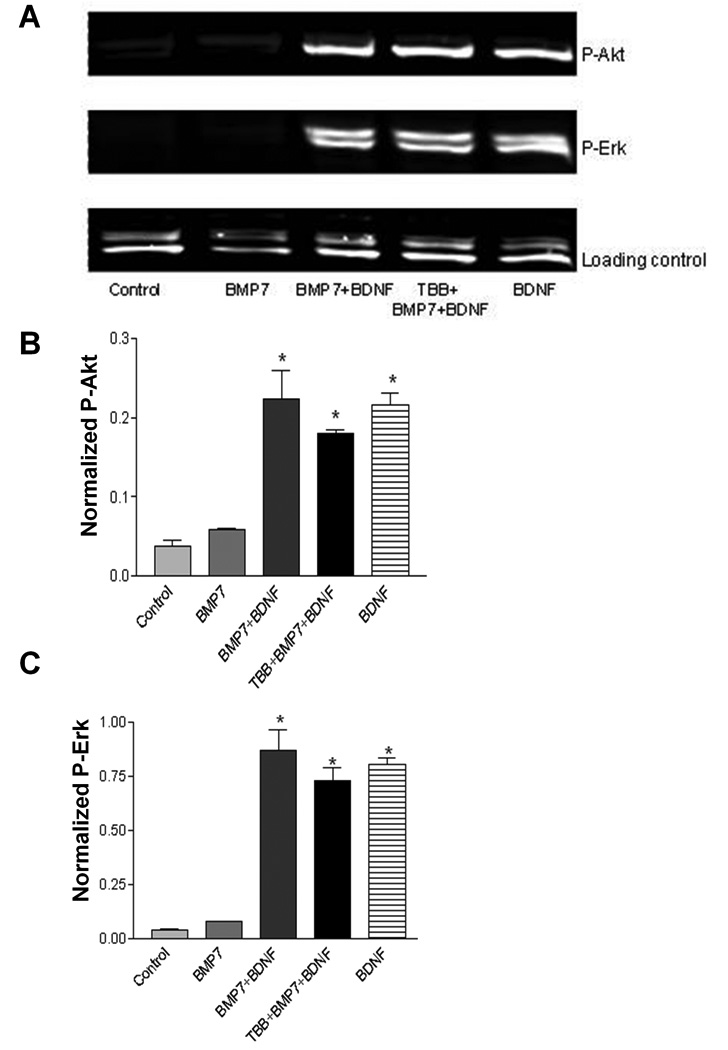
A) Western blot of cell lysates exposed for 15 min to BMP7, BDNF, or their combination. One experimental group was pretreated for 1 h with 20 µM TBB prior to trophic factor addition. B and C) Quantification of blots for P-Akt (B) and P-Erk (C), normalized to the loading control (TATA box binding protein, TBP). * indicates significant difference from control (p<0.001), one-way ANOVA followed by Dunnett’s test.
Fig. 5A summarizes immunocytochemical evidence that the increase in nuclear P-Smad produced by (BMP7 + BDNF) under hypoglycemic conditions was not reduced by wortmannin or by U0126. This increase in nuclear P-Smad was also unaffected by BAPTA-AM (25 µM), indicating that it is unlikely that Ca2+-dependent activation of calmodulin kinase (Blanquet & Lamour, 1997) is required.
Fig. 5B shows that the (BMP7 + BDNF)-induced increase in nuclear P-Smad was reduced by the CK2 inhibitor, TBB. These results were confirmed using Western blots under both normoglycemic (Fig. 6A) and hypoglycemic (Fig. 6B) conditions. Similar results were obtained with another CK2 inhibitor, DRB (20 µM, Supporting Fig. 5). DRB also significantly reduced the increase in nuclear P-Smad produced by BMP7 alone. This additional action might reflect a non-specific effect of DRB. Alternatively, inhibition of CK2 might reduce responses to BMP7 alone, as well as to the BDNF + BMP7 combination. Taken together, the results of Fig. 4–Fig. 6 indicate that classical BDNF-activated PI3K/Akt, Mek/Erk, and calcium-calmodulin pathways are not required for BDNF’s enhancement of BMP7-induced Smad phosphorylation and suggest that CK2 activity is required for this synergistic trophic factor effect.
