

Abstract
We report here the first structure of a member of the IgA protease family at 1.75Å resolution. This protease is a founding member of the Type V (autotransporter) secretion system and is considered a virulence determinant among the bacteria expressing the enzyme. The structure of the enzyme fits that of a classical autotransporter in which several unique domains necessary for protein function are appended to a central, 100 Å long β-helical domain. The N-terminal domain of the IgA protease is found to possess a chymotrypsin-like fold. However, this catalytic domain contains a unique loop D that extends over the active site acting as a lid, gating substrate access. The data presented provide a structural basis for the known ability of IgA proteases to only cleave the P/S/T rich hinge peptide unique to IgA1 in the context of the intact fold of the immunoglobulin. Based upon the structural data as well as molecular modeling, a model is presented that suggests the unique, extended loop D in this IgA protease sterically occludes the active site binding cleft in the absence of immunoglobulin binding. Only in the context of binding of the IgA1 immunoglobulin Fc domain in a valley formed between the N-terminal protease domain and another domain appended to the β-helix spine (domain-2) is the lid stabilized in an open conformation. The stabilization of this open conformation through Fc association subsequently allows access of the hinge peptide to the active site resulting in recognition and cleavage of the substrate.
Introduction
Immunoglobulin A1 proteases (IgAP1) are a family of serine (E.C. 3.4.21.72 ) and metallo (E.C 3.4.24.13) endopeptidases, that specifically cleave immunoglobulin A (IgA1) of human beings and great apes. The IgA proteases were originally discovered in 1973 (1) and the enzyme of the human pathogen Neisseria gonorrhoeae was later utilized as the model system for the elucidation of the Type V (autotransporter) protein secretion pathway of bacteria (2, 3). Proteins of the Type V pathway are virulence factors of pathogenic gram-negative bacteria. Briefly their pathway of secretion (reviewed in (4, 5)) involves the targeting of the synthesized protein to the periplasm via recognition of an unusually long N-terminal signal sequence by the sec complex. Upon translocation to the periplasm, the C-terminal β-domain forms a β-barrel structure that inserts into the outer membrane and is thought to act as a pore allowing for translocation of the N-terminal passenger domain to the external environment. Presently there is some controversy surrounding the exact mechanism by which this translocation occurs (4, 6-10). Following transport, in some members of the family (including IgAP), the N-terminal passenger domain is proteolytically excised from the C-terminal pore domain and released from the membrane. One group of passenger domain proteins is the serine protease autotransporters of the Enterobacteriaceae (SPATE) family of serine proteases of which the serine protease isoforms of IgAP are members. The enzymes of this family of related proteases are essential virulence factors in the bacteria that express them, however the individual members do not appear to have any related biological functions (11, 12).
After the discovery of IgAP, a number of important human bacterial pathogens were found to produce the enzyme; these include Haemophilus influenzae, Streptococcus pneumoniae, Streptococcus sanguis, Neisseria gonorrhoeae and Neisseria meningitides. The enzymes of Streptococcus sp. are suggested to have a metalloprotease activity rather than the serine protease activity of the other members (13-16). Regardless of the proposed chemical mechanism for cleavage, it has been documented that all IgAP family members cleave a proline, serine and threonine (P/S/T) rich duplicated octapeptide that upon comparison to other immunoglobulins is unique to the structure of IgA1 from humans and great apes (14, 15, 17-19) and is located in the hinge region (Figure 1) of the immunoglobulin heavy chain that lies between the Fab and Fc domains. Further, bacteria of Neisseria and Haemophilus sp. are known to express different isozymes of IgAP. These isozymes are highly homologous and are distinct gene products. While the different isozymes mediate the cleavage of the same P/S/T rich hinge region of IgA1 and demonstrate a requirement for cleavage C-terminal to a proline residue within this hinge region, the specific site of proteolysis differs amongst isozymes (Figure 1). Because IgA is the principle immunoglobulin in human secretions and on mucosal surfaces, specific cleavage of secretory IgA1 supports the idea that these enzymes are virulence factors, and this activity influences the ability of these bacteria to colonize and infect mucosal tissues in humans. Hydrolysis of the IgA hinge is known to interrupt certain defensive properties of mucosal and serum IgA such as bacterial agglutination, a function that is dependent on the intact protein with its multiple antigenic binding sites. Further, it has been suggested that because the Fab cleavage products of secretory IgA have unmodified antigen-binding capacity (20), at mucosal sites the Fab can bind to bacteria, thus masking surface epitopes from intact, functional secretory IgA (21).
Figure 1.
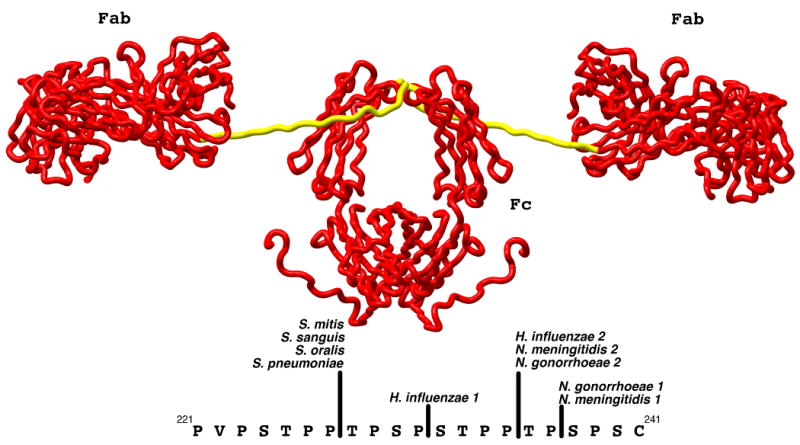
Model of the structure of human IgA1 (PDB:1IGA) as determined from x-ray and neutron solution scattering and homology modeling (45). The Fab and Fc domains are rendered as red tubes and labeled while the hinge domain is rendered in yellow. The sequence of the hinge peptide and the location of the cleavage sites by various members of the IgA protease family are shown in the lower portion of the figure. The numbering utilized for the sequence of IgA1 is that of Torano and Putnam (76).
Haemophilus influenzae has several attributes that lead to seemingly harmless colonization, but this microorganism often causes localized or generalized infection. Among its virulence factors is IgA protease, whose levels are higher in H. influenzae bacteria causing disease than in H. influenzae harmlessly colonizing human mucosal surfaces (22). This information, when taken in concert with the observation that both serine-type and metallo-type IgA proteases that have identical substrate specificity are expressed in common human pathogens strongly implicates these enzymes in the pathogenesis of disease mediated by the organisms expressing IgA protease isozymes. It should also be noted that H. influenzae has other virulence factors including a polyribosyl ribitol phosphate (PRP) capsule that makes it resistant to phagocytosis, and certain proteins on the cell surface that promote adherence to host tissue cells. Part of the difficulty in directly assigning a role to IgA proteases in the infectivity processes arises from the lack of an effective model system of infection that includes human IgA1 and its susceptible hinge region, a protein found only in humans and great apes. Further complicating the understanding of the role that IgAP plays in the pathogenicity of these bacteria is the difficulty in biochemically characterizing the substrate preferences of this enzyme family. With the exception of the type 2 enzyme from N. gonorrhoeae, the ability of IgAPs to cleave small peptide substrates based upon the primary sequence of the hinge region of human IgA1 has not been successful (23, 24). This is in contrast to the observation that peptidyl-boronic acids based upon this same hinge sequence are able to inactivate IgAPs from H. influenzae and N. gonorrhoeae (25). Whereas IgA1 is the best characterized substrate of IgAP family members, there have been reports of additional substrates that may also have a role in the infectivity pathway of these bacteria. These substrates include synaptobrevin, human lysosome-associate lamp-1 and the TNF-α type II receptor, all having been shown to be cleaved by the enzyme from Neisseria (26-29). Another substrate for IgAP is the immature precursor form of the enzyme. As previously noted, the Type V pathway of bacterial protein secretion requires a proteolysis event in order for the passenger domain to be released from its C-terminal transporter domain. This has been shown to be an autoproteolysis event as mutation of the active site serine (S288) to a valine results in the full length H. influenzae IgAP remaining attached to the bacterial outer membrane (30, 31). Due to the predicted role of this enzyme in the virulence of the aforementioned bacterial pathogens, the mechanism of specificity utilized by the IgAP family is of great interest. In addition, recent work has demonstrated a possible therapeutic role for the enzyme in the treatment of IgA nephropathy (32) further emphasizing the importance of establishing a structure-function relationship for proteolysis mediated by this enzyme family.
In order to discover how this family of serine proteases mediates the specific cleavage of the hinge peptide of IgA1 we have undertaken crystallographic studies aimed at determining the three-dimensional structure of a serine protease isozyme of IgAP. Here we present the first structure of an IgAP, a type-1 enzyme from H. influenzae at 1.75Å. Analysis of the structural data provides insight into the mechanism by which this enzyme achieves proteolytic specificity, and the results are consistent with the current biochemical data. The structural studies demonstrate that the general structural topology of IgAP, while having modest overall sequence identity to another SPATE family member, haemoglobin protease (HbP), is very similar to that enzyme. HbP is the only other SPATE family member with a known structure (33). In contrast to HbP, IgAP contains a unique ‘lid’ element that is positioned directly above the catalytic serine and is a unique insertion element in loop D of the chymotrypsin fold. We suggest that this lid, in conjunction with the valley formed between IgAP's domain-2 and the N-terminal protease domain can explain the selectivity exhibited by IgAP. We propose that interactions between the Fc domain of IgA1 and the unique loop D results in the Fc domain holding loop D away from the active site, and this open conformation provides the hinge peptide access to the active site, allowing for effective cleavage only in the context of intact IgA1.
Results and Discussion
General Structural Fold
The overall structural topology of IgAP is quite similar to that of HbP (Figure 2) consisting of an N-terminal trypsin/chymotrypsin-like protease domain (domain-1), a long β-helix ‘spine’ and a small domain previously termed domain-2 (33) that is an insert in a loop at the end of one of the β-sheets forming the β-helix. As discussed below, while the overall topology is similar, in IgAP the N-terminal protease domain and domain-2 possess structures that are subtly different than the equivalent domains in HbP. There are also differences in the smaller subdomains (domains-3 and -4) present in the valley between the β-helical spine and the N-terminal protease domain (Figure 2).
Figure 2.
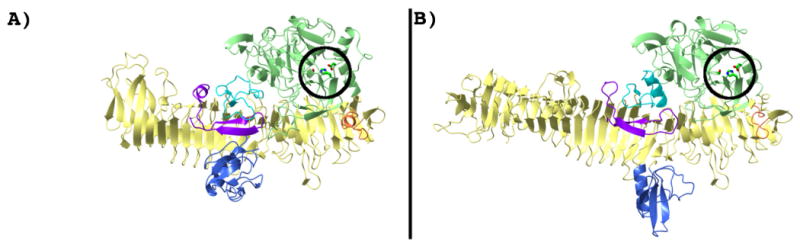
Structures of IgAP (A) and HbP (B) (PDB:1WXR). Both proteins are shown in an identical orientation and scale. The N-terminal protease domain (domain-1; 26-337) is rendered in green and the residues of the catalytic triad are circled and rendered as stick models. Domains -2, -3 and -4 are rendered in blue (564-657), cyan (710-743) and purple (786-819), respectively. In addition, a small loop insertion that is variable amongst IgAP isozymes (residues 370-387) is rendered in orange. The remaining structure, (residues 338-369, 388-563, 658-709, 744-785 and 820-989) is rendered in light yellow. All residue numbering given is that of IgAP.
The β-helical ‘Spine’
The most prominent feature of the structure of IgAP is the long, ∼100Å β-helical domain that forms the spine of the protein to which the other domains are appended. This structural feature has been suggested to be a general design principle of classical autotransporters (34). The central spine is also observed in the HbP structure (33), but as illustrated in Figure 2, the β-helical domain of that protein is even longer (∼125 Å) with the overall structure of the β-helix differing most significantly toward the C-terminal end of the secreted protein. Regardless, despite only 31% sequence identity in the β-helical region from 339-899 (IgAP) the domains have a DALI similarity score of 25.7, and the spines in this region superpose with a 1.9 Å RMSD. Similar to HbP, IgAP is observed to contain stacked asparagine residues on the external face of the β-helix. In addition, the external faces of the β-helix in IgAP contain stacked serine and threonine residues (Figure 3). In contrast to HbP but in a fashion similar to the pectate lyases (in which the β-helix was first discovered (35)) IgAP has significant stacking of leucine and isoleucine residues forming the hydrophobic β-helical core (Figure 3).
Figure 3.
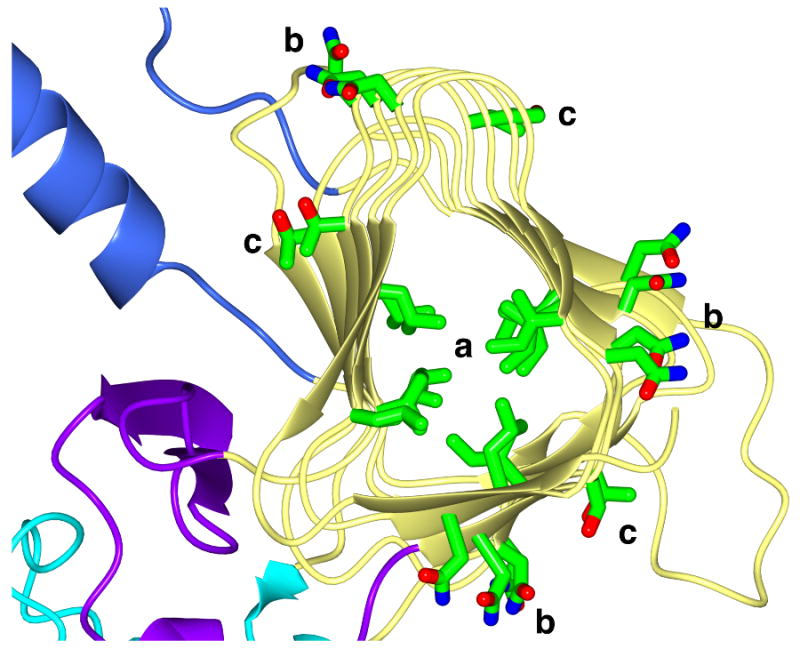
Polar and hydrophobic residue stacking in the β-helix domain of IgAP. Representative stacking interactions of Ile and Leu residues (a) in the hydrophobic core as well as Asn (b) and Ser/Thr (c) stacking on the surface of the β-helix is shown. For clarity, only the region of the β-helix from residues 562-864 is depicted. The coloring of the molecule is identical to that in Figure 2.
It is of interest to note that IgAP contains two cysteine residues (C792 and C803) and they form a disulfide bond acting to stabilize the 2-stranded β-sheet found in domain-4. The observation of this native disulfide in H. influenzae IgAP is in agreement with studies on N. gonorrhoeae suggesting the ability of folded passenger domains to be transported (36). However, these results contrast with reports suggesting that disulfide bond formation in the passenger domain inhibits transport across the outer membrane of N. gonorrhoeae (37). If, as expected, the formation of this disulfide bond is catalyzed while the protein resides in the periplasm, this structural feature may be useful in defining the mechanism of Type V autotransport across the bacterial outer membrane. Any model proposed for passenger domain transport by the Type V system would need to specify a pore large enough to accommodate the secondary structure dictated by both the formation of the C792-C803 disulfide bond and the secondary structure it stabilizes.
Domain-1: The N-terminal chymotrypsin-like domain
Not surprisingly, with an overall sequence identity of 38% the N-terminal domain of IgAP is quite similar in fold to the N-terminal domain found in HbP, the only other SPATE family member with a known structure (33). As seen in the case of HbP, a DALI structural similarity search on the residues comprising the globular N-terminal domain (A26 through D338) in IgAP (Figure 4) demonstrates that this domain possesses a high structural similarity to the trypsin/chymotrypsin family of serine proteases. DALI structural analysis of this region results in Z-scores of 30.6, and 2.7 for HbP (1.4 Å RMSD), and elastase/α-chymotrypsin (2.7 Å RMSD, 15% identity), respectively. As discussed in detail below, while the N-terminal protease domain of IgAP has a similar global fold, and the positions of the catalytic triad and oxy-anion hole are conserved, it contains several unique insertions in the loops of the chymotrypsin fold that have been demonstrated to play a significant role in the substrate selectivity in different members of this enzyme family (reviewed in (38, 39) (Figure 4)). The preservation of the oxy-anion hole in IgAP is further supported by our finding of an acetate molecule bound at this location in the structure. The differences in these loop structures in IgAP when compared to other members of the chymotrypsin family suggest that like other chymotrypsin-like proteases, these unique changes, encompassing both changes to the primary subsites for recognition of the cleaved peptide as well as loop changes remote from direct subsite contacts provide the ability of IgAP to recognize and mediate cleavage C-terminal to the P12 proline residue of the P/S/T rich hinge. However, as explained in the section below, unlike other chymotrypsin family members we think that the IgAP structural data provides evidence as to why, with respect to cleavage of the IgA1 hinge, proteolysis requires other structural elements of the IgA1 immunoglobulin.
Figure 4.

A comparison of the N-terminal chymotrypsin-like domains of IgAP (A), and HbP (B) (PDB:1WXR (33)) and the structure of elastase (C) (PDB:1PPF (77)). The structural differences in IgAP are highlighted by differential coloring of the molecules according to the loop nomenclature utilized for the chymotrypsin family. The upper and lower figures in each panel are related to each other by a 90° rotation about the horizontal axis as indicated by the horizontal black bar. The structures in the lower portion of each panel are rendered as surfaces colored according to electrostatic potential. The residues of the catalytic triad in each enzyme are rendered as grey stick models. A portion of the bound inhibitor (C-T-L-E-Y; P3-P2′) in the elastase-turkey ovomucoid inhibitor complex is rendered as a green stick model colored by atom type to illustrate the subsite locations and active site cleft. In the upper portion of each panel the molecules are rendered as ribbon diagrams and colored according to the following color scheme: Loop A; dark blue 81-91 (34-41), Loop B; red 99-106 (56-64), Loop C; yellow 144-164 (97-103), Loop D; magenta 205-243 (143-149), Loop E; cyan 109-134 (74-80), Loop 1; orange 261-282 (185-188), Loop 2 black 311-319 (217-225) and Loop 3 green 246-257 (169-174). The N-terminal region, residues 26-78 (16-29) that forms a discrete domain in IgAP and HbP but is absent in elastase is colored light blue. The residue numbering given is that for IgAP with the corresponding numbering for elastase/chymotrypsin given in parentheses.
