

Abstract
Background
The extent to which mitochondrial DNA (mtDNA) content (also termed mtDNA copy number) in normal human cells is influenced by genetic factors has yet to be established. In addition, whether inherited variation of mtDNA content in normal cells contributes to cancer susceptibility remains unclear. Renal cell carcinoma accounts for 85% of all renal cancers. No studies have investigated the association between mtDNA content and the risk of renal cell carcinoma.
Methods
We first used a classic twin study design to estimate the genetic contribution to the determination of mtDNA content. mtDNA content was measured by quantitative real-time polymerase chain reaction in peripheral blood lymphocytes from 250 monozygotic twins, 92 dizygotic twins, and 33 siblings (ie, individual siblings of a pair of twins). We used biometric genetic modeling to estimate heritability of mtDNA content. We then used a case–control study with 260 case patients with renal cell carcinoma and 281 matched control subjects and multivariable logistic regression analysis to examine the association between mtDNA content in peripheral blood lymphocytes and the risk of renal cell carcinoma. All statistical tests were two-sided.
Results
The heritability (ie, proportion of phenotypic variation in a population that is attributable to genetic variation among individuals) of mtDNA content was 65% (95% confidence interval [CI] = 50% to 72%; P < .001). Case patients with renal cell carcinoma had a statistically significantly lower mtDNA content (1.18 copies) than control subjects (1.29 copies) (difference = 0.11, 95% CI = 0.03 to 0.17; P = .006). Low mtDNA content (ie, less than the median in control subjects) was associated with a statistically significantly increased risk of renal cell carcinoma, compared with high content (odds ratio = 1.53, 95% CI = 1.07 to 2.19). In a trend analysis, a statistically significant dose–response relationship was detected between lower mtDNA content and increasing risk of renal cell carcinoma (P for trend <.001).
Conclusions
mtDNA content appears to have high heritability. Low mtDNA content appears to be associated with increased risk of renal cell carcinoma.
CONTEXT AND CAVEATS
Prior knowledge
The extent to which the mitochondrial DNA (mtDNA) content of normal human cells is influenced by genetic factors is yet to be established, and whether inherited variation of mtDNA content in normal cells contributes to cancer susceptibility remains unclear.
Study design
A classic twin study design was used to estimate the genetic contribution of mtDNA content among individuals. A case–control study with 260 case patients with renal cell carcinoma and 281 control subjects used to examine the association between mtDNA content in peripheral blood lymphocytes and the risk of renal cell carcinoma.
Contribution
mtDNA content appears to have high heritability (ie, proportion of phenotypic variation in a population that is attributable to genetic variation among individuals). Low mtDNA content appears to be associated with increased risk of renal cell carcinoma.
Implications
Additional research into the association between mtDNA content and the risk of renal cell carcinoma and other cancers is warranted.
Limitations
The moderate sample size of the case–control study limits its statistical power. The case–control study was restricted to white individuals, which limits the generalizability of its results.
From the Editors
Human mitochondrial DNA (mtDNA) is a maternally inherited genome consisting of a 16 569–base-pair circular double-stranded DNA molecule that encodes 13 polypeptides of the respiratory chain, 22 transfer RNAs, and 2 ribosomal RNAs (1). Each mitochondrion contains 2–10 mtDNA molecules. The number of mtDNA copies in a cell ranges from several hundred to more than 10 000 copies, depending on the cell type. For example, the mtDNA copy number per cell is 223–854 in peripheral blood mononuclear cells (2), 323–1111 in human progressive spermatozoa (3), 1075–2794 in muscle cells (4), 1200–10 800 in neurons (5), and up to 25 000 in liver cells (6). Cao et al. (7) reported that the mtDNA copy number in primordial germ cells of mice fits a normal distribution at each developmental stage. However, Mizumachi et al. (8) found that mtDNA content follows a higher skewed distribution in prostate cancer cells than in normal cells. Moreover, two previous studies (9,10) reported a non-normal copy number distribution of mtDNAs extracted from human peripheral leukocytes or whole blood (only leukocytes in whole blood contain mtDNA). However, because different assays were used to quantify mtDNA copy number, the absolute values reported in those studies are not comparable. The mtDNA content normally has a steady-state level in each specific tissue that is related to the energy demand of the host cells (11). Interindividual variations of mtDNA copy number in cells exist in the general population (12).
The factors that regulate mtDNA homeostasis are not fully understood. It is likely that both environmental and genetic factors play important roles. Over the past decade, several nucleus-encoded mtDNA-regulating factors have been identified, including DNA polymerase γ subunits A and B, mitochondrial RNA polymerase, mitochondrial transcription factor A, and single-stranded DNA-binding protein (13). The classic twin study, by comparing similarities between monozygotic twins (who are genetically identical) and dizygotic twins (who share half their genes on average), is an ideal model to determine the relative contribution of genetic and environmental factors to interindividual variability in a given phenotypic biomarker. This model has been previously used to investigate the genetic heritability of recognized phenotypic markers for cancer and other complex diseases (14,15). Therefore, one of the primary objectives of our study was to apply the classic twin study design to determine the heritability of mtDNA content in peripheral blood lymphocytes.
In recent years, a decrease in mtDNA copy number (termed mtDNA depletion) has been reported in many types of cancers, including ovarian (16), gastric (17), hepatocellular (18), and breast (19) cancers, indicating that the decreased mtDNA copy number may contribute to tumorigenesis. Renal cell carcinoma is the most common adult malignant disease of the kidney and has been increasing worldwide for the past several decades (20). A variety of genetic defects may be involved in the initiation and progression of renal cell carcinoma, including deletions and translocations of chromosome 3p and alteration of mtDNA (21,22). In a previous small study, Meierhofer et al. (23) reported a statistically significant reduction of mitochondrial enzyme activities and mtDNA copy number in 34 of 37 renal cell carcinoma tissues as compared with adjacent noncancerous tissues. However, to our knowledge, no studies have evaluated the association between constitutive mtDNA content in peripheral blood lymphocytes and risk of renal cell carcinoma. Therefore, the second objective of our study was to provide proof-of-principle evidence within the context of an ongoing case–control study of renal cell carcinoma for the hypothesis that individuals with lower mtDNA content are at increased risk of renal cell carcinoma.