The combination of BDNF and BMP7 increases CK2 activity under hypoglycemic conditions
Fig. 7A presents both immunocytochemical and Western blot evidence that both catalytic subunits of CK2 (α and α’) are present in septal neurons. Most CK2 immunostaining was cytoplasmic. No changes in the distribution of either CK2 subunit were observed following a 1 h exposure to trophic factors.
Fig. 7. Localization of catalytic subunits of CK2 in septal neurons and activation of CK2 by BDNF and BMP7.
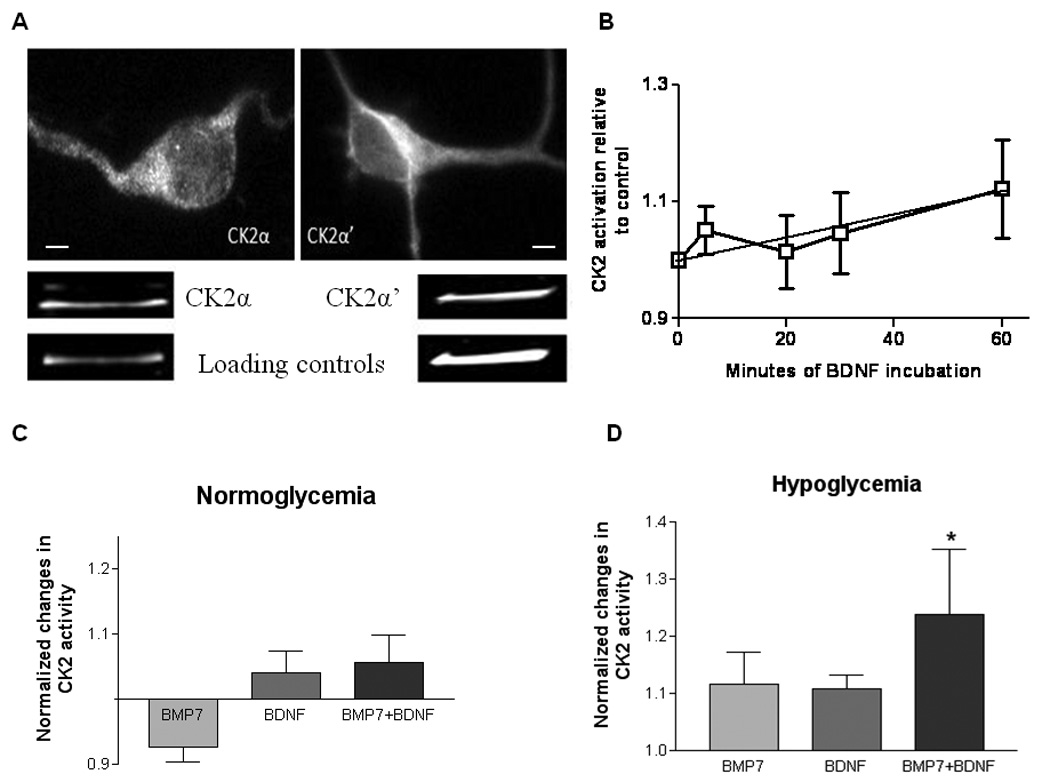
A) Immunocytochemical staining (top) and immunoblotting (bottom) for CK2α and CK2α’. Loading markers were neurofilament 200 for CK2α and TBP for CK2α’. Both assays were performed under normoglycemic conditions with no added trophic factors; there was no obvious change in subcellular localization during hypoglycemia, whether in the presence or absence of trophic factors. B) CK2 activity assayed after different durations of exposure to BDNF. Linear regression analysis of values averaged from 5 experiments (normalized to untreated controls) showed a significant increase over 60 min of BDNF incubation (r=0.53, p<0.01). C, D) Effects of a 1 h exposure to BMP7, BDNF or a BMP7+BDNF combination on CK2 activity, measured under normoglycemic (C) and hypoglycemic (D, 3 h) conditions. Experimental values were corrected by subtracting values measured in the absence of cell lysates, then normalized to values measured in the absence of trophic factors. The activity of control lysates was ~5.4×10−6 pmol/µg protein/min. CK2 inhibitors (TBB and TBBz) inhibited this control activity by 78% and 89%, respectively (not shown). *significant difference from control, one-way ANOVA followed by Dunnett’s test (p<0.05, n=10 experiments). None of the other differences or intergroup comparisons was significant. Calibration bars in A: 2 µm
Fig. 7B plots the summed results of 5 experiments in which septal cell lysates were assayed for their ability to phosphorylate a peptide substrate specific for CK2 following 5–60 min exposures to BDNF. CK2 activity increased within 1 h of BDNF exposure, compared to non-treated controls. This upregulation of CK2 activity was maintained after 2 h in BDNF.
Since stress can also activate CK2 (Sayed et al., 2000; Davis et al., 2002) and since BMP7 + BDNF combinations produce a greater increase in P-Smad levels during hypoglycemia (Fig. 3, Fig. 6), we compared CK2 activities measured following a 1 h exposure to BMP7, BDNF or (BMP7 + BDNF) under normoglycemic (Fig. 7C) and hypoglycemic (Fig. 7D) conditions. The only significant change was a modest (24%) increase in CK2 activity with the BMP7 + BDNF combination under hypoglycemic conditions. These results, combined with the effects of CK2 inhibitors described above, are consistent with the hypothesis that CK2 plays an active and/or permissive role in the synergistic trophic effects of BMP7 and BDNF.
Discussion
Results of this study confirm and extend evidence for BMP signaling in the basal forebrain. Previous work demonstrated the presence of BMP9 and BMP receptors in the basal forebrain and Smad phosphorylation in response to BMP9, and presented evidence that BMP9 upregulates and maintains the cholinergic phenotype (Lopez-Coviella et al., 2000, 2006). BMP7 studied in the present paper interacts with a different subset of receptors than BMP9 (Macias-Silva et al., 1998; Brown et al., 2005; David et al., 2007).
Pathways mediating synergy between BMP7 and BDNF signaling
Both immunocytochemical and Western blot evidence demonstrates that BMP7 (like BMP 9) increases both nuclear and cytoplasmic levels of P-Smads 1/5/8 in septal cultures (Fig. 3 & Fig. 6). The rapid increase in P-Smad levels (within 30–60 min of exposure to BMP7) suggests that this response was mediated by BMP activation of neuronal receptors rather than an indirect effect mediated via other cells (e.g. glia). This idea is consistent with the presence of BMP receptors and BMP-responsive Smads in septal neurons (Lopez-Coviella et al., 2006; and supporting Fig. 2).
BMP7-induced increases in nuclear and cytoplasmic Smad phosphorylation were enhanced by BDNF, especially during hypoglycemia (Fig. 3). The decline in nuclear P-Smad measured 90–120 min after exposure to BMP7 + BDNF under hypoglycemic conditions (Fig. 2C) may have been due to Smad dephosphorylation, as reported for TGF-β-responsive Smads 2 and 3 (Inman et al. 2002). BDNF-initiated signaling pathway(s) may have sped the disappearance of nuclear P-Smad. For example, phosphorylation of Smad1 in its central linker region by Erk2 may decrease P-Smad by targeting it to the proteasome (Sapkota et al., 2007). This might explain why BMP7 effects on neurite outgrowth from sympathetic ganglion neurons were enhanced by the Mek1/2 inhibitors PD98059 and U0126 (Althini et al., 2004; Kim et al., 2004a).
Although BDNF enhanced BMP7-induced intracellular signaling, the converse was not true. The BDNF-induced increase in phosphorylation of Akt and Erk1/2 was not increased by BMP 7. Thus, it is unlikely that BMP7 induced a rapid general up-regulation of TrkB-mediated signaling during the brief (1 h) exposures studied here. However, neurotrophin-mediated signaling might well be altered during more prolonged exposures to BMP7, such as that used by Nonner et al. (2001) to document preservation of cholinergic function by BMP + neurotrophin combinations during hypoglycemia. Schnitzler et al. (2008) found that a 3 day exposure to BMP9 increased expression of both mRNA and protein for the p75 neurotrophin receptor in basal forebrain neurons.
CK2 activity contributes to the synergistic effects of BMP7 and BDNF on Smad phosphorylation
Inhibitors of PI3K/Akt or Mek1/2/Erk did not reduce Smad phosphorylation by BMP7+BDNF (Fig. 5A). However, the CK2 inhibitor, TBB, reduced both the BDNF-induced increase in BMP7-stimulated Smad phosphorylation (Fig. 5B & Fig. 6), and the stress-protective effects of BMP+neurotrophin combinations during both hypoglycemic and phosphatase inhibitor stresses (unpublished observations). BMP7 + BDNF also increased CK2 activity during hypoglycemia (Fig. 7). These findings suggest a role for CK2 in the synergistic stress protection mediated by this trophic factor combination.
Blanquet (1998) and Arevalo & Rodriguez-Tébar (2006) recorded marked upregulation of CK2 activity in hippocampal tissue in response to neurotrophins. However, after phosphorylation events mediated by other kinases were eliminated from the assay, the growth factor-induced increase in CK2 activity we measured was relatively small, only 24% with a combination of BMP7+BDNF during hypoglycemia (Fig. 7D). Under these same conditions, the CK2-dependent (TBB-inhibitable) increase in nuclear P-Smad was ~38% (Fig. 6B). We used a brief (1 h) trophic factor exposure to minimize effects due to altered gene expression; it is possible that CK2 activity might increase more at longer intervals and/or with longer trophic factor exposures (Blanquet, 1998). It is also possible that increases in CK2 activity might be greater within localized cellular regions, if different subpopulations of CK2 coexist within cells and are subject to discrete modes of regulation (Olsten & Lichtfield 2004).
Small changes in total CK2 activity might have significant functional consequences. Studies using other cells and trophic factors have documented cooperative effects between Smads 1 or 2 and other families of transcription factors (e.g. Germain et al., 2000; Fukuda & Taga, 2005; Chen et al. 2008). It is thus conceivable that binding of BDNF to TrkB might activate a transcription factor that could interact with e.g. a BMP7-activated Smad1/Smad4 complex. Cooperative interactions between transcription factors might be important since Smads by themselves bind DNA with low affinity (Shi et al., 1998).
Possible relevance to protection against hypoglycemia in vivo
Neurotrophins protect cultured septal and hippocampal neurons during a hypoglycemic stress (Cheng & Mattson, 1991; Nonner et al., 1996). Given individually, both BMP7 (Perides et al. 1995) and BDNF (Cheng et al., 1997; Han & Holtzman, 2000; Galvin et al., 2003) protect the brains of rat pups against another type of energy stress, transient hypoxia/ischemia. BMP7 or BMP6 (Lin et al., 1999; Wang et al. 2001; Cox et al. 2004; Shen et al. 2008) and BDNF (Ferrer et al., 2001) also protect adult rats during transient brain ischemia in vivo. The ability of Smads to induce transcription of ATF3, a stress-activated gene (reviewed in Hai et al., 1999), also suggests a possible role for Smads in cellular responses to stress (Kang et al., 2003). The finding that BDNF and BMP7 have synergistic protective effects against neuronal damage from hypoglycemia in vitro (Nonner et al., 2001) raises the possibility dthat this trophic factor combination might be helpful in minimizing hypoglycemia-induced neuronal damage in newborns. Our demonstration that hypoglycemia enhances the increase in P-Smad produced by brief exposure to BMP7 and BDNF (Fig. 3) suggests a mechanism that might contribute to the synergistic stress-protective effects of this trophic factor combination. Additional studies will be needed to clarify the relationship between this short-term effect and the longer-term protective effects described in Nonner et al. (1996); (2001)
In summary, BMP+neurotrophin combinations such as BMP7+BDNF exert synergistic stress-protective effects in cultures of embryonic septal neurons. For the brief (1 h) trophic factor exposures studied here, evidence suggests that this synergy was due more to BDNF-mediated enhancement of BMP7-initiated signaling pathways than to BMP7-mediated enhancement of classical BDNF-initiated signaling pathways. BMP7-initiated phosphorylation of Smad1/5/8 in both nuclear and cytoplasmic compartments was enhanced by BDNF, especially under hypoglycemic conditions. Studies using inhibitors of CK2 and an assay for CK2 activity suggest an important role for this enzyme in the synergistic trophic effects of BMP7 and BDNF.
Supplementary Material
Acknowledgments
We thank Doris Nonner for dissection of embryos, Dr. Ellen Barrett for help with the manuscript, Dr. Carlos Moraes for help with the CK2 assay and Dr. Gavriel David for help with the computer programs. This work was supported by NIH grant NS 12207. Ms. Chaverneff was supported by a Lois Pope Life Fellowship, a University of Miami Center on Aging Forrest Fellowship and American Heart Association predoctoral fellowship 0515116B.
Non-standard abbreviations
- ActR
activin receptor
- Akt
protein kinase B
- Alk
activin receptor-like kinase
- BDNF
brain-derived neurotrophic factor
- BMP
bone morphogenetic protein
- BMPR
BMP receptor
- BSA
bovine serum albumin
- ChAT
choline acetyl transferase
- CK2
casein kinase 2
- DAPI
4',6-diamidino-2-phenylindole
- DRB
5,6-dichloro-1-β-D-ribofuranosylbenzimidazole
- Erk
extracellular signal–regulated kinase
- MAPK
mitogen-activated protein kinase
- NGF
nerve growth factor
- NT-3, 4/5
neurotrophin 3, neurotrophin 4/5
- p75NTR
p75 neurotrophin receptor
- PI3K
phosphatidylinositol 3-kinase
- Smad
Sma- and Mad (mothers against decapentaplegic)-related proteins
- TBB
4,5,6,7-tetrabromobenzotriazole
- TBBz
4,5,6,7-tetrabromobenzimidazole
- Trk
tyrosine kinase receptor
References
- Althini S, Usoskin D, Kylberg A, Kaplan PL, Ebendal T. Blocked MAP kinase activity selectively enhances neurotrophic growth responses. Mol. Cell. Neurosci. 2004;25(2):345–354. doi: 10.1016/j.mcn.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Angley C, Kumar M, Dinsio KJ, Hall AK, Siegel RE. Signaling by bone morphogenetic proteins and Smad1 modulates the postnatal differentiation of cerebellar cells. J. Neurosci. 2003;23(1):260–268. doi: 10.1523/JNEUROSCI.23-01-00260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo MA, Rodriguez-Tebar A. Activation of casein kinase II and inhibition of phosphatase and tensin homologue deleted on chromosome 10 phosphatase by nerve growth factor/p75NTR inhibit glycogen synthase kinase-3beta and stimulate axonal growth. Mol. Biol. Cell. 2006;17(8):3369–3377. doi: 10.1091/mbc.E05-12-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal JK, Massie B, Miller FD, Kaplan DR. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron. 2000;27(2):265–277. doi: 10.1016/s0896-6273(00)00035-0. [DOI] [PubMed] [Google Scholar]
- Bengtsson H, Soderstrom S, Kylberg A, Charette MF, Ebendal T. Potentiating interactions between morphogenetic protein and neurotrophic factors in developing neurons. J. Neurosci. Res. 1998;53(5):559–568. doi: 10.1002/(SICI)1097-4547(19980901)53:5<559::AID-JNR6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Blanquet PR. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 2000;60(3):211–246. doi: 10.1016/s0301-0082(99)00026-x. [DOI] [PubMed] [Google Scholar]
- Blanquet PR. Neurotrophin-induced activation of casein kinase 2 in rat hippocampal slices. Neuroscience. 1998;86(3):739–749. doi: 10.1016/s0306-4522(98)00087-6. [DOI] [PubMed] [Google Scholar]
- Blanquet PR, Lamour Y. Brain-derived neurotrophic factor increases Ca2+/calmodulin-dependent protein kinase 2 activity in hippocampus. J. Biol. Chem. 1997;272(39):24133–24136. doi: 10.1074/jbc.272.39.24133. [DOI] [PubMed] [Google Scholar]
- Brown MA, Zhao Q, Baker KA, et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005;280(26):25111–25118. doi: 10.1074/jbc.M503328200. [DOI] [PubMed] [Google Scholar]
- Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003;23(3):908–915. doi: 10.1128/MCB.23.3.908-915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CM, Rutherford MA, Boardman JP, Cowan FM. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics. 2008;122(1):65–74. doi: 10.1542/peds.2007-2822. [DOI] [PubMed] [Google Scholar]
- Chalazonitis A, D'Autreaux F, Guha U, et al. Bone morphogenetic protein-2 and −4 limit the number of enteric neurons but promote development of a TrkC-expressing neurotrophin-3-dependent subset. J. Neurosci. 2004;24(17):4266–4282. doi: 10.1523/JNEUROSCI.3688-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wang S, Yang D, Xiao R, Mattson MP. Calmodulin mediates brain-derived neurotrophic factor cell survival signaling upstream of Akt kinase in embryonic neocortical neurons. J. Biol. Chem. 2003;278(9):7591–7599. doi: 10.1074/jbc.M207232200. [DOI] [PubMed] [Google Scholar]
- Cheng B, Mattson MP. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 1994;640(1–2):56–67. doi: 10.1016/0006-8993(94)91857-0. [DOI] [PubMed] [Google Scholar]
- Cheng B, Mattson MP. NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron. 1991;7(6):1031–1041. doi: 10.1016/0896-6273(91)90347-3. [DOI] [PubMed] [Google Scholar]
- Cox S, Harvey BK, Sanchez JF, Wang JY, Wang Y. Mediation of BMP7 neuroprotection by MAPK and PKC IN rat primary cortical cultures. Brain Res. 2004;1010(1–2):55–61. doi: 10.1016/j.brainres.2004.02.068. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998;18(8):2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008;102(8):914–922. doi: 10.1161/CIRCRESAHA.107.165530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Davis AT, Wang H, Zhang P, Ahmed K. Heat shock mediated modulation of protein kinase CK2 in the nuclear matrix. J. Cell. Biochem. 2002;85(3):583–591. doi: 10.1002/jcb.10158. [DOI] [PubMed] [Google Scholar]
- Egea J, Espinet C, Soler RM, Dolcet X, Yuste VJ, Encinas M, Iglesias M, Rocamora N, Comella JX. Neuronal survival induced by neurotrophins requires calmodulin. J. Cell Biol. 2001;154(3):585–597. doi: 10.1083/jcb.200101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MM, Zhang H, Hayden MR, Pelech SL, Raymond LA. Protective up-regulation of CK2 by mutant huntingtin in cells co-expressing NMDA receptors. J. Neurochem. 2008;104(3):790–805. doi: 10.1111/j.1471-4159.2007.05016.x. [DOI] [PubMed] [Google Scholar]
- Fan MM, Zhang H, Hayden MR, Pelech SL, Raymond LA. Protective up-regulation of CK2 by mutant huntingtin in cells co-expressing NMDA receptors. J. Neurochem. 2008;104(3):790–805. doi: 10.1111/j.1471-4159.2007.05016.x. [DOI] [PubMed] [Google Scholar]
- Farkas LM, Jaszai J, Unsicker K, Krieglstein K. Characterization of bone morphogenetic protein family members as neurotrophic factors for cultured sensory neurons. Neuroscience. 1999;92(1):227–235. doi: 10.1016/s0306-4522(98)00735-0. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Marti E, Ambrosio S, Arenas E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001;101(3):229–238. doi: 10.1007/s004010000268. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Taga T. Cell fate determination regulated by a transcriptional signal network in the developing mouse brain. Anat. Sci. Int. 2005;80(1):12–18. doi: 10.1111/j.1447-073x.2005.00097.x. [DOI] [PubMed] [Google Scholar]
- Germain S, Howell M, Esslemont GM, Hill CS. Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes Dev. 2000;14(4):435–451. [PMC free article] [PubMed] [Google Scholar]
- Gratacos E, Checa N, Perez-Navarro E, Alberch J. Brain-derived neurotrophic factor (BDNF) mediates bone morphogenetic protein-2 (BMP-2) effects on cultured striatal neurones. J. Neurochem. 2001;79(4):747–755. doi: 10.1046/j.1471-4159.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7(4–6):321–335. [PMC free article] [PubMed] [Google Scholar]
- Hanna DE, Rethinaswamy A, Glover CV. Casein kinase II is required for cell cycle progression during G1 and G2/M in Saccharomyces cerevisiae. J. Biol. Chem. 1995;270(43):25905–25914. doi: 10.1074/jbc.270.43.25905. [DOI] [PubMed] [Google Scholar]
- Hu BR, Wieloch T. Casein kinase II activity in the postischemic rat brain increases in brain regions resistant to ischemia and decreases in vulnerable areas. J. Neurochem. 1993;60(5):1722–1728. doi: 10.1111/j.1471-4159.1993.tb13396.x. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell. 2002;10(2):283–294. doi: 10.1016/s1097-2765(02)00585-3. [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell. 2003;11(4):915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000;10(3):381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kaufman LM, Barrett JN. Serum factor supporting long-term survival of rat central neurons in culture. Science. 1983;220(4604):1394–1396. doi: 10.1126/science.6857258. [DOI] [PubMed] [Google Scholar]
- Kawamoto JC, Barrett JN. Cryopreservation of primary neurons for tissue culture. Brain Res. 1986;384(1):84–93. doi: 10.1016/0006-8993(86)91222-9. [DOI] [PubMed] [Google Scholar]
- Kim IJ, Drahushuk KM, Kim WY, Gonsiorek EA, Lein P, Andres DA, Higgins D. Extracellular signal-regulated kinases regulate dendritic growth in rat sympathetic neurons. J. Neurosci. 2004;24(13):3304–3312. doi: 10.1523/JNEUROSCI.3286-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Maurer A, Zhu Y, Aichele D, Pinna LA, Krieglstein J. Protein kinase CK2 phosphorylates BAD at threonine-117. Neurochem. Int. 2004;45(5):747–752. doi: 10.1016/j.neuint.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Lein PJ, Beck HN, Chandrasekaran V, Gallagher PJ, Chen HL, Lin Y, Guo X, Kaplan PL, Tiedge H, Higgins D. Glia induce dendritic growth in cultured sympathetic neurons by modulating the balance between bone morphogenetic proteins (BMPs) and BMP antagonists. J. Neurosci. 2002;22(23):10377–10387. doi: 10.1523/JNEUROSCI.22-23-10377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SZ, Hoffer BJ, Kaplan P, Wang Y. Osteogenic protein-1 protects against cerebral infarction induced by MCA ligation in adult rats. Stroke. 1999;30(1):126–133. doi: 10.1161/01.str.30.1.126. [DOI] [PubMed] [Google Scholar]
- Lopez-Coviella I, Berse B, Krauss R, Thies RS, Blusztajn JK. Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science. 2000;289(5477):313–316. doi: 10.1126/science.289.5477.313. [DOI] [PubMed] [Google Scholar]
- Lopez-Coviella I, Mellott TM, Kovacheva VP, Berse B, Slack BE, Zemelko V, Schnitzler A, Blusztajn JK. Developmental pattern of expression of BMP receptors and Smads and activation of Smad1 and Smad5 by BMP9 in mouse basal forebrain. Brain Res. 2006;1088(1):49–56. doi: 10.1016/j.brainres.2006.02.073. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998;273(40):25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J. Neurosci. 1999;19(22):9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17(3):349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Merlio JP, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51(3):513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Nishida E. Evaluating CK2 activity with the antibody specific for the CK2-phosphorylated form of a kinase-targeting cochaperone Cdc37. Mol. Cell. Biochem. 2008;316(1–2):127–134. doi: 10.1007/s11010-008-9818-1. [DOI] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Barrett JN. Neurotrophin effects on survival and expression of cholinergic properties in cultured rat septal neurons under normal and stress conditions. J. Neurosci. 1996;16(21):6665–6675. doi: 10.1523/JNEUROSCI.16-21-06665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Kaplan P, Barrett JN. Bone morphogenetic proteins (BMP6 and BMP7) enhance the protective effect of neurotrophins on cultured septal cholinergic neurons during hypoglycemia. J. Neurochem. 2001;77(2):691–699. doi: 10.1046/j.1471-4159.2001.00273.x. [DOI] [PubMed] [Google Scholar]
- Nonner D, Panickar K, Barrett EF, Barrett JN. Bone morphogenetic proteins and neurotrophins provide complementary protection of septal cholinergic function during phosphatase inhibitor-induced stress. J. Neurochem. 2004;91(1):77–87. doi: 10.1111/j.1471-4159.2004.02687.x. [DOI] [PubMed] [Google Scholar]
- Perides G, Jensen FE, Edgecomb P, Rueger DC, Charness ME. Neuroprotective effect of human osteogenic protein-1 in a rat model of cerebral hypoxia/ischemia. Neurosci. Lett. 1995;187(1):21–24. doi: 10.1016/0304-3940(95)11327-s. [DOI] [PubMed] [Google Scholar]
- Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell. 2007;25(3):441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Sarno S, Ruzzene M, Frascella P, Pagano MA, Meggio F, Zambon A, Mazzorana M, Di Maira G, Lucchini V, Pinna LA. Development and exploitation of CK2 inhibitors. Mol. Cell. Biochem. 2005;274(1–2):69–76. doi: 10.1007/s11010-005-3079-z. [DOI] [PubMed] [Google Scholar]
- Sayed M, Kim SO, Salh BS, Issinger OG, Pelech SL. Stress-induced activation of protein kinase CK2 by direct interaction with p38 mitogen-activated protein kinase. J. Biol. Chem. 2000;275(22):16569–16573. doi: 10.1074/jbc.M000312200. [DOI] [PubMed] [Google Scholar]
- Schnitzler AC, Lopez-Coviella I, Blusztajn JK. Differential modulation of nerve growth factor receptor (p75) and cholinergic gene expression in purified p75-expressing and non-expressing basal forebrain neurons by BMP9. Brain Res. 2008 doi: 10.1016/j.brainres.2008.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Luo Y, Kuo CC, Wang Y. BMP7 reduces synergistic injury induced by methamphetamine and ischemia in mouse brain. Neurosci. Lett. 2008;442(1):15–18. doi: 10.1016/j.neulet.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. 1998;94(5):585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- Silva A, Montague JR, Lopez TF, Mudd LM. Growth factor effects on survival and development of calbindin immunopositive cultured septal neurons. Brain Res. Bull. 2000;51(1):35–42. doi: 10.1016/s0361-9230(99)00188-4. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J. Comp. Neurol. 1994;350(4):587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Koh S, Tayrien MW, Loy R. Basal forebrain magnocellular neurons stain for nerve growth factor receptor: correlation with cholinergic cell bodies and effects of axotomy. J. Neurosci. Res. 1987;17(2):111–118. doi: 10.1002/jnr.490170204. [DOI] [PubMed] [Google Scholar]
- Tam EW, Widjaja E, Blaser SI, Macgregor DL, Satodia P, Moore AM. Occipital lobe injury and cortical visual outcomes after neonatal hypoglycemia. Pediatrics. 2008;122(3):507–512. doi: 10.1542/peds.2007-2002. [DOI] [PubMed] [Google Scholar]
- Tedeschi B, Barrett JN, Keane RW. Astrocytes produce interferon that enhances the expression of H-2 antigens on a subpopulation of brain cells. J. Cell Biol. 1986;102(6):2244–2253. doi: 10.1083/jcb.102.6.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez ME, Ebendal T. Messenger RNAs for trk and the low-affinity NGF receptor in rat basal forebrain. Neuroreport. 1991;2(10):593–596. doi: 10.1097/00001756-199110000-00010. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang CF, Morales M, et al. Bone morphogenetic protein-6 reduces ischemia-induced brain damage in rats. Stroke. 2001;32(9):2170–2178. doi: 10.1161/hs0901.095650. [DOI] [PubMed] [Google Scholar]
- Yan J, Barrett JN. Purification from bovine serum of a survival-promoting factor for cultured central neurons and its identification as selenoprotein-P. J. Neurosci. 1998;18(21):8682–8691. doi: 10.1523/JNEUROSCI.18-21-08682.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267(5206):2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mehler MF, Song Q, Kessler JA. Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. J. Neurosci. 1998;18(9):3314–3326. doi: 10.1523/JNEUROSCI.18-09-03314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zien P, Duncan JS, Skierski J, Bretner M, Litchfield DW, Shugar D. Tetrabromobenzotriazole (TBBt) and tetrabromobenzimidazole (TBBz) as selective inhibitors of protein kinase CK2: evaluation of their effects on cells and different molecular forms of human CK2. Biochim. Biophys. Acta. 2005;1754(1–2):271–280. doi: 10.1016/j.bbapap.2005.07.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.