As illustrated in Figures 4 and 5, a surface rendering of the protease domain demonstrates that changes in the identity of the amino acid residues comprising the active site region of IgAP, when compared to other chymotrypsin-like proteases such as trypsin and chymotrypsin, results in the occlusion of the subsite pockets forming the IgAP active site. This is reminiscent of elastase, which prefers the cleavage of short chain aliphatics at the P1 subsite. While the occlusion of the subsites is reminiscent of elastase, as is shown in Figure 4, the active site of IgAP provides a shallower and more narrow substrate-binding surface than observed for either elastase or HbP.
Figure 5.
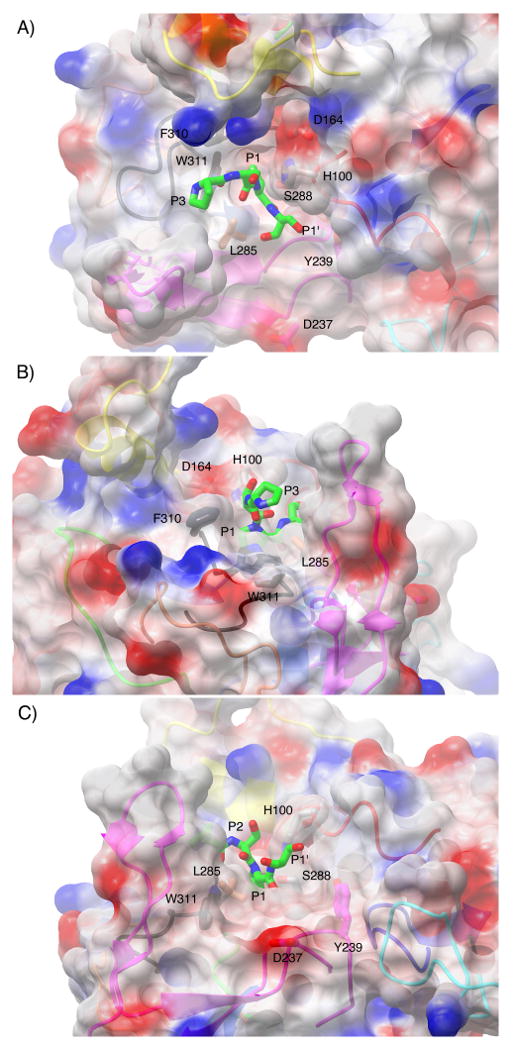
Surface representation of the active site cleft of IgAP shown in a view from above (A), from the N-terminal direction of the hinge peptide (B) and from the C- terminal direction of the hinge peptide (C). The electrostatic surface is rendered semitransparent with the secondary structure of the protease domain that gives rise to the surface being rendered as a ribbon diagram and colored in an equivalent fashion to Figure 4. Those side chains that contribute to the formation of unique surface architecture of the active site are labeled and colored by atom type with the carbon atoms colored according to the loop region of which they are members. The catalytic triad is colored by atom type and rendered in grey. The P3-P1′ region (229P-S-P-S232) of the hinge peptide was manually modeled into the active site of IgAP based upon the position of the equivalent region in the elastase-turkey ovomucoid inhibitor complex.
The filling of the subsites is particularly evident in the S1 pocket where the deep channel present in trypsin that is required for P1 lysine and arginine binding is filled by Y308 in IgAP (Figure 6). This generates an S1 pocket that is quite shallow, creating a steric restriction that seems ideally suited to fit proline that is a feature of all IgAP isozymes regardless of the site of cleavage in the hinge region (Figure 1). Further insight into substrate recognition at the active site subsites of the N-terminal domain comes from modeling of the P3-P2′ residues of the hinge based upon the available structure of elastase in complex with turkey ovomucoid inhibitor (Figures 5 and 6). Inspection of this model reveals that while F310 is conserved between elastase and IgAP (F215 in elastase), in IgAP F310 is found in an altered conformation. If this same orientation for the side chain of F310 exists in the actual IgAP enzyme-substrate (E-S) complex it would prevent the binding of the P4 residue of the substrate in a position equivalent to that of the P4 residue of elastase substrates (Figures 4 and 5). This potential steric occlusion of the S4 subsite by F310 indicates that the substrate peptide would be directed toward the crevice between this residue and the extended loop D, altering the path taken by the substrate when compared to elastase and other chymotrypsin-like proteases (Figure 5B).
Figure 6.
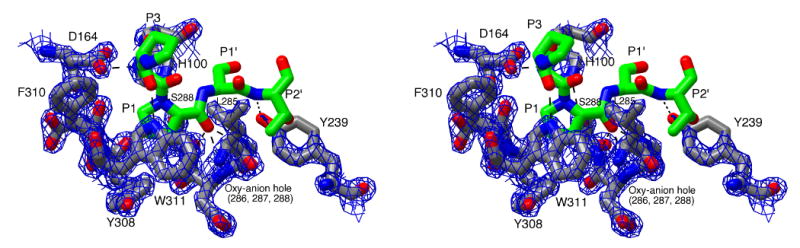
Stereoview of the active site of IgAP illustrating the recognition of the hinge peptide. The P3-P2′ region (229P-S-P-S-T233) of the hinge peptide was manually modeled into the active site of IgAP based upon the position of the equivalent region in the elastase-turkey ovomucoid inhibitor complex and is colored by atom type with the carbon atoms being rendered in green. The catalytic triad and residues of IgAP involved in substrate recognition are colored by atom type with the carbon atoms being rendered in grey. Potential hydrogen bonds between the substrate peptide and the enzyme and members of the catalytic triad (D164, H100, S288) are illustrated by dashed lines. 2FO-FC density for the protein rendered at 2.5σ is shown as a blue mesh.
In contrast to the changes in the topology of the subsites, the modeled E-S complex suggests that backbone interactions between the hinge peptide and the enzyme are preserved. The preservation of these interactions would therefore allow for correct substrate positioning to be facilitated through enzyme-substrate hydrogen bonding in a fashion typical of most proteases; the recognition of the substrate peptide as an extended β-strand (40) (Figure 6). While loop A of the protease domain is significantly truncated in IgAP (Figure 4), the phenolic oxygen of Y239 is in an identical position to the backbone carbonyl of F41 found in the structure of elastase and mediates the same hydrogen bond with the amide nitrogen of the P2′ residue. Further comparison of the subsite architecture of IgAP with that of elastase shows that the change in substrate positioning dictated by the potential narrowing of the active site through the positioning of F310 in IgAP (described above) would eliminate the interaction between the P3 residue carbonyl and the backbone amide of V216 present in elastase (W311 in IgAP). However, additional structural inspection reveals that the enzyme-P3 backbone interaction could be preserved in IgAP through the side chain of W311. While W311 functions to form the wall of a smaller P1 binding pocket tailored to fit proline, this side chain position also results in the W311 indole nitrogen being ideally oriented to act as a hydrogen bond donor to the backbone carbonyl of the P3 residue, thus preserving the favorable interaction (Figures 5 and 6). Lastly, the modeled position of the peptide results in the orientation of the serine residues at P2 and P1′ away from the surface of the enzyme, consistent with the ability of the enzyme to hydrolyze the hinge peptide in the context of its native O-linked glycosylated state (41).
Domains -2, -3 and -4
As mentioned above, and described previously in the context of the structure of HbP (33) domain-2 forms a discrete domain (residues 564-657) off of the stalk of the β-helix, entering and exiting as an extension of two β-strands forming a sheet in the β-helix (Figures 2 and 3). Structural inspection demonstrates that domain-2 in IgAP extends from the β-helix at a different angle than the similar domain in HbP, likely due to crystal packing interactions with the N-terminal protease domain of an adjacent molecule in the crystalline lattice (Figure 7). This difference between the orientation of domain-2 relative to the β-helical spine found in the structures of HbP and IgAP suggests the potential for independent movement of domain-2 relative to the β-helical spine in the absence of crystal lattice contacts. As discussed below, the potential dynamic nature of domain-2 may be important in IgAP function. While similar in its overall fold to the domain in HbP (DALI score of 8.2, 21 % sequence identity, 2.3 Å RMSD), in itself similar to chitinase, the domain in IgAP differs slightly in structure due predominantly to a loop insertion of ∼12 residues in the loop between the two β-strands at the N-terminus of the domain. This domain also possesses structural similarity to the family of integrase proteins, phospholipase (PDB: 1ZLB, Z=3.9), extracellular matrix protein anosmin-1 (PDB:1ZLG, Z=3.6), and N-terminal domain l-integrase (PDB:1KJK, Z=3.4) with similar RMSD values of 2.0-2.5 Å.
Figure 7.
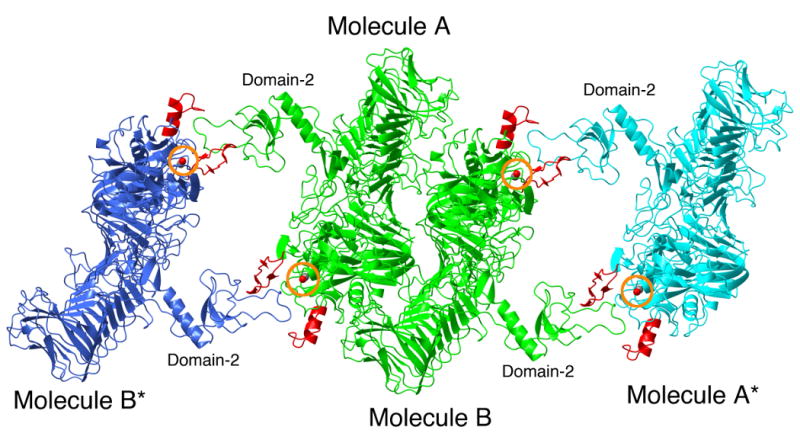
Crystal packing interactions in the structure of IgAP. The crystallographic dimer (NCS related molecules A and B) present in the asymmetric unit are rendered in green and labeled. Molecules A* and B* are related to molecules A and B by crystallographic symmetry and are rendered in cyan and blue, respectively. Loops C and D of the protease domains of all molecules are rendered in red. For the purpose of orientation, the side chain oxygen atom of the active site serine residue is visible as a red ball and circled in orange.
As mentioned above, the interaction of domain-2 with the N-terminal protease domain of a symmetry related molecule is of interest as it suggests the ability of this domain to function in protein-protein interactions and by extrapolation, that domain-2 may contribute to substrate recognition by forming a binding surface for IgA1. In the crystals of IgAP (Table 1) as in all crystals, the crystal is composed of identical repeating microscopic units (unit cells) that are stacked together in a three-dimensional array to form the macroscopic crystal. In the monoclinic (P21) crystals of IgAP each one of these unit cells is composed of two smaller asymmetric units (ASU). The ASU is the smallest repeating unit of the crystal lattice. All other molecules in the unit cell can be generated by applying rotation and translation operators on the molecules contained in the ASU as dictated by the crystallographic symmetry. Each ASU of the IgAP crystals contains two independent molecules of IgAP that are not related to each other by crystallographic symmetry and are termed non-crystallographic symmetry (NCS) related molecules. In the case of IgAP these two molecules have essentially identical structures, however by definition they experience different crystallographic environments. Due to the molecular packing of the individual molecules of IgAP in the unit cell it is observed that domain-2 of the two independent molecules in one ASU (molecules A and B) interact with the unique loop D of the protease domain in the adjacent ASU (Figure 7). This interaction between domain-2 and loop D of the protease domain places the extended loop of domain-2 of molecule A directly over the active site of the symmetry related molecule B in the adjacent ASU. This packing of the protein molecules places domain-2 in the cleft between the extended loops C and D that in the context of chymotrypsin-like proteases, are unique to IgAP. Importantly, it is this crystallographic packing interaction between domain-2 and loop D that we propose stabilizes the conformations of loops C and D observed in the crystal structure.
Table 1.
Data and Model Statistics for the 1.75Å structure of Haemophilus influenzae IgAP.
| wavelength (Å) | 0.86 |
| space group | P21 |
| unit cell dimensions |
a = 94.39Å b = 131.87 Å c = 111.81 Å α = γ = 90.0° β = 113.11° |
| resolution limit (Å) | 50.0-1.75 |
| no. of unique reflections | 251,843 |
| Completenessb (%; all data) | 99.9(99.0) |
| Redundancya | 7.4(5.2) |
| I/σ(I)a | 16.7(2.0) |
| Rmergea,b | 0.08(0.67) |
| no. of ASU molecules | 2 |
| solvent content (%) | 53 |
| Rfreea,c (%) | 19.0(30.5) |
| Rworka,d (%) | 16.2(27.9) |
| estimated coordinate error based on maximum likelihood (Å) | 0.059 |
| bond length rmsd (Å) | 0.013 |
| bond angle rmsd (deg) | 1.430 |
| Ramachandran statistics (most favored, additionally allowed, generously allowed, disallowed) (%) | 88.0, 11.6, 0.4, 0.0 |
Values in parentheses represent statistics for data in the highest-resolution shell. The highest-resolution shell comprises data in the range of 1.81–1.75 Å.
Rmerge = Σ|Iobs - Iavg|/ΣIobs
See Brunger (78) for a description of Rfree.
Rwork = Σ‖Fobs| -|Fcalc‖/Σ|Fobs|
In addition to domain-2, IgAP and HbP contain two other small domains we refer to as domain-3 and domain-4 (Figure 2). These domains are similar to domain-2 in that they are insertions in the β-strands of the β-helix that extend away from the β-helical spine and are located at the base of domain-2, in the cleft between the N-terminal protease domain and the β-helical spine (Figure 2). In comparing the domains in IgAP to those found in HbP it is observed that both domains are projections from the same points in the β-helical spine however, the sequence and structure of the domains differ between the two enzymes. Due to the location of these elements on a face of the enzyme that places them in line with the subsite binding cleft of the protease domain we think that these three subdomains (domains-2, -3, and -4) are involved in forming a unique structure that is involved in recognition and binding of the Fc domain of IgA1.
Docking results: The IgA1(Fc)-IgAP Complex
In order to test the above hypothesis we undertook computational docking of the Fc domain of IgA1 that has previously been determined by crystallography (42) with the structure of IgAP presented here. Consistent with the model proposed above, two of the top four results of the computational protein-protein docking study place the Fc molecule in the valley between the protease domain and domain-2 (Figure 8A and 8B). The two plausible orientations obtained are the result of a similar positioning of either the A or B chain, which are identical, of the Fc domain adjacent to the protease domain. Further, these two solutions were the only solutions of the ten solutions reported that gave a plausible orientation of the N-terminal end of the Fc domain relative to the active site of the protease (Figure 8C and 8D). As illustrated in Figure 8, these two orientations place the N-terminus (C241) of the Fc domain in a general orientation and at a reasonable length that could result in the correct positioning of the hinge region with respect to the active site serine residue. Further, the docked model suggests a plausible mechanism for the stabilization of loop D in the observed conformation by loop D acting as a 2-strand extension of the β-sheet forming the β-barrel of the Fc domain of IgA1. This β-sheet interaction also suggests a general mechanism of substrate recognition for IgAPs. Certainly the region of the β-helix of IgAP that requires self-cleavage for release from the bacterial membrane fulfills this substrate requirement. Further inspection of the docked structures demonstrate the two modeled conformations for the Fc domain differ in whether contacts with the N-terminal protease domain or domain-2 are maximized, giving rise to the slightly different orientations of the Fc molecule relative to the protease. As shown in Figure 8A, the first model maximizes the interface between the protease domain and the Fc-CH2 region of the Fc domain at the expense of contacts between the CH2 and CH3 region of the Fc domain with domain-2 of IgAP. In the second model shown in Figure 8B, the converse is true. This disparity in the interfaces between the docked Fc component and domain-2 and the protease domain of IgAP in the two models again supports the idea that domain-2 is a dynamic element of IgAP structure as mentioned above. The two subtly different modeled conformations of the IgAP and the Fc region of IgA1 suggests that conformational changes in the protease, most likely in the orientation of domain-2 relative to the protease domain and the β-helical spine, are necessary upon IgA1 binding to maximize both the Fc-domain-2 and Fc-protease domain interfaces.
Figure 8.
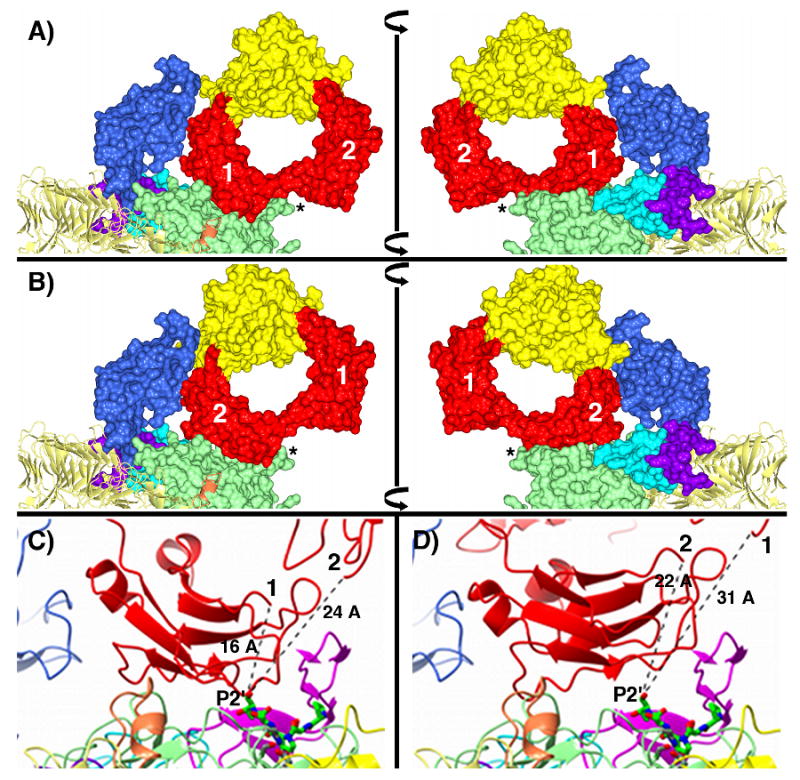
Theoretical models of the IgAP-IgA1-Fc binary complex. The two different solutions discussed in the text are shown in panels A and B. In all panels the domain coloring scheme for IgAP is identical to that in Figure 2 and the N-terminal protease domain, and domains -2, -3 and -4 are rendered as molecular surfaces. In panels A and B, the CH2 and CH3 regions of the Fc domain are rendered as molecular surfaces and colored red and yellow, respectively. The left and right figures in panels A and B are related by a 180° rotation about the vertical axis indicated by the vertical black bar. The two CH2 domains are further annotated with the numbers 1 and 2 corresponding to chain A and chain B of the Fc domain and loop D of the protease domain is indicated by an *. In panels C and D the individual loops of the protease domain are colored according to the scheme in Figure 4 and the Fc domain of IgA1 is rendered as a red ribbon diagram. In panels C and D, the manually docked hinge peptide (229P-S-P-S-T233) is rendered as a green stick model colored by atom type and the N-terminal cysteine (C241) of the Fc domain is labeled either 1 (C241, chain A) or 2 (C241, chain B) as are the approximate distances between the C-terminal P2′ residue of the docked hinge peptide and the N-terminus of C241 of chains A and B of the Fc domain.
Support for the location of the Fc binding site on IgAP indicated by the crystallographic and docking studies presented here is provided by studies that demonstrate structures in the Fc domain are required for cleavage of IgA1 (41) and further studies that suggest elements of the CH3 region of the Fc domain are involved in recognition and cleavage of IgA1 by IgAP (43). As is shown in Figure 8 A and B, the docked orientation of the Fc domain is consistent with these previous results as the models presented here place the CH3 domain in an orientation where its tertiary structure and individual amino acid contacts would play a role in substrate recognition through the interface formed with domain-2 of IgAP. Further experimental support for the docking studies comes from data that suggests that a minimum spacing between the Fab and Fc domains provided by the hinge peptide must be present and this length is approximately ten amino acids (44). Measuring a linear distance across the active site face of IgAP from the Fc CH2 region to the far side of the protease domain generates a distance of ∼30 Å. Based upon this distance constraint, the modeled binding site for the Fc domain is again consistent with the biochemical studies as the measured ‘width’ could be spanned by an extended peptide linker of ten amino acids providing a structural basis for the observed biochemical studies. More simply, if the Fab and Fc domains are visualized as two balls attached to opposite ends of a length of string (the hinge peptide), the string length must be long enough to provide a minimum separation distance such that the Fab domain does not run into steric conflicts with the surface of the IgAP protease domain when the Fc domain is ‘tethered’ to the binding site that is suggested by the docking studies.
One discrepancy between the computational models above relative to the literature is the necessity for a ‘bent’ hinge region that is in contrast to the extended, linear structure of the IgA hinge suggested by molecular modeling and solution scattering data (45, 46). We suggest the cautious interpretation of the docking results. Inspection of the docked model suggests that a rotation of the Fc domain relative to the IgAP structure would allow for a more linear exit of the hinge peptide from the active site, more consistent with the current solution scattering data. Clearly, as discussed above, the actual structures of IgAP and IgA1 in the enzyme-substrate complex will differ from the individual structures utilized for the docking studies and these conformational changes could possibly result in a more linear hinge. What we do conclude is that the docking studies support the idea that the Fc domain of IgA1 binds in the valley between domain-2 and the protease domain. This orientation also illustrates that the β-barrel of the Fc domain is capable of interacting with the unique loop insertion in loop D, stabilizing the 2-stranded β-sheet of this loop in a similar fashion to the stabilization of this conformation of loop D by domain-2 through crystal packing interactions. While we do not suggest that the modeling of the IgA1-Fc-IgAP complex excludes an extended conformation in solution for the IgA1 hinge, this extended conformation is likely only one of the conformations that the immunoglobulin can adopt, and formation of the enzyme-substrate complex could possibly result in the stabilization of a more bent orientation for the hinge than that determined for the molecule in solution. If the modeling of the N-terminal region of the substrate peptide is correct (Figures 4-6), the interaction of IgA1 with IgAP could stabilize a slightly bent conformation of the hinge that deviates from that calculated for the most energetically favored/most highly populated conformation as determined by the aforementioned techniques. The potential for conformational sampling of the hinge structure is echoed by Boehm et al. in their work as is the possibility for subtle changes in hinge structure due to the absence of O-linked glycoslyation in their modeling of the hinge domain (45).
Based upon the location of the Fc binding site suggested by the computational docking studies we think it is reasonable to predict that the Fab domain interacts with the ‘non-prime’ face of the protease domain in the general area depicted in Figure 5B. If the Fab domain binds in this region it is reasonable to speculate that the Fab region could make contacts with loop C, stabilizing this structural element. The crystallographic data suggest that the enlarged loop C found in IgAP is a mobile element as the quality of the density in this loop is poorer than the well-ordered regions of the protein. This mobility is reflected in an elevated average B-factor value for loop C (chain A = 38 Å2, chain B = 51 Å2) when compared to the average B-factor for the rest of the protein (chain A = 18 Å2, chain B = 21 Å2). Therefore, in a fashion similar to that proposed for loop D, the stabilization of loop C through interactions with IgA1 could prevent loop C from occupying a conformation that prevents hinge peptide binding to the subsites. Further, as in other chymotrypsin-like proteases, the aspartate residue of the catalytic triad (D164 in IgAP) lies at the base of loop C (Figure 4), and therefore any motion of loop C could influence catalytic function as it would disrupt the formation of the triad and subsequently reduce the nucleophilicity of the active site serine residue for the acylation half reaction and/or reduce the ability of H100 to activate the hydrolytic water molecule in the deacylation step. Therefore, the mobility of loop C in the absence of IgA1 binding could decrease catalytic function through the perturbation of the D164-H100 pair, reducing the rate of peptide hydrolysis.
Mechanism of IgAP substrate recognition
Based upon the structural and modeling data presented here and the previously reported biochemical studies, we present two possible models to explain the unique ability of IgAP to hydrolyze the hinge region of IgA1 only when it is in the context of the entire IgA1 molecule.
The interactions between the hinge peptide and the protease at the active site, dominated by five hydrogen bonds between the enzyme and the P3, P1 and P2′ residues is not sufficient to provide a tight binding complex that is stable enough for catalysis to proceed. Thus elements of Fc structure are needed to provide interactions capable of forming a complex stable enough to allow for the positioning of the peptide substrate via the aforementioned hydrogen bonds between the enzyme and P3, P1 and P2′ backbone.
The unique, extended loops C and D and domain-2 are dynamic elements and in the absence of interactions with IgA1, one or all of these elements occlude the active site of the protease, reducing the ability of the enzyme to interact with small peptide ligands based upon the sequence of the hinge domain. The binding of the Fc domain in the valley between domain-2 and the protease domain results in interactions of the Fc domain with domain-2 and loop D that stabilizes an open conformation, similar to that observed in the crystal structure which is stabilized by interactions between loop D and domain-2 of a symmetry related molecule in the crystal lattice. In a similar fashion, the binding of the Fab domain to the opposite face of the protease could stabilize loop C preventing active site occlusion and/or stabilizing the conformation of D164 to optimally participate as a member of the catalytic triad. This stabilized, open conformation of the active site allows access of the hinge peptide to the active site, whereby the aforementioned hydrogen bonds between the hinge peptide and the active site provide a stable complex necessary for proteolytic cleavage.
Based upon the crystallographic data and the previous observations of chymotrypsin-like proteases having 5-enzyme substrate bonds between positions P1 and P3 (39) we think that the potential interactions available between the backbone of the hinge peptide and the enzyme are sufficient for IgAP to form a tight, specific complex with the hinge peptide allowing for proteolysis in the absence of the remaining structure of IgA1. This conclusion is also supported by the ability of peptidyl boronic acids based upon the hinge sequence to irreversibly inactivate IgAP isozymes (25). In our view this information suggests that an alternative mechanism is necessary to explain the general lack of cleavage of hinge peptides by these enzymes in the absence of the remaining structure of the immunoglobulin. We think that model 2 is the most reasonable model that is consistent with all of the data. The key importance of loops C and D in positioning of the IgA1 substrate relative to the active site serine is also supported by the homology model of the H. influenzae IgAP-2 isozyme that has an altered cleavage site and is dominated by truncation of these two loops (Figure 1, supporting information). In a more general fashion, this idea that part of the IgA1 structure acts as a ‘spacer’ in positioning of the hinge peptide for cleavage has been suggested previously based upon biochemical characterization of the cleavage of hinge variants of IgA1 by IgAPs (47). Extending upon this model, rather than the C-terminal half of the hinge peptide acting as a spacer as previously suggested, we think that the specific interactions between the Fc region and IgAP, through binding in the valley between domain-2 and the N-terminal protease domain and through interactions with loop D, results in the establishment of the sequence register of the hinge at the active site that allows for positioning of the peptide for cleavage. As discussed above, a minimal length for the hinge is necessary so the distance between the Fc and Fab domains is large enough to span the N-terminal protease domain, avoiding the steric conflict between the protease domain and the Fab segment that would occur with shorter hinge segments. Importantly, all the structural data is consistent with the previous body of biochemical studies including those demonstrating that elements in the Fc region are necessary for recognition and cleavage of the hinge peptide of IgA1 by H. influenzae and N. gonorrhoeae IgAPs (41, 43).
In the context of the proposed model, the ability of boronic acids to inactivate IgAP while small peptide molecules based upon the hinge sequence fail to act as substrates appears to be unanswered. However, in order to be stabilized in the open conformation, in the absence of IgA1 binding loop C and/or loop D would be required to sample an open conformation. This type of behavior is well documented in enzymes such as RNaseA where NMR studies demonstrate that the substrate bound conformation is sampled in the absence of substrate binding and is the basis for the conformational selection model of enzyme-ligand association (48, 49). Thus we suggest that the intrinsic sampling of the open conformation would allow small peptide based ligands access to active site in the absence of IgA1 binding. However the ability of small peptide substrates to bind at the active site during this sampling of the open conformation would be a direct competition between the on rate for the substrate and the transition rate between open and closed conformations in the absence of IgA1 binding. Therefore, differing on-rates between peptide ligands, whether they are small molecule substrates or peptidyl boronic acids could explain the absence of activity with peptidyl substrates and the inactivation of the enzyme by peptidyl boronic acids. An additional possibility is that even if both peptidyl substrates and boronic acid inhibitors are able to associate with the active site upon the enzyme sampling the open conformation, the closed conformation may subsequently become further stabilized in this enzyme-ligand complex due to new interactions between the bound ligand and the mobile structural elements. Therefore, establishing the enzyme-substrate-like complex could further reduce the ability of the enzyme to sample the open conformation. The direct consequence of this stabilization would be that substrate turnover would not be observed in the kinetic assay. Alternatively, the proposed mobility of loop C in the absence of IgA1 binding and the potential impact this would have on the the catalytic triad, could explain the lack of hydrolysis observed with peptidyl substrates versus the peptidyl boronic acids. By design, the potent boron electrophile of the boronic acid inhibitors results in the alkylation of the active site serine by these inhibitors being more facile than the acylation step of a natural peptide substrate and any effects of disruption of the catalytic triad on the deacylation process due to the mobility of loop C would not be relevant in the inactivation of the enzyme by boronic acids. Clearly further biochemical and structural studies will clarify how the discrepancy between the inactivity of IgAP against peptidyl substrates and its inactivation by peptidyl boronic acids fits within the context of our proposed model.
Conclusion
Not surprisingly, the active site architecture of the N-terminal protease domain of IgAP is consistent with the divergent evolution of a molecule from a chymotrypsin-like progenitor that has minimal subsite-substrate contacts but that is highly selective for the cleavage of substrates containing a P1 proline residue within the context of a P/S/T rich octapeptide repeat that corresponds to the hinge peptide of IgA1. However, in contrast to traditional chymotrypsin-like protease substrate recognition we think that specific proteolytic recognition of the substrate in the context of the intact IgA1 immunoglobulin is regulated primarily by the unique loop insertions in loop C and loop D and with domain-2. The structural data is consistent with a model whereby domain-2 forms a specific binding site for the Fc domain of IgA1 adjacent to the active site of the N-terminal protease domain and loop D of IgAP. We speculate that in the presence of specific interactions between loop-D and the Fc domain of the immunoglobulin (or other substrates that are capable of similar interactions) the loop occupies an open conformation, resulting in a access of the substrate peptide to the active site binding cleft. We further speculate that due to the modeled position of the hinge peptide and the Fc domain of IgA1 that a similar stabilization of loop C is possible through interactions with the Fab domain. Upon stabilization of this open conformation for the protease domain, the hinge peptide is recognized and cleaved through a process typical of chymotrypsin-like proteases.
Clearly, further testing of this model of substrate recognition is needed. However, we suggest that this hypothesis is both consistent with the current structural knowledge and the previous biochemical data. Further, homology modeling of a type 2 IgAP isozyme demonstrating alteration of the specificity loops (Figure1, supporting information) can explain both the differences in the position of the proline after which cleavage occurs as well as the ability of certain isozymes with more truncated loops to catalyze the proteolysis of small peptide substrates (23). Lastly, the structure of IgAP clearly illustrates the diversity in substrate recognition that is possible within a chymotrypsin-like scaffold through modifications to distal loop elements allowing for the evolution of selectivity in this enzyme superfamily beyond that of traditional subsite-substrate interactions.
Materials and Methods
Materials
All chemicals utilized were of the highest commercially available purity.
IgAP Expression and Purification
IgAP was expressed in H. influenzae Rd 6His strain as a secreted fusion protein having a C-terminal 6-Histidine tag, as previously described (32). Bacteria-free fermentor broth (∼10L) was concentrated to ∼400 mL using a Pelicon device and precipitated with 60% saturated ammonium sulfate. The pellet was subsequently resuspended in 25 mL of 25 mM HEPES, pH 7.5, 10 mM imidazole, and applied to a 50 mL bed volume of Ni-NTA agarose (Qiagen). The column was washed with several column volumes of 25 mM HEPES pH 7.5, 10 mM imidazole followed by 25 mM HEPES pH 7.5, 50 mM imidazole. The protein was eluted with a gradient of imidazole (50 – 90 mM, in 25 mM HEPES, pH 7.5) using a BioRad BioLogic Duo Flow FPLC. Fractions containing intact IgAP were identified by SDS gel electrophoresis. These fractions were pooled, concentrated to ∼10 mL under nitrogen pressure in an Amicon concentrator and passed through a 25 mL bed volume of DE-52 anion exchange resin equilibrated in 25 mM HEPES, pH 7.5 to remove yellow pigment carried over from the fermentation media. The wash-through was collected and concentrated in an Amicon concentrator to a volume of approximately 2 mL and applied to a 26/60 Sephacryl S-200 column (GE Biosciences) equilibrated in 25 mM HEPES, pH 7.5. The fractions containing IgAP were concentrated to a final protein concentration of 7 mg/mL based upon a predicted extinction coefficient (ε280) of 1.04 mL mg-1 and a molecular mass of 108,671 Da, both calculated using the ProtParam tool (http://ca.expasy.org/tools/protparam.html) (50). The protein was used immediately for crystallization studies as it was found that the protein suffered from proteolytic degradation when stored for extended periods at 4 °C.
Crystallization
Initial crystallization conditions for Haemophilus influenzae IgAP were determined by submission of the protein to the high-throughput crystallization facility at the Hauptman Woodward Institute, Buffalo, NY (51). Crystals of IgAP used for data collection were grown by the hanging-drop method at 4 °C by mixing 4 μL of protein [containing 7 mg/mL IgAP in 25 mM HEPES, pH 7.5] with 2 μL mother liquor [0.1 M sodium acetate, pH 5.0, 0.1 M potassium dihydrogen phosphate and 10% PEG 20,000]. The crystals were cryoprotected by transferring them in succession to 20 μL drops containing 40, 50 and 60 % saturated sodium malonate prior to cryocooling in liquid nitrogen (52).
Data Collection
Data on the cryocooled crystals at 100 K were collected at the Stanford Synchrotron Radiation Laboratory, Beamline 11-1, Menlo Park, CA. All data were integrated and scaled with HKL-2000 (53). Data statistics are presented in Table 1.
Structure Determination and Refinement
The structure of IgAP was solved by the molecular replacement method. In this procedure, the program Chainsaw (54) was utilized to create a molecular replacement model using the N-terminal domain of hemoglobin protease (PDB 1WXR, 36% sequence identity over 536 residues). This model was subsequently used as the search model in Phaser (55). The Phaser solution contained two copies of this domain in the ASU. This solution was utilized as the input for automated model building using ARP/WARP 7.0 (56-59). The output from ARP/wARP was a model that was ∼80% complete for both chains. Subsequent correction and further building of the model was carried out utilizing COOT (60). The final model lacks the C-terminal 25 residues with both models ending at P989. In addition, density in chain A for the region 967-969 was not suitable to build this region and is missing from the final model.
After model building, heteroatom and water addition were carried out using COOT. A final round of TLS refinement was performed in Refmac5 (61) in the CCP4 suite (62). A total of 5 groups/chain were utilized. Refinements using greater than 5 groups/chain did not significantly improve R/Rfree. The optimum TLS groups were determined by submission of the pdb file to the TLSMD server (http://skuld.bmsc.washington.edu/∼tlsmd/index.html; (63-65)). Tight NCS restraints were utilized during the initial rounds of refinement and were removed during the final stages of refinement. Both chains in the model have excellent stereochemistry as determined by PROCHECK (66). Final model statistics are presented in Table 1.
IgA-Fc domain-IgAP Docking
Docking of the Fc portion of human IgA1 was accomplished by submission of the structure of the monomer (chain B) of IgAP and the structure of the Fc domain of human IgA1 (PDB:1OW0, chains A and B (42)) to the Cluspro server (http://nrc.bu.edu/cluster/) (67, 68) utilizing DOT (http://www.sdsc.edu/CCMS/DOT/) to carry out the docking (69).
Three-dimensional Structure Similarity Comparisons
Structural similarity searches were performed by submission of the relevant protein domains of IgAP to the DALI server (http://ekhidna.biocenter.helsinki.fi/dali_server) (70).
Homology modeling of IgAP-2
The sequence of a type 2 IgA protease (IgAP-2; Q93T34) from H. influenzae var. aegyptius (71) was aligned with the sequence for the type 1 IgA protease (IgAP-1; P44969) corresponding to the enzyme utilized in the crystallographic studies using ClustalW (72, 73). These two proteins share 69% sequence identity over the 964 residues of the secreted IgAP-1 isozyme visible in the crystal structure. This alignment and the IgAP-1 structure were used as the input to the automodel script for the program Modeler 9 version 5 (74) to produce a 3-dimensional homology model of the entire IgAP-2 isozyme.
Supplementary Material
Acknowledgments
National Center for Research Resources P20 RR016443 (T.H.) and P20 RR17708 (T.H.), the Tufts Clinical and Translation Science Institute, NIH NCRR UL1 RR025752 (A.G.P.) and NIH R21 DK071675 (A.G.P.). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We acknowledge the invaluable technical help of Dr. Anne Kane and Charles Sutera III, B.S.
Footnotes
Coordinates and structure factors have been deposited in the RCSB protein databank (http://www.rcsb.org/pdb) under the accession code 3H09.
Abbreviations: ASU, asymmetric unit; HbP, E. coli hemoglobin protease; IgAP, Haemophilus influenzae immunoglobulin A1 protease; IgAP-1, Haemophilus influenzae immunoglobulin A1 protease type 1; IgAP-2, Haemophilus influenzae immunoglobulin A1 protease type 2; IgA1, human immunoglobulin isotype 1; NCS, non-crystallographic symmetry; PEG, polyethylene glycol; RMSD, root-mean-square-deviation; SPATE, serine protease autotransporters of the Enterobacteriaceae; TLS, translation/libration/screw.
The nomenclature utilized is that of Schechter and Berger (75) in which the substrate amino acid residues are designated NH3+-Pn … P2, P1, P1′, P2′…Pn′-COO- where the scissile bond is located between the P1 and P1′ residues. Conversely, the binding sites for the Pn residues at the enzyme active site are designated Sn … S2, S1, S1′, S2′… Sn′.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mehta SK, Plaut AG, Calvanico NJ, Tomasi TB., Jr Human immunoglobulin A: production of an Fc fragment by an enteric microbial proteolytic enzyme. J Immunol. 1973;111:1274–1276. [PubMed] [Google Scholar]
- 2.Jose J, Jahnig F, Meyer TF. Common structural features of IgA1 protease-like outer membrane protein autotransporters. Mol Microbiol. 1995;18:378–380. doi: 10.1111/j.1365-2958.1995.mmi_18020378.x. [DOI] [PubMed] [Google Scholar]
- 3.Pohlner J, Halter R, Beyreuther K, Meyer TF. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature. 1987;325:458–462. doi: 10.1038/325458a0. [DOI] [PubMed] [Google Scholar]
- 4.Dautin N, Bernstein HD. Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu Rev Microbiol. 2007;61:89–112. doi: 10.1146/annurev.micro.61.080706.093233. [DOI] [PubMed] [Google Scholar]
- 5.Henderson IR, Navarro-Garcia F, Nataro JP. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 1998;6:370–378. doi: 10.1016/s0966-842x(98)01318-3. [DOI] [PubMed] [Google Scholar]
- 6.Klauser T, Pohlner J, Meyer TF. Extracellular Transport of Cholera-Toxin B-Subunit Using Neisseria Iga Protease Beta-Domain -Conformation-Dependent Outer-Membrane Translocation. Embo Journal. 1990;9:1991–1999. doi: 10.1002/j.1460-2075.1990.tb08327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klauser T, Pohlner J, Meyer TF. Selective Extracellular Release of Cholera Toxin-B Subunit by Escherichia-Coli - Dissection of Neisseria Iga-Beta-Mediated Outer-Membrane Transport. Embo Journal. 1992;11:2327–2335. doi: 10.1002/j.1460-2075.1992.tb05292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oomen CJ, van Ulsen P, Van Gelder P, Feijen M, Tommassen J, Gros P. Structure of the translocator domain of a bacterial autotransporter. Embo Journal. 2004;23:1257–1266. doi: 10.1038/sj.emboj.7600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skillman KM, Barnard TJ, Peterson JH, Ghirlando R, Bernstein HD. Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Molecular Microbiology. 2005;58:945–958. doi: 10.1111/j.1365-2958.2005.04885.x. [DOI] [PubMed] [Google Scholar]
- 10.Veiga E, Sugawara E, Nikaido H, de Lorenzo V, Fernandez LA. Export of autotransported proteins proceeds through an oligomeric ring shaped by C-terminal domains. Embo Journal. 2002;21:2122–2131. doi: 10.1093/emboj/21.9.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta PR, Cappello R, Navarro-Garcia F, Nataro JP. Functional comparison of serine protease autotransporters of enterobacteriaceae. Infect Immun. 2002;70:7105–7113. doi: 10.1128/IAI.70.12.7105-7113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henderson IR, Nataro JP. Virulence functions of autotransporter proteins. Infect Immun. 2001;69:1231–1243. doi: 10.1128/IAI.69.3.1231-1243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulks MH, Kornfeld SJ, Plaut AG. Specific proteolysis of human IgA by Streptococcus pneumoniae and Haemophilus influenzae. J Infect Dis. 1980;141:450–456. doi: 10.1093/infdis/141.4.450. [DOI] [PubMed] [Google Scholar]
- 14.Mulks MH, Plaut AG, Feldman HA, Frangione B. IgA proteases of two distinct specificities are released by Neisseria meningitidis. J Exp Med. 1980;152:1442–1447. doi: 10.1084/jem.152.5.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plaut AG, Gilbert JV, Artenstein MS, Capra JD. Neisseria gonorrhoeae and neisseria meningitidis: extracellular enzyme cleaves human immunoglobulin A. Science. 1975;190:1103–1105. doi: 10.1126/science.810892. [DOI] [PubMed] [Google Scholar]
- 16.Plaut AG, Bachovchin WW. IgA-specific prolyl endopeptidases: serine type. Methods Enzymol. 1994;244:137–151. doi: 10.1016/0076-6879(94)44012-3. [DOI] [PubMed] [Google Scholar]
- 17.Batten MR, Senior BW, Kilian M, Woof JM. Amino acid sequence requirements in the hinge of human immunoglobulin A1 (IgA1) for cleavage by streptococcal IgA1 proteases. Infect Immun. 2003;71:1462–1469. doi: 10.1128/IAI.71.3.1462-1469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilian M, Mestecky J, Schrohenloher RE. Pathogenic species of the genus Haemophilus and Streptococcus pneumoniae produce immunoglobulin A1 protease. Infect Immun. 1979;26:143–149. doi: 10.1128/iai.26.1.143-149.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulks MH, Kornfeld SJ, Frangione B, Plaut AG. Relationship between the specificity of IgA proteases and serotypes in Haemophilus influenzae. J Infect Dis. 1982;146:266–274. doi: 10.1093/infdis/146.2.266. [DOI] [PubMed] [Google Scholar]
- 20.Mansa B, Kilian M. Retained Antigen-Binding Activity of Fab-Alpha Fragments of Human Monoclonal Immunoglobulin Al (Igal) Cleaved by Igal Protease. Infection and Immunity. 1986;52:171–174. doi: 10.1128/iai.52.1.171-174.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mistry D, Stockley RA. Iga1 Protease. International Journal of Biochemistry & Cell Biology. 2006;38:1244–1248. doi: 10.1016/j.biocel.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitovski S, Dunkin KT, Howard AJ, Sayers JR. Nontypeable Haemophilus influenzae in carriage and disease: a difference in IgA1 protease activity levels. JAMA. 2002;287:1699–1705. doi: 10.1001/jama.287.13.1699. [DOI] [PubMed] [Google Scholar]
- 23.Wood SG, Burton J. Synthetic Peptide-Substrates for the Immunoglobulin-A1 Protease from Neisseria-Gonorrhoeae (Type-2) Infection and Immunity. 1991;59:1818–1822. doi: 10.1128/iai.59.5.1818-1822.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulks MH, Shoberg RJ. Bacterial Immunoglobulin a(1) Proteases. Bacterial Pathogenesis, Pt A. 1994;235:543–554. doi: 10.1016/0076-6879(94)35169-4. [DOI] [PubMed] [Google Scholar]
- 25.Bachovchin WW, Plaut AG, Flentke GR, Lynch M, Kettner CA. Inhibition of IgA1 proteinases from Neisseria gonorrhoeae and Hemophilus influenzae by peptide prolyl boronic acids. J Biol Chem. 1990;265:3738–3743. [PubMed] [Google Scholar]
- 26.Ayala P, Lin L, Hopper S, Fukuda M, So M. Infection of epithelial cells by pathogenic neisseriae reduces the levels of multiple lysosomal constituents. Infect Immun. 1998;66:5001–5007. doi: 10.1128/iai.66.10.5001-5007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beck SC, Meyer TF. IgA1 protease from Neisseria gonorrhoeae inhibits TNFalpha-mediated apoptosis of human monocytic cells. FEBS Lett. 2000;472:287–292. doi: 10.1016/s0014-5793(00)01478-2. [DOI] [PubMed] [Google Scholar]
- 28.Hauck CR, Meyer TF. The lysosomal/phagosomal membrane protein h-lamp-1 is a target of the IgA1 protease of Neisseria gonorrhoeae. FEBS Lett. 1997;405:86–90. doi: 10.1016/s0014-5793(97)00163-4. [DOI] [PubMed] [Google Scholar]
- 29.Lin L, Ayala P, Larson J, Mulks M, Fukuda M, Carlsson SR, Enns C, So M. The Neisseria type 2 IgA1 protease cleaves LAMP1 and promotes survival of bacteria within epithelial cells. Mol Microbiol. 1997;24:1083–1094. doi: 10.1046/j.1365-2958.1997.4191776.x. [DOI] [PubMed] [Google Scholar]
- 30.Plaut AG, Qiu JZ, St Geme JW. Human lactoferrin proteolytic activity: analysis of the cleaved region in the IgA protease of Haemophilus influenzae. Vaccine. 2000;19:S148–S152. doi: 10.1016/s0264-410x(00)00296-6. [DOI] [PubMed] [Google Scholar]
- 31.Qiu J, Hendrixson DR, Baker EN, Murphy TF, St Geme JW, 3rd, Plaut AG. Human milk lactoferrin inactivates two putative colonization factors expressed by Haemophilus influenzae. Proc Natl Acad Sci U S A. 1998;95:12641–12646. doi: 10.1073/pnas.95.21.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamm ME, Emancipator SN, Robinson JK, Yamashita M, Fujioka H, Qiu JZ, Plaut AG. Microbial IgA protease removes IgA immune complexes from mouse glomeruli In Vivo: Potential therapy for IgA nephropathy. American Journal of Pathology. 2008;172:31–36. doi: 10.2353/ajpath.2008.070131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JRH. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. Journal of Biological Chemistry. 2005;280:17339–17345. doi: 10.1074/jbc.M412885200. [DOI] [PubMed] [Google Scholar]
- 34.Dautin N, Bernstein HD. Protein secretion in gram-negative bacteria via the autotransporter pathway. Annual Review of Microbiology. 2007;61:89–112. doi: 10.1146/annurev.micro.61.080706.093233. [DOI] [PubMed] [Google Scholar]
- 35.Yoder MD, Keen NT, Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;260:1503–1507. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]
- 36.Veiga E, de Lorenzo V, Fernandez LA. Probing secretion and translocation of a beta-autotransporter using a reporter single-chain Fv as a cognate passenger domain. Molecular Microbiology. 1999;33:1232–1243. doi: 10.1046/j.1365-2958.1999.01571.x. [DOI] [PubMed] [Google Scholar]
- 37.Jose J, Kramer J, Klauser T, Pohlner J, Meyer TF. Absence of periplasmic DsbA oxidoreductase facilitates export of cysteine-containing passenger proteins to the Escherichia coli cell surface via the Iga(beta) autotransporter pathway. Gene. 1996;178:107–110. doi: 10.1016/0378-1119(96)00343-5. [DOI] [PubMed] [Google Scholar]
- 38.Perona JJ, Craik CS. Structural Basis of Substrate-Specificity in the Serine Proteases. Protein Science. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. Journal of Biological Chemistry. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 40.Tyndall JDA, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites. Chemical Reviews. 2005;105:973–999. doi: 10.1021/cr040669e. [DOI] [PubMed] [Google Scholar]
- 41.Chintalacharuvu KR, Chuang PD, Dragoman A, Fernandez CZ, Qiu JZ, Plaut AG, Trinh KR, Gala FA, Morrison SL. Cleavage of the human immunoglobulin A1 (IgA1) hinge region by IgA1 proteases requires structures in the Fc region of IgA. Infection and Immunity. 2003;71:2563–2570. doi: 10.1128/IAI.71.5.2563-2570.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human Fc alpha RI and its complex with IgA1-Fc. Nature. 2003;423:614–620. doi: 10.1038/nature01685. [DOI] [PubMed] [Google Scholar]
- 43.Senior BW, Woof JM. Sites in the CH3 domain of human IgA1 that influence sensitivity to bacterial IgA1 proteases. J Immunol. 2006;177:3913–3919. doi: 10.4049/jimmunol.177.6.3913. [DOI] [PubMed] [Google Scholar]
- 44.Senior BW, Woof JM. The influences of hinge length and composition on the susceptibility of human IgA to cleavage by diverse bacterial IgAl proteases. Journal of Immunology. 2005;174:7792–7799. doi: 10.4049/jimmunol.174.12.7792. [DOI] [PubMed] [Google Scholar]
- 45.Boehm MK, Woof JM, Kerr MA, Perkins SJ. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: A study by X-ray and neutron solution scattering and homology modelling. Journal of Molecular Biology. 1999;286:1421–1447. doi: 10.1006/jmbi.1998.2556. [DOI] [PubMed] [Google Scholar]
- 46.Perkins S, Bonner A. Structure determinations of human and chimaeric antibodies by solution scattering and constrained molecular modelling. Biochemical Society Transactions. 2008;36:37–42. doi: 10.1042/BST0360037. [DOI] [PubMed] [Google Scholar]
- 47.Senior BW, Woof JM. Effect of mutations in the human immunoglobulin A1 (IgA1) hinge on its susceptibility to cleavage by diverse bacterial IgAl proteases. Infection and Immunity. 2005;73:1515–1522. doi: 10.1128/IAI.73.3.1515-1522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beach H, Cole R, Gill ML, Loria JP. Conservation of mus-ms enzyme motions in the apo- and substrate-mimicked state. J Am Chem Soc. 2005;127:9167–9176. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- 49.Kovrigin EL, Loria JP. Enzyme dynamics along the reaction coordinate: critical role of a conserved residue. Biochemistry. 2006;45:2636–2647. doi: 10.1021/bi0525066. [DOI] [PubMed] [Google Scholar]
- 50.Gasteiger E, Hoohland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 51.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 52.Holyoak T, Fenn TD, Wilson MA, Moulin AG, Ringe D, Petsko GA. Malonate: a versatile cryoprotectant and stabilizing solution for salt-grown macromolecular crystals. Acta Crystallogr D Biol Crystallogr. 2003;59:2356–2358. doi: 10.1107/s0907444903021784. [DOI] [PubMed] [Google Scholar]
- 53.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 54.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. Journal of Applied Crystallography. 2008;41:641–643. [Google Scholar]
- 55.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen SX, Ben Jelloul M, Long F, Vagin A, Knipscheer P, Lebbink J, Sixma TK, Lamzin VS, Murshudov GN, Perrakis A. ARP/wARP and molecular replacement: the next generation. Acta Crystallographica Section D-Biological Crystallography. 2008;64:49–60. doi: 10.1107/S0907444907047580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen SX, Morris RJ, Fernandez FJ, Ben Jelloul M, Kakaris M, Parthasarathy V, Lamzin VS, Kleywegt GJ, Perrakis A. Towards complete validated models in the next generation of ARP/wARP. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2222–2229. doi: 10.1107/S0907444904027556. [DOI] [PubMed] [Google Scholar]
- 58.Joosten K, Cohen SX, Emsley P, Mooij W, Lamzin VS, Perrakis A. A knowledge-driven approach for crystallographic protein model completion. Acta Crystallographica Section D-Biological Crystallography. 2008;64:416–424. doi: 10.1107/S0907444908001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris RJ, Zwart PH, Cohen S, Fernandez FJ, Kakaris M, Kirillova O, Vonrhein C, Perrakis A, Lamzin VS. Breaking good resolutions with ARP/wARP. Journal of Synchrotron Radiation. 2004;11:56–59. doi: 10.1107/s090904950302394x. [DOI] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 61.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 62.Bailey S. The Ccp4 Suite - Programs for Protein Crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Cryst D. 2005;61:465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 64.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallographica Section D-Biological Crystallography. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 65.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. Journal of Applied Crystallography. 2006;39:109–111. [Google Scholar]
- 66.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 67.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Research. 2004;32:W96–W99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 69.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Ten Eyck LF. Protein docking using continuum electrostatics and geometric fit. Protein Engineering. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 70.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGillivary G, Smoot LM, Actis LA. Characterization of the IgA1 protease from the Brazilian purpuric fever strain F3031 of Haemophilus influenzae biogroup aegyptius. FEMS Microbiol Lett. 2005;250:229–236. doi: 10.1016/j.femsle.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 72.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 73.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 75.Schechter I, Berger A. On Active Site of Proteases. 3. Mapping Active Site of Papain - Specific Peptide Inhibitors of Papain. Biochemical and Biophysical Research Communications. 1968;32:898–902. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 76.Torano A, Putnam FW. Complete amino acid sequence of the alpha 2 heavy chain of a human IgA2 immunoglobulin of the A2m (2) allotype. Proc Natl Acad Sci U S A. 1978;75:966–969. doi: 10.1073/pnas.75.2.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bode W, Wei AZ, Huber R, Meyer E, Travis J, Neumann S. X-Ray Crystal-Structure of the Complex of Human-Leukocyte Elastase (Pmn Elastase) and the 3rd Domain of the Turkey Ovomucoid Inhibitor. Embo Journal. 1986;5:2453–2458. doi: 10.1002/j.1460-2075.1986.tb04521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brunger AT. Free R-Value - a Novel Statistical Quantity for Assessing the Accuracy of Crystal-Structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.