Subjects and Methods
Study Population
We carried out this study with available blood samples collected in a twin study. This resource has been used to evaluate the genetic component of mutagen sensitivity, a phenotypic cancer susceptibility marker (24). The procedures for twin recruitment have been described elsewhere (25,26). Briefly, potential twins were identified from the Stanford Research Institute (SRI) Northern California Twin Registry and recruited by the Center for Health Sciences, SRI International. Self-reported zygosity was further verified by comparing members of each twin pair on 10–12 highly polymorphic microsatellite markers, as described previously (27). Blood samples were collected at the Clinical Research Center, University of California, San Francisco. For each participant, a 20-mL blood sample was delivered to the Department of Epidemiology, The University of Texas M. D. Anderson Cancer Center, within 24 hours of blood collection for laboratory assay. A total of 375 individuals (including 250 monozygotic twins, 92 dizygotic twins, and 33 siblings) were enrolled in this heritability analysis. The siblings were individual siblings related to the twins. Twins and siblings were free of renal cell carcinoma or any other form of cancer. The twin study was approved by the Institutional Review Boards of SRI International; The University of California, San Francisco; and M. D. Anderson Cancer Center. All study participants provided written informed consent.
For the case–control study of renal cell carcinoma, patients with histologically confirmed renal cell carcinoma were consecutively recruited at the Departments of Urology and Genitourinary Medical Oncology of The University of Texas M. D. Anderson Cancer Center by use of daily reviews of computerized appointment schedules. All case patients were diagnosed within 1 year of enrollment and had not received previous chemotherapy or radiotherapy before enrollment. There were no age, sex, or disease-stage restrictions on recruitment. Healthy control subjects without a history of cancer except nonmelanoma skin cancer were identified and recruited through random digit dialing methods. In this method, randomly selected household phone numbers were generated to select potential control volunteers in the same residency areas as the case patients according to telephone directory listings. Control subjects were required to have lived for at least 1 year in the same county or socioeconomically matched surrounding counties in which the case patient resided. The control subjects were frequency matched to the case patients by age (±5 years), sex, ethnicity, and county of residence. After written informed consent was obtained, all study participants completed a 45-minute in-person interview that was administered by M. D. Anderson Cancer Center staff interviewers. The interview elicited information on demographics, smoking history, family history of cancer, occupational history and exposures, and medical history. At the conclusion of the interview, a 40-mL blood sample was drawn into coded heparinized tubes and delivered to the laboratory for analysis. This analysis was restricted to white subjects because of small sample sizes of subjects of other ethnicities. A total of 260 case patients and 281 healthy control subjects were included in the case–control study.
Determination of mtDNA Content by Real-Time Quantitative Polymerase Chain Reaction
Genomic DNA was extracted from whole blood for all the samples by use of QIAamp DNA Mini kits (Qiagen, Valencia, CA). Relative mtDNA copy number was measured by a quantitative real-time polymerase chain reaction (PCR)-based method as previously described, with some modifications (28,29). In brief, two pairs of primers were designed and used in the two steps of relative quantification for mtDNA content. One primer pair was used for the amplification of the MT-ND1 gene in mtDNA. The primer sequences were as follows: forward primer (ND1-F), 5′-CCCTAAAACCCGCCACATCT-3′; reverse primer (ND1-R), 5′-GAGCGATGGTGAGAGCTAAGGT-3′. Another primer pair was used for the amplification of the single-copy nuclear gene human globulin (HGB). The primer sequences were as follows: forward primer (HGB-1), 5′-GTGCACCTGACTCCTGAGGAGA-3′; reverse primer (HGB-2), 5′-CCTTGATACCAACCTGCCCAG-3′. In the first step, the ratio of mtDNA copy number to HGB copy number was determined for each sample from standard curves. This ratio is proportional to the mtDNA copy number in each cell. The ratio for each sample was then normalized to a calibrator DNA in order to standardize between different runs. The calibrator DNA is a genomic DNA sample from a healthy control subject to be used for comparison of results of different independent assays. The PCR mixture in a total volume of 14 μL contained 1× SYBR Green Mastermix (Applied Biosystems; Foster City, CA), 215 nM ND1-R (or HGB-1) primer, 215 nM ND1-F (or HGB-2) primer, and 4 ng of genomic DNA. The thermal cycling conditions for the mtDNA (MT-ND1 gene) amplification were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute; and for the HGB amplification were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 56°C for 1 minute. All samples were assayed in duplicate on a 384-well plate with an Applied Biosystems 7900 Sequence Detection System. The PCRs for mtDNA and HGB were always performed on separate 384-well plates with the same samples in the same well positions to avoid possible position effect. A standard curve of a diluted reference DNA, one negative control, and one calibrator DNA were included in each run. For each standard curve, one reference DNA sample was serially diluted 1:2 to produce a seven-point standard curve between 0.3125 and 20 ng of DNA. The R2 for each standard curve was 0.99 or greater. Standard deviations for the cycle of threshold (Ct) value were accepted at 0.25. Otherwise, the test was repeated. To further assess intra-assay variation, we assayed nine blood DNA samples from healthy control subjects three times on the same day. To further evaluate interassay variation, we evaluated the same blood DNA samples from the nine control subjects on different days. All the laboratory personnel performing the experiments described above were blinded to the zygosity status of the twin pairs or the case–control status of the DNA samples.
Statistical Analysis
The distributions of mtDNA in both the twin study and the case–control study of renal cell carcinoma were unimodal but with moderate deviation from a normal distribution. We performed the analyses with raw data and with log-transformed data. Because the results from these analyses were similar, we chose, for simplicity, to present the results from the raw data only.
In the twin study, the differences in mtDNA content in different subgroups defined by age, sex, and smoking status were assessed by Student t test or analysis of variance. Generalized estimating equations were used to account for dependence among family members (30). Intraclass correlations were determined for monozygotic twins and dizygotic pairs (including dizygotic twins and twin–sibling pairs). In families with both monozygotic pairs and siblings, we randomly picked one member of the pair to form a twin–sibling pair. The heritability of mtDNA content was estimated by the methods of biometric genetic modeling as previously described (31,32). According to this model, the observed quantitative trait of an individual (ie, mtDNA content) was modeled as a function of both fixed and random components. The biometrical genetic model, in its simplest form, is used to partition variation of an observed phenotypic trait into three variance components: genetic, shared or common environment (ie, environmental influences shared by family members), and nonshared or unique environment (ie, environmental influences from a random environmental component that is not shared by family members). In this study, we were particularly interested in the genetic effects of mtDNA, so both unrestricted and restricted models in which genetic variance (σ2) was constrained to 0 were fitted. The null hypothesis was that there is no genetic contribution to mtDNA content. The 95% confidence intervals (CIs) of the heritability were obtained by applying a bootstrapping procedure with 1000 repeated samplings.
In the case–control analysis, the Pearson χ2 test was used to examine the differences in the distribution of case patients and control subjects in terms of sex, smoking status, hypertension history, and body mass index (<25, ≥25 and <30, and ≥30 kg/m2). Student t test was used to test for differences in age, pack-years of smoking, and mtDNA content as continuous variables. To assess the association between mtDNA content and the risk of renal cell carcinoma, data were dichotomized by the use of the median from the distribution of the mtDNA content in control subjects. To assess the dose–response trend, we applied a spline modeling procedure implemented in R software (Version 2.5) to identify potential cutoff points at which optimal thresholds to assess dose–response pattern were identified. A trend test was performed to test for a linear trend in the odds ratios (ORs) by use across these cutoff points. Unconditional multivariable logistic regression analysis was performed to estimate the odds ratio and 95% confidence interval, adjusting for age, sex, smoking status, hypertension history, and body mass index. All statistical analyses were performed with the Stata 8.0 statistical software package (Stata Corp., College Station, TX).
Smoking status and pack-years of smoking were defined as previously described (33). A never smoker was defined as a person who had never smoked or smoked fewer than 100 cigarettes in his or her lifetime. A former smoker was defined as a person who had stopped smoking at least 1 year before the diagnosis of cancer (for case patients) or 1 year before the interview (for control subjects). A current smoker was someone who continued smoking or who had stopped smoking less than 1 year before the diagnosis of cancer (case patients) or before the interview (control subjects). The number of pack-years was calculated as the average number of cigarettes smoked per day divided by 20 cigarettes and then multiplied by smoking years. All statistical tests were two-sided with a statistical significance level of .05.
Results
Reproducibility of mtDNA Content Assay
The quantitative real-time PCR technique was used to determine the mtDNA content of each sample. We assayed nine replicate samples to assess the reproducibility. To assess intra-assay variation, we assayed each of the nine blood DNA samples three times on the same day. The coefficient of variance for each sample ranged from 2.7% to 6.7%, with an average of 4.5%, suggesting high intra-assay reliability. To further evaluate interassay variation, we assayed the same blood DNA samples from the same nine subjects on two separate days. The coefficient of variation ranged from 0.2% to 7.6%, with an average of 5.5%, indicating high interassay reliability. However, we were unable to evaluate the intra-individual variability of mtDNA content through serial blood samplings because we did not collect serial samples.
Heritability of mtDNA Content in Peripheral Blood Lymphocytes
The twin study population of 375 participants included 284 (75.7%) white subjects, 12 (3.2%) African American subjects, 46 (12.3%) Hispanic subjects, 16 (4.3%) Asian subjects, and 17 (4.5%) individuals of other ethnicities. The mean age was 42.4 years (range = 19–80 years). There were 261 (69.6%) females and 114 (30.4%) males. The majority of subjects were never smokers (61.6%); the percentages of former and current smokers were 22.4% and 16%, respectively. The mean relative mtDNA content was 1.19 copies (95% CI = 1.16 to 1.22 copies), and the median was 1.12 copies (range = 0.57–2.80 copies). The mtDNA content was not associated with age, sex, or ethnicity (data not shown). However, ever smokers had a higher mtDNA content than never smokers (1.25 copies, 95% CI = 1.19 to 1.31 copies, vs 1.15 copies, 95% CI = 1.12 to 1.19 copies).
The correlation between monozygotic twins and the correlation between dizygote pairs was assessed by calculating intraclass correlation coefficients. Figure 1, A and B, shows the scatter plots of the mtDNA content in monozygotic and dizygote pairs (pooled dizygotic twin pairs and monozygotic twin–sibling pairs). The intraclass correlation coefficient of mtDNA content for monozygotic twins (r = 0.60, P < .001) was statistically significantly higher than that for the dizygote pairs (r = 0.33, P < .01), indicating a strong genetic effect on mtDNA level. We then used biometric genetic modeling to estimate the heritability of mtDNA content (Table 1). The null hypothesis of no genetic contribution was rejected because the restricted model (which assumed no genetic influence) fitted the data statistically significantly worse than the full model (P < .001), indicating that the genetic component contributed statistically significantly to the determination of mtDNA content. The model generated a heritability estimate of 65% (95% CI = 50% to 72%, P < .001).
Figure 1.
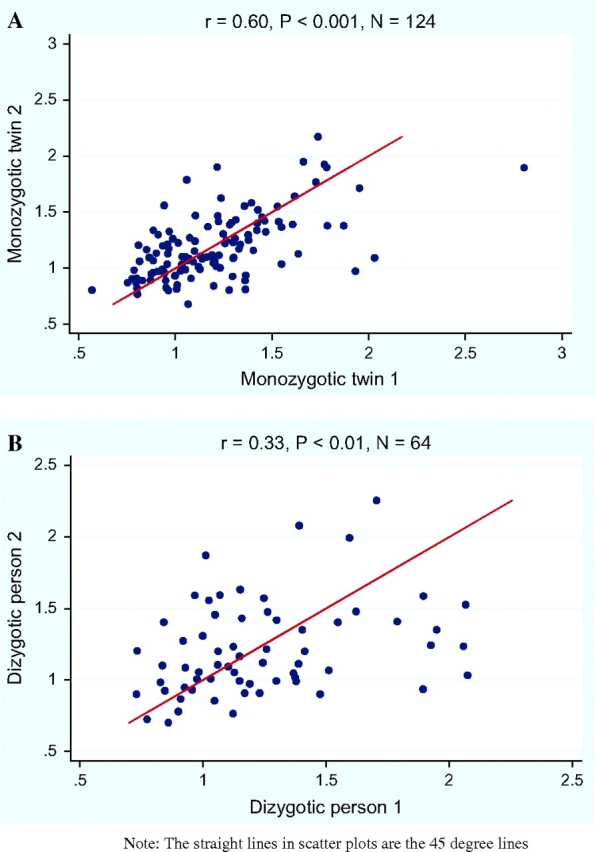
Intraclass correlation of mitochondrial DNA (mtDNA) content. A) Monozygotic twins. B) Dizygote pairs (pooled dizygotic twin pairs and monozygotic twin–sibling pairs). The first person within the dizygote pairs is defined as dizygotic person 1 and the second person within the pair is defined as dizygotic person 2. The straight lines in the scatter plots are the 45° line. All statistical tests were two-sided. The x- and y-axes represent mtDNA content for individuals 1 and 2, in both panels.
Table 1.
Contribution of genetic and environmental components to mitochondrial DNA content*
| Full model | Restricted model | |
| Log likelihood value | 281.895 | 274.855 |
| χ2 statistic | 12.775 | |
| df | 1 | |
| P value (two-sided) | <.001 | |
| Smoking coefficient† | 0.033 | 0.043 |
| Variances‡ | ||
| Genetic | 0.073 | Fixed |
| Shared environmental | 0.000 | 0.038 |
| Nonshared environmental | 0.038 | 0.056 |
| Heritability, % (95% CI) | 65 (50 to 72) |
The biometric genetic model was used to determine the contribution of genetic and environment components to mtDNA content. In the full model, variation of mtDNA content was partitioned into three variance components: genetic, shared or common environment, and nonshared or unique environment. In the restricted model, the variance due to genetic component was constrained to zero. The null hypothesis was that there is no genetic contribution to mtDNA content. CI = confidence interval; mtDNA = mitochondrial DNA.
Smoking coefficient represents the effects of smoking status, which is the covariate in the model.
The variance was partitioned into genetic, shared environment, and nonshared environment in the full model, whereas the genetic component was constrained to zero in the restricted model.
Risk Estimates for Renal Cell Carcinoma by mtDNA Content
The characteristics of the 260 case patients with renal cell carcinoma and 281 control subjects are summarized in Table 2. The case patients and control subjects were well matched on sex (P = .35) and age (59.2 vs 59.5 years; P = .73). There were no statistically significant differences between the case patients and the control subjects in terms of smoking status (P = .10) or pack-years of smoking (29.0 vs 31.1 pack-years; P = 0.55), although the percentage of ever smokers was higher among the control subjects than among the case patients. However, the case patients had a statistically significantly higher percentage of self-reported hypertension history (55% vs 40%; P < .001) and also a higher percentage of obese subjects (for body mass index ≥30 kg/m2; 38% vs 31%; P = .006) than control subjects, which is not surprising because both hypertension and obesity are established risk factors for renal cell carcinoma.
Table 2.
Distribution of selected characteristics of case patients with renal cell carcinoma and healthy control subjects*
| Variable | Case patients (n = 260) | Control subjects (n = 281) | P value† |
| Sex, No. (%) | |||
| Male | 172 (66) | 175 (62) | |
| Female | 88 (34) | 106 (38) | .348 |
| Smoking status, No. (%) | |||
| Never | 136 (52) | 126 (45) | |
| Former | 93 (36) | 107 (38) | |
| Current | 30 (12) | 48 (17) | .099 |
| Hypertension history, No. (%) | |||
| Yes | 144 (55) | 111 (40) | |
| No | 116 (45) | 170 (60) | <.001 |
| Body mass index, No. (%) | |||
| <25 kg/m2 | 52 (20) | 90 (32) | |
| ≥25 and <30 kg/m2 | 109 (42) | 104 (37) | |
| ≥30 kg/m2 | 99 (38) | 87 (31) | .006 |
| Age, mean (SD), y | 59.20 (10.57) | 59.52 (10.91) | .730 |
| Mean pack-years of smoking‡ (SD) | 28.95 (25.96) | 31.08 (31.93) | .547 |
| mtDNA content, copy number | |||
| Mean (SD) | 1.18 (0.43) | 1.29 (0.44) | .006 |
| Median (range) | 1.14 (0.44–4.67) | 1.22 (0.41–4.24) | .001 |
mtDNA = mitochondrial DNA.
Differences in the distribution of sex, smoking status, hypertension history, and body mass index were tested with the Pearson χ2 test. Student t test was used to test for differences in age, pack-years of smoking, and mtDNA content. All statistical tests were two-sided.
Pack-years of smoking was assessed for ever smokers only.
The relative mean mtDNA content was statistically significantly lower in case patients with renal cell carcinoma (1.18 copies) than control subjects (1.29 copies) (difference = 0.11 copies, 95% CI = 0.03 to 0.17 copies; P = .006). The median values among case patients and control subjects were 1.14 copies (range = 0.44–4.67 copies) and 1.22 copies (range = 0.41–4.24 copies) (Table 2), respectively. Men had a lower mtDNA content than women among both case patients (1.15 vs 1.25 copies; P = .07) and control subjects (1.23 vs 1.38 copies; P = .006). Never smokers had a statistically significantly lower mtDNA content than ever smokers among both case patients (1.13 vs 1.23 copies; P = .057) and control subjects (1.23 vs 1.35 copies; P = .027) (Table 3). The mtDNA content was not modified by age, history of hypertension, family history of cancer (cancer in first-degree relatives), or obesity.
Table 3.
mtDNA content by host characteristics of case patients with renal cell carcinoma and control subjects*
| Case patients |
Control subjects |
|||
| Variable | No. of patients | mtDNA content (95% CI) | No. of subjects | mtDNA content (95% CI) |
| Sex | ||||
| Male | 172 | 1.15 (1.09 to 1.20) | 175 | 1.23 (1.17 to 1.30) |
| Female | 88 | 1.25 (1.15 to 1.37) | 106 | 1.38 (1.29 to 1.46) |
| P value† | .070 | .006 | ||
| Age, y | ||||
| <60 | 124 | 1.19 (1.12 to 1.29) | 129 | 1.30 (1.22 to 1.38) |
| ≥60 | 136 | 1.17 (1.11 to 1.23) | 152 | 1.27 (1.20 to 1.34) |
| P value† | .693 | .535 | ||
| Hypertension history | ||||
| Yes | 144 | 1.17 (1.11 to 1.23) | 111 | 1.26 (1.19 to 1.32) |
| No | 116 | 1.19 (1.11 to 1.29) | 170 | 1.31 (1.23 to 1.38) |
| P value† | .702 | .353 | ||
| Family history of any cancer‡ | ||||
| Yes | 143 | 1.15 (1.07 to 1.23) | 147 | 1.30 (1.23 to 1.37) |
| No | 87 | 1.21 (1.13 to 1.29) | 100 | 1.27 (1.18 to 1.37) |
| P value† | .327 | .631 | ||
| Family history of RCC‡ | ||||
| Yes | 15 | 1.33 (1.09 to 1.58) | 4 | 1.32 (0.84 to 1.81) |
| No | 215 | 1.18 (1.12 to 1.24) | 243 | 1.29 (1.23 to 1.35) |
| P value† | .179 | .868 | ||
| Body mass index, kg/m2 | ||||
| <25 | 52 | 1.16 (1.09 to 1.58) | 90 | 1.25 (1.16 to 1.33) |
| ≥25 to <30 | 109 | 1.22 (1.13 to 1.31) | 104 | 1.35 (1.24 to 1.45) |
| ≥30 | 99 | 1.15 (1.09 to 1.23) | 87 | 1.25 (1.18 to 1.33) |
| P value† | .481 | .187 | ||
| Smoking status | ||||
| Never | 125 | 1.13 (1.07 to 1.21) | 155 | 1.23 (1.17 to 1.29) |
| Ever | 134 | 1.23 (1.15 to 1.31) | 126 | 1.35 (1.26 to 1.44) |
| P value† | .057 | .027 | ||
RCC = renal cell carcinoma; CI = confidence interval; mtDNA = mitochondrial DNA.
Analysis of variance was used to test the differences in body mass index. Student t test was used to test differences in other variables. All statistical tests were two-sided.
Family history data were not available for some study subjects.
After dichotomization at the 50th percentile value (or median) of mtDNA content among control subjects, individuals with low mtDNA content were at a statistically significantly increased risk for renal cell carcinoma (adjusted OR = 1.53, 95% CI = 1.07 to 2.19) (Table 4). Spline modeling identified two cutoff points (optimal thresholds to assess a dose–response trend): 0.814 and 1.216. Subjects were then categorized by use of these two cutoff points. Compared with individuals with the highest category of mtDNA content, individuals in the medium and lowest categories were at an increased risk of renal cell carcinoma (adjusted OR for the medium and lowest categories = 1.43, 95% CI = 0.98 to 2.07, and 4.15, 95% CI = 2.03 to 8.48, respectively), with a statistically significant dose–response trend (P for trend <.001) (Table 4).
Table 4.
Risk of renal cell carcinoma as estimated by mtDNA content*
| mtDNA copy number | Case patients, No. (%) | Control subjects, No. (%) | Adjusted OR† (CI) |
| By median‡ | |||
| ≥1.219 | 102 (39) | 138 (49) | 1.0 (reference) |
| <1.219 | 158 (61) | 143 (51) | 1.53 (1.07 to 2.19) |
| By spline modeling cutoff points | |||
| ≥1.216 | 102 (39) | 143 (51) | 1.0 (reference) |
| 0.814–1.215 | 123 (47) | 125 (44) | 1.43 (0.98 to 2.07) |
| <0.814 | 35 (14) | 13 (5) | 4.15 (2.03 to 8.48) |
| P for trend <.001 |
mtDNA = mitochondrial DNA; OR = odds ratio; CI = confidence interval.
Odds ratios were adjusted by age, sex, smoking status, hypertension history, and body mass index. The trend in odds ratios was tested by use of the test for linear trend. All statistical tests were two-sided.
Median is based on levels in control subjects.
Stratified analyses by smoking status were also performed (data not shown). Among ever smokers, individuals with low mtDNA content (less than the median content in control subjects) were at a nonstatistically significantly increased risk for renal cell carcinoma (adjusted OR = 1.46, 95% CI = 0.87 to 2.43). When ever smokers were categorized by cutoff points identified by the spline modeling, compared with the reference group with the highest mtDNA content, individuals in the medium and lowest categories were at increased risk (adjusted OR for the medium and lowest categories = 1.25, 95% CI = 0.74 to 2.11, and 6.26, 95% CI = 2.29 to 17.09, respectively), with a statistically significant dose–response trend (P for trend = .002). Among never smokers, when the median split was used to dichotomize the group, lower mtDNA content was associated with increased risk (OR = 1.67, 95% CI = 1.00 to 2.81). In dose–response analysis, compared with the group of nonsmokers with the highest mtDNA content, the groups with the medium and lowest categories had increased risks (adjusted OR for the medium and lowest categories = 1.73, 95% CI = 1.01 to 2.96, and 2.29, 95% CI = 0.82 to 6.37, respectively). The interaction between mtDNA content and smoking was, however, not statistically significant (P for interaction = .17).
Discussion
The three major findings of this study were as follows: 1) Using the classic twin study design, we demonstrated that genetic factors were statistically significantly associated with variation in mtDNA content in peripheral blood lymphocytes. 2) Using a case–control study, we found that low mtDNA content was associated with a statistically significantly increased renal cell carcinoma risk. These results indicate that mtDNA content may be useful to identify subsets of individuals at increased risk of renal cell carcinoma. 3) Results of both case–control and twin studies indicated that mtDNA content is associated with smoking.
Although a number of protein factors encoded by nuclear genes have been shown to be involved in the replication, transcription, and maintenance of mtDNA, the extent to which mtDNA content is influenced by genetic factors has not been clearly established. Curran et al. (34) in a family-based study among Mexican Americans showed that mtDNA content has a genetic component. However, such a study design cannot distinguish between shared environmental and/or genetic factors (24). A twin study is the most powerful tool for direct assessment of a genetic component (35), and we have now demonstrated high heritability of mtDNA content with such a classic twin study design.
The genetic loci and biologic mechanisms that regulate mtDNA content remain to be elucidated. Curran et al. (34) found evidence that two genomic regions (10q11 and a small region on mtDNA) may harbor genes influencing variations in mtDNA content. Furthermore, they identified a total of 829 genes that had a statistically significant correlation with mtDNA content by analyzing genome-wide quantitative transcriptional profiles. In addition, a p53-binding sequence has been identified in mtDNA, indicating that p53 might be involved in the regulation of mtDNA replication (36). Other factors such as Ras and p66shc have also been reported to contribute to the regulation of mtDNA content (37). Further studies are warranted to identify genes that are responsible for mtDNA content regulation.
Although we estimated that the strong association between renal cell carcinoma and mtDNA content evident in twins was due solely to genetic factors, some of the additive genetic variance could reflect stronger intrauterine association among monozygotic twins than among dizygotic twins. This possibility would have to be evaluated in a different design such as a half-sib study with different mothers.
In the case–control study of renal cell carcinoma, we found that lower mtDNA content was associated with a 1.56-fold increased risk of renal cell carcinoma. To the best of our knowledge, this is the first molecular epidemiological study to evaluate mtDNA content in lymphocytes as a susceptibility biomarker for cancer. Reduced mtDNA content (or mtDNA depletion) has been previously detected in tumor tissue samples and has been associated with tumor progression. For example, Yamada et al. (38) reported that mtDNA content was statistically significantly decreased in hepatocellular carcinoma as compared with the corresponding noncancerous liver tissues and that patients with low mtDNA content had shorter 5-year survival rates than those with higher mtDNA content. Wu et al. (17) found that mtDNA depletion existed in gastric cancer and was associated with poor differentiation of cancer cells. In addition, Meierhofer et al. (23) reported a decrease in mtDNA content in 34 (91%) of 37 renal cell carcinomas compared with control kidney tissue. Selvanayagam et al. (39) likewise noted a decrease of mtDNA content and mRNA coding for NADH dehydrogenase subunit 3 in eight (61%) of 13 renal cell carcinomas compared with adjacent normal tissue. This phenomenon was also observed in five of six renal carcinoma cell lines (ie, SKRC29, SKRC42, CAK11, CAK12, SW 839, and ACHN) (39). Thus, mtDNA depletion may be an important phenotype associated with neoplastic transformation of renal cells.
The mechanism underlying the role of mtDNA depletion in tumorigenesis remains under investigation. Warburg (40) initiated research on the alteration of mitochondrial respiratory function in cancer more than half a century ago. He hypothesized that “injury” to the respiratory machinery was a critical event in carcinogenesis. Subsequently, a number of reports showed that mutations and depletion of mtDNA were common events in various types of malignancies (19,41,42). Chandel et al. (43) demonstrated that mtDNA depletion made cells resistant to apoptosis. Murine C2C12 skeletal myoblasts with depleted mtDNA exhibit an invasive phenotype and the overexpression of several tumor-specific markers (34). Biswas et al. (44) reported that depletion of mtDNA activated nuclear factor-kappa B (NFκB) and/or Rel factors through inactivation of NFκB inhibitor β. It has been demonstrated that mtDNA depletion alters mitochondrial gene expression and causes deficiency in oxidative phosphorylation and enhanced production of reactive oxidative species in aerobic metabolism, resulting in disruption of cellular functions (42). However, further studies are warranted to fully elucidate the functional mechanism of mtDNA depletion.
Our case–control data indicated a non–statistically significantly higher percentage of smokers among control subjects than among case patients and a non–statistically significantly higher number of pack-years of smoking among control subjects. Although tobacco smoking is considered to be a risk factor for renal cell carcinoma, the data are still inconsistent. For example, a recent meta-analysis (45) with 24 studies examining smoking and renal cell carcinoma observed 12 non–statistically significant associations. Likewise, a systematic review (46) of 11 studies examining risk factors for renal cell cancer found an association between smoking and renal cell carcinoma among only seven of the studies. More studies on smoking intensity, duration, and duration since quitting are warranted to further elucidate the association between smoking and renal cell carcinoma.
In the current study, stratified analyses indicated that mtDNA profile may be modified by sex or smoking status. Specifically, women had statistically significantly higher mtDNA content than men among both case patients and control subjects. In the twin study, there was a similar trend (1.15 copies, 95% CI = 1.11 to 1.21 copies, vs 1.20 copies, 95% CI = 1.17 to 1.24 copies), although statistical significance was not reached (P = .169). The biologic plausibility of this association is not clear. In addition, our results also showed that ever smokers had higher mtDNA content than never smokers among both case patients with renal cell carcinoma and healthy control subjects, as well as in twin study participants. Consistent with our findings, a previous study (47) reported increased mtDNA content in response to cigarette smoking. This increase was thought to be a compensatory mechanism for oxidative damage to mtDNA and respiratory chain components caused by smoking.
Our study had several limitations. Although our data indicated that mtDNA content was associated with a statistically significant increased risk for renal cell carcinoma, the cause–effect relationship between mtDNA content and cancer is subject to the scrutiny of reverse causation. This is a limitation inherent in case–control study design. Future prospective epidemiological studies are needed to investigate the hypothesis that mtDNA depletion predisposes individuals to increased cancer risk. Another limitation of the current case–control study is the moderate sample size, which limits the statistical power to detect interactions between mtDNA content and environmental risk factors in renal cell carcinoma etiology. Future studies with larger sample size are warranted to further investigate the complex interplay between mtDNA content and other risk factors. Finally, our analysis was restricted to white subjects; data from other ethnic groups would be valuable for comparison.
In summary, our study provides evidence that decreased mtDNA content is statistically significantly associated with risk of renal cell carcinoma. Our study also demonstrates a strong genetic component for mtDNA content that warrants additional research into the utility of mtDNA content as a genetic marker for cancer susceptibility. These results also indicate that phenotypic screening for mtDNA content might be a useful screening test for renal cell carcinoma.
Funding
National Cancer Institute (CA085576 and CA098897 to X.W., DA11170 to G.E.S.).
Footnotes
We thank Mary R. McElroy, Ruth E. Krasnow, and Jill Rubin for their efforts with twin subject recruitment and data collection. The authors had full responsibility for the design of the study, the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
References
- 1.Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88(1):41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- 2.Gahan ME, Miller F, Lewin SR, et al. Quantification of mitochondrial DNA in peripheral blood mononuclear cells and subcutaneous fat using real-time polymerase chain reaction. J Clin Virol. 2001;22(3):241–247. doi: 10.1016/s1386-6532(01)00195-0. [DOI] [PubMed] [Google Scholar]
- 3.Diez-Sanchez C, Ruiz-Pesini E, Lapena AC, et al. Mitochondrial DNA content of human spermatozoa. Biol Reprod. 2003;68(1):180–185. doi: 10.1095/biolreprod.102.005140. [DOI] [PubMed] [Google Scholar]
- 4.Barthelemy C, Ogier de Baulny H, Diaz J, et al. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Ann Neurol. 2001;49(5):607–617. [PubMed] [Google Scholar]
- 5.Cavelier L, Jazin EE, Eriksson I, et al. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995;29(1):217–224. doi: 10.1006/geno.1995.1234. [DOI] [PubMed] [Google Scholar]
- 6.Berdanier CD, Everts HB. Mitochondrial DNA in aging and degenerative disease. Mutat Res. 2001;475(1–2):169–183. doi: 10.1016/s0027-5107(01)00068-9. [DOI] [PubMed] [Google Scholar]
- 7.Cao L, Shitara H, Horii T, et al. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet. 2007;39(3):386–390. doi: 10.1038/ng1970. [DOI] [PubMed] [Google Scholar]
- 8.Mizumachi T, Muskhelishvili L, Naito A, et al. Increased distributional variance of mitochondrial DNA content associated with prostate cancer cells as compared with normal prostate cells. Prostate. 2008;68(4):408–417. doi: 10.1002/pros.20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiao MM, Lin TK, Kuo FY, et al. Early stage of biliary atresia is associated with significant changes in 8-hydroxydeoxyguanosine and mitochondrial copy number. J Pediatr Gastroenterol Nutr. 2007;45(3):329–334. doi: 10.1097/MPG.0b013e3180cc2c0f. [DOI] [PubMed] [Google Scholar]
- 10.Gemma C, Sookoian S, Alvarinas J, et al. Mitochondrial DNA depletion in small- and large-for-gestational-age newborns. Obesity (Silver Spring) 2006;14(12):2193–2199. doi: 10.1038/oby.2006.257. [DOI] [PubMed] [Google Scholar]
- 11.Capps GJ, Samuels DC, Chinnery PF. A model of the nuclear control of mitochondrial DNA replication. J Theor Biol. 2003;221(4):565–583. doi: 10.1006/jtbi.2003.3207. [DOI] [PubMed] [Google Scholar]
- 12.Moraes CT. What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 2001;17(4):199–205. doi: 10.1016/s0168-9525(01)02238-7. [DOI] [PubMed] [Google Scholar]
- 13.Scarpulla RC. Nuclear control of respiratory chain expression in mammalian cells. J Bioenerg Biomembr. 1997;29(2):109–119. doi: 10.1023/a:1022681828846. [DOI] [PubMed] [Google Scholar]
- 14.Boyd NF, Dite GS, Stone J, et al. Heritability of mammographic density, a risk factor for breast cancer. N Engl J Med. 2002;347(12):886–894. doi: 10.1056/NEJMoa013390. [DOI] [PubMed] [Google Scholar]
- 15.Bataille V, Snieder H, MacGregor AJ, Sasieni P, Spector TD. Genetics of risk factors for melanoma: an adult twin study of nevi and freckles. J Natl Cancer Inst. 2000;92(6):457–463. doi: 10.1093/jnci/92.6.457. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Liu VW, Xue WC, Cheung AN, Ngan HY. Association of decreased mitochondrial DNA content with ovarian cancer progression. Br J Cancer. 2006;95(8):1087–1091. doi: 10.1038/sj.bjc.6603377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CW, Yin PH, Hung WY, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer. 2005;44(1):19–28. doi: 10.1002/gcc.20213. [DOI] [PubMed] [Google Scholar]
- 18.Lee HC, Li SH, Lin JC, Wu CC, Yeh DC, Wei YH. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat Res. 2004;547(1–2):71–78. doi: 10.1016/j.mrfmmm.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 19.Tseng LM, Yin PH, Chi CW, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer. 2006;45(7):629–638. doi: 10.1002/gcc.20326. [DOI] [PubMed] [Google Scholar]
- 20.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer Statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 21.Baldewijns MM, van Vlodrop IJ, Schouten LJ, Soetekouw PM, de Bruine AP, van Engeland M. Genetics and epigenetics of renal cell cancer. Biochim Biophys Acta. 2008;1785(2):133–155. doi: 10.1016/j.bbcan.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Simonnet H, Alazard N, Pfeiffer K, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23(5):759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- 23.Meierhofer D, Mayr JA, Foetschl U, et al. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis. 2004;25(6):1005–1010. doi: 10.1093/carcin/bgh104. [DOI] [PubMed] [Google Scholar]
- 24.Wu X, Spitz MR, Amos CI, et al. Mutagen sensitivity has high heritability: evidence from a twin study. Cancer Res. 2006;66(12):5993–5996. doi: 10.1158/0008-5472.CAN-06-1007. [DOI] [PubMed] [Google Scholar]
- 25.Swan GE, Benowitz NL, Jacob P, III, et al. Pharmacogenetics of nicotine metabolism in twins: methods and procedures. Twin Res. 2004;7(5):435–448. doi: 10.1375/1369052042335269. [DOI] [PubMed] [Google Scholar]
- 26.Lin J, Swan GE, Shields PG, et al. Mutagen sensitivity and genetic variants in nucleotide excision repair pathway: genotype-phenotype correlation. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2065–2071. doi: 10.1158/1055-9965.EPI-06-1041. [DOI] [PubMed] [Google Scholar]
- 27.Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44(3):388–396. [PMC free article] [PubMed] [Google Scholar]
- 28.Kaaman M, Sparks LM, van Harmelen V, et al. Strong association between mitochondrial DNA copy number and lipogenesis in human white adipose tissue. Diabetologia. 2007;50(12):2526–2533. doi: 10.1007/s00125-007-0818-6. [DOI] [PubMed] [Google Scholar]
- 29.Santos TA, El Shourbagy S, St John JC. Mitochondrial content reflects oocyte variability and fertilization outcome. Fertil Steril. 2006;85(3):584–591. doi: 10.1016/j.fertnstert.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Liang KY, Beaty TH. Statistical designs for familial aggregation. Stat Methods Med Res. 2000;9(6):543–562. doi: 10.1177/096228020000900603. [DOI] [PubMed] [Google Scholar]
- 31.Amos CI, Zhu DK, Boerwinkle E. Assessing genetic linkage and association with robust components of variance approaches. Ann Hum Genet. 1996;60(pt 2):143–160. doi: 10.1111/j.1469-1809.1996.tb01184.x. [DOI] [PubMed] [Google Scholar]
- 32.de Andrade M, Amos CI, Thiel TJ. Methods to estimate genetic components of variance for quantitative traits in family studies. Genet Epidemiol. 1999;17(1):64–76. doi: 10.1002/(SICI)1098-2272(1999)17:1<64::AID-GEPI5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 33.Xing J, Spitz MR, Lu C, et al. Deficient G2-M and S checkpoints are associated with increased lung cancer risk: a case-control analysis. Cancer Epidemiol Biomarkers Prev. 2007;16(7):1517–1522. doi: 10.1158/1055-9965.EPI-07-0111. [DOI] [PubMed] [Google Scholar]
- 34.Curran JE, Johnson MP, Dyer TD, et al. Genetic determinants of mitochondrial content. Hum Mol Genet. 2007;16(12):1504–1514. doi: 10.1093/hmg/ddm101. [DOI] [PubMed] [Google Scholar]
- 35.Boomsma D, Busjahn A, Peltonen L. Classical twin studies and beyond. Nat Rev Genet. 2002;3(11):872–882. doi: 10.1038/nrg932. [DOI] [PubMed] [Google Scholar]
- 36.Heyne K, Mannebach S, Wuertz E, Knaup KX, Mahyar-Roemer M, Roemer K. Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett. 2004;578(1–2):198–202. doi: 10.1016/j.febslet.2004.10.099. [DOI] [PubMed] [Google Scholar]
- 37.Trinei M, Berniakovich I, Pelicci PG, Giorgio M. Mitochondrial DNA copy number is regulated by cellular proliferation: a role for Ras and p66(Shc) Biochim Biophys Acta. 2006;1757(5–6):624–630. doi: 10.1016/j.bbabio.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 38.Yamada S, Nomoto S, Fujii T, et al. Correlation between copy number of mitochondrial DNA and clinico-pathologic parameters of hepatocellular carcinoma. Eur J Surg Oncol. 2006;32(3):303–307. doi: 10.1016/j.ejso.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Selvanayagam P, Rajaraman S. Detection of mitochondrial genome depletion by a novel cDNA in renal cell carcinoma. Lab Invest. 1996;74(3):592–599. [PubMed] [Google Scholar]
- 40.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 41.Yin PH, Lee HC, Chau GY, et al. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br J Cancer. 2004;90(12):2390–2396. doi: 10.1038/sj.bjc.6601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HC, Yin PH, Lin JC, et al. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci. 2005;1042:109–122. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- 43.Chandel NS, Schumacker PT. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett. 1999;454(3):173–176. doi: 10.1016/s0014-5793(99)00783-8. [DOI] [PubMed] [Google Scholar]
- 44.Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol. 2003;161(3):507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunt JD, van der Hel OL, McMillan GP, Boffetta P, Brennan P. Renal cell carcinoma in relation to cigarette smoking: meta-analysis of 24 studies. Int J Cancer. 2005;114(1):101–108. doi: 10.1002/ijc.20618. [DOI] [PubMed] [Google Scholar]
- 46.Dhote R, Pellicer-Coeuret M, Thiounn N, Debre B, Vidal-Trecan G. Risk factors for adult renal cell carcinoma: a systematic review and implications for prevention. BJU Int. 2000;86(1):20–27. doi: 10.1046/j.1464-410x.2000.00708.x. [DOI] [PubMed] [Google Scholar]
- 47.Masayesva BG, Mambo E, Taylor RJ, et al. Mitