

Abstract

The catalytic, diastereoselective coupling of α-silyloxy aldehydes and alkynylsilanes catalyzed by a nickel(0) N-heterocyclic carbene complex provides an effective entry to anti-1,2-diols. The scope of couplings and extent of diastereoselection are excellent across a range of substrates.
Diastereoselective nucleophilic additions to α-alkoxy aldehydes provide an attractive method for preparation of 1,2-diols. Synthesis of anti-1,2-diols can be achieved by additions to chiral aldehydes following the Felkin model for diastereoselection, although control of stereochemistry can often be a challenge. A number of studies involving alkenylation of α-alkoxy aldehydes illustrate that anti selectivity can sometimes be achieved, but that the results are variable depending upon the precise structure of both the chiral aldehyde and vinyl organometallic.1,2 Several attractive alternative strategies that rely upon aldol technology have also been developed to avoid these limitations. 3
The nickel-catalyzed addition of aldehydes and alkynes has emerged as a useful way to prepare allylic alcohols in a variety of contexts.4,5 Several reports of nickel-catalyzed reductive couplings of this class involving highly stereoselective additions to chiral α-alkoxy aldehydes have appeared.6,7 Absent among the reports involving nickel catalysis is the intermolecular diastereoselective addition of non-aromatic alkynes, terminal alkynes, and silyl alkynes. Additionally, intermolecular additions involving α-alkoxyaldehydes bearing an unbranched substituent at the α-carbon proceeded with modest diastereoselectivity.6c Given the utility of catalytic couplings of aldehydes and alkynes involving nickel(0) / N-heterocyclic carbene catalysts recently disclosed from our group,8 we have examined this catalyst formulation in diastereoselective additions of alkynes to α-silyloxyaldehydes. The reaction scope and extent of diastereoselection are excellent for substrate combinations and substitution patterns not previously reported, and an attractive entry to anti-1,2-diols is thus provided.
Although our group previously demonstrated that the structure of an N-heterocyclic carbene ligand can play an important role in determining yield and regioselectivity in nickel-catalyzed aldehyde / alkyne reductive couplings, the current study focused exclusively on reactions involving Ni(COD)2 and the N-heterocyclic carbene (IMes) derived from imidazolium 1 in THF. From our prior studies, we recognized that silane structure was important in order to select for the desired three component coupling over undesired hydrosilylation of the aldehyde or alkyne moieties. We thus compared the catalytic addition of a TBS-protected silyloxy aldehyde with trimethylsilyl(phenyl)acetylene using various silanes. Due to competing hydrosilylation processes with unhindered silanes, chemical yields with (i-pr)3SiH were superior to (t-Bu)Me2SiH and Et3SiH (Table 1, entries 1–3). We next compared (i-pr)3SiH-mediated couplings of TMS-propyne with various α-oxyaldehydes (Table 1, entries 4–7). Both diastereoselectivities and yields were only modestly impacted in comparing α-benzyloxy to various α-silyloxy groups, and the t-butyldimethylsilyloxy group was selected for further study given the ease of installation and removal and the slightly superior yield and diastereoselectivity compared with other options examined. In order to compare the addition reaction of alkynyl silanes vs. terminal alkynes, phenyl acetylene was examined as a reaction partner. This terminal alkyne underwent addition in low yield (Table 1, entry 8), clearly illustrating that alkynylsilanes were a preferred synthetic equivalent of terminal alkynes. Interestingly, the syn diastereomer was slightly favored starting from this terminal alkyne.9 1-Phenylpropyne underwent efficient coupling with an aliphatic aldehyde, although diastereoselectivities were modest in comparison to couplings with alkynylsilanes (Table 1, entry 9). Analysis of the above examples suggested that (i-pr)3SiH as the reducing agent, (t-Bu)Me2Si as the α-hydroxy protecting group on the aldehyde, and trimethylsilyl alkynes would be a good combination for further study (Table 1, entries 3 and 6). Notably, exhaustive deprotection of the tris-silylated products is straightforward, or chemoselective deprotections are also possible if partially deprotected structures are desired.
Table 1.
Optimization of diastereoselectivity.
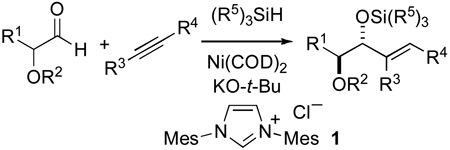 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | R4 | R53Si | % yield | anti:syn |
| 1 | CH3 | TBS | TMS | Ph | TEs | 36 | 89:11 |
| 2 | CH3 | TBS | TMS | Ph | TBS | 48 | 88:12 |
| 3 | CH3 | TBS | TMS | Ph | TIPS | 75 | 89:11 |
| 4 | CH3 | CH2Ph | TMS | Ph | TIPS | 81 | 78:22 |
| 5 | CH3 | TES | TMS | Me | TIPS | 75 | 81:19 |
| 6 | CH3 | TBS | TMS | Me | TIPS | 81 | 88:12 |
| 7 | CH3 | TIPS | TMS | Me | TIPS | 73 | 88:12 |
| 8 | n-pent | TBS | H | Ph | TIPS | 20 | 41:59 |
| 9 | n-pent | TBS | Me | Ph | TIPS | 82 | 73:27 |
Abbreviations: TMS = Me3Si, TES = Et3Si, TBS = (t-Bu)Me2Si, TIPS = (i-pr)3Si.
Using this optimized set of reaction parameters, several combinations of α-silyloxyaldehydes and alkynes were examined. Both aliphatic and aromatic alkynyl silanes underwent couplings in high yield and good diastereoselectivity (Table 2, entries 1–3).10 Next, a series of siloxyl aldehydes that possess unbranched substituents at the α-position were examined. This structural modification of extending the length of the α-alkyl substituent (R1) in the aldehyde resulted in a marked improvement in diastereoselectivity. As illustrated by the examples provided (Table 2, entries 4–9), diastereoselectivities and yields were uniformly outstanding for this class of aldehydes with both aliphatic and aromatic alkynyl silanes. The combination of silyloxy aldehydes and silyl alkynes was most effective with unbranched R1 groups. The participation of more hindered aldehydes requires modifications of protecting group and alkyne structure, thus making the procedure reported herein complementary to previously reported variants.6
Table 2.
Examination of Reaction Scope.
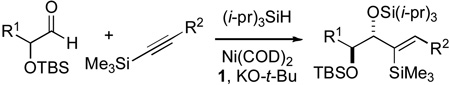 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | % yield | diastereo-selectivity |
| 1 | CH3 | CH3 | 81 | 88:12 |
| 2 | CH3 | Ph | 75 | 89:11 |
| 3 | CH3 | p-(CH3O)C6H4 | 85 | 89:11 |
| 4 | n-pentyl | p-(CH3O)C6H4 | 80 | > 98:2 |
| 5 | (CH2)2Ph | CH3 | 85 | > 98:2 |
| 6 | n-pentyl | CH3 | 85 | > 98:2 |
| 7 | (CH2)2Ph | n-butyl | 85 | > 98:2 |
| 8 | n-pentyl | n-butyl | 78 | > 98:2 |
| 9 | n-pentyl | Ph | 80 | > 98:2 |
The anti stereochemical assignment of the products was made by exhaustive silyl deprotection followed by acetonide protection of products 2a and 2b (from Table 2, entries 2 and 8). NOE analysis of products 3a and 3b thus provided unambiguous assignment of stereochemistry (eq 1). The stereochemistry of other products was assigned by analogy.11
 |
(1) |
Additionally, conservation of enantiopurity in a coupling was illustrated by the conversion of the aldehyde 4 (>98% ee), derived from (S)-ethyl lactate,12 into product 5 (>98% ee) (Scheme 1).
Scheme 1.

Conservation of enantioselectivity
The mechanistic basis for the outstanding diastereoselectivities in the specific substrate combinations described (Table 2) is unclear. One can simply argue that the Felkin model for diastereoselectivity is operative.13 However, we are reluctant to draw close analogies between aldehyde addition reactions of classical metallated nucleophiles in comparison to nickel couplings, which we have proposed to proceed via the formation of a nickel metallacycle derived from a carbonyl π-complex. The requisite orbital interactions, steric considerations, and trajectories of approach for a classical nucleophile adding to a carbonyl are clearly different from the requirements for formation and oxidative cyclization of a late metal-aldehyde π-complex. Additionally, we have illustrated that the mechanism of nickel-catalyzed aldehyde-alkyne reductive couplings is ligand dependent,8a thus adding further ambiguity to any mechanistic model. Nonetheless, the classic Felkin model serves as a useful predictor of stereochemistry in this reaction even though the underlying basis for the effect will require further study.
In summary, the nickel-catalyzed three-component coupling of α-silyloxy aldehydes, alkynyl silanes, and (i-pr)3SiH provides protected allylic alcohol products in excellent yield and diastereoselectivity. This particular combination of substrates, reagents, and catalyst significantly expands the scope of 1,2-anti diols that are available by alternate procedures. Application of the process in complex synthetic problems is in progress.
Supplementary Material
Experimental procedures and copy of spectral data for all new compounds is provided. These materials are available free of charge via the Internet at http://pubs.acs.org
Acknowledgment
Support for this work was provided by the National Institutes of Health (GM 57014).
References
- 1.For alkenylations of α-alkoxy aldehydes, seeSpino C, Granger M-C, Boisvert L, Beaulieu C. Tetrahedron Lett. 2002;43:4183.Iio H, Mizobuchi T, Tsukamoto M, Tokoroyama T. Tetrahedron Lett. 1986;27:6373.
- 2.For alkenylations of other classes of chiral aldehydes, seeWipf P, Xu W. Tetrahedron Lett. 1994;35:5197.Garner P, Park JM, Malecki E. J. Org. Chem. 1988;53:4395.Coleman RS, Carpenter AJ. Tetrahedron Lett. 1992;33:1697.
- 3.(a) Northrup AB, MacMillan DWC. Science. 2004;305:1752. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]; (b) Notz W, List B. J. Am. Chem. Soc. 2000;122:7386. [Google Scholar]; (c) Evans DA, Gage JR, Leighton JL, Kim AS. J. Org. Chem. 1992;57:1961. [Google Scholar]; (d) Crimmins MT, McDougall PJ. Org. Lett. 2003;5:591. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; (e) Yamashita Y, Ishitani H, Shimizu H, Kobayashi S. J. Am. Chem. Soc. 2002;124:3292. doi: 10.1021/ja016293t. [DOI] [PubMed] [Google Scholar]; (f) Andrus MB, Soma Sekhar BBV, Meredith EL, Dalley NK. Org, Lett. 2000;2:3035. doi: 10.1021/ol0002166. [DOI] [PubMed] [Google Scholar]; (g) Dixon DJ, Ley SV, Polara A, Sheppard T. Org. Lett. 2001;3:3749. doi: 10.1021/ol016707n. [DOI] [PubMed] [Google Scholar]
- 4.(a) Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065. [Google Scholar]; (b) Lozanov M, Montgomery J. J. Am. Chem. Soc. 2002;124:2106. doi: 10.1021/ja0175845. [DOI] [PubMed] [Google Scholar]; (c) Ni Y, Amarasinghe KKD, Montgomery J. Org. Lett. 2002;4:1743. doi: 10.1021/ol025812+. [DOI] [PubMed] [Google Scholar]; (d) Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; (e) Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 5.For reviews, seeMontgomery J. Angew. Chem. Int. Ed. 2004;43:3890. doi: 10.1002/anie.200300634.Montgomery J. Acc. Chem. Res. 2000;33:467. doi: 10.1021/ar990095d.Miller KM, Molinaro C, Jamison TF. Tetrahedron: Asymmetry. 2003;14:3619.Ikeda S-i. Angew. Chem., Int. Ed. 2003;42:5120. doi: 10.1002/anie.200301673.
- 6.(a) Tang X-Q, Montgomery J. J. Am. Chem. Soc. 1999;121:6098. [Google Scholar]; (b) Tang X-Q, Montgomery J. J. Am. Chem. Soc. 2000;122:6950. [Google Scholar]; (c) Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org. Lett. 2005;7:2937. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For an emerging body of work involving titanium-promoters, seeBahadoor AB, Flyer A, Micalizio GC. J. Am. Chem. Soc. 2005;127:3694. doi: 10.1021/ja050039+.Bahadoor AB, Micalizio GC. Org. Lett. 2006;8:1181. doi: 10.1021/ol0600786.
- 8.(a) Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]; (b) Knapp-Reed B, Mahandru GM, Montgomery J. J. Am. Chem. Soc. 2005;127:13156. doi: 10.1021/ja054590i. [DOI] [PubMed] [Google Scholar]; For an earlier example of the unique role of N-heterocyclic carbene ligands in nickel-catalyzed reductive couplings, seeSato Y, Sawaki R, Mori M. Organometallics. 2001;20:5510.
- 9.The minor isomer from tetrabutyl ammonium fluoride-mediated exhaustive desilylation of the product from Table 1, entry 8 was identical to that derived from desilylation of the major product from Table 2, entry 9
- 10.Entries 1 and 2 in Table 2 are duplicated from the optimization studies described in Table 1
- 11.Additionally, stereochemistry of the example from Table 1, entry 4 was confirmed by conversion of that product into the dibenzylated material, which was identical to the dibenzylated compound derived from the Table 1, entry 3 product. See Supporting Information for details
- 12.Marshall JA, Yanik MM, Adams ND, Ellis KC, Chobanian HR. Org. Synth. 2004;81:157. [Google Scholar]
- 13.Chérest M, Felkin H, Prudent N. Tetrahedron Lett. 1968;9:2199. [Google Scholar]; For a discussion of silyloxy-based effects, seeChen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1992;114:1778.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and copy of spectral data for all new compounds is provided. These materials are available free of charge via the Internet at http://pubs.acs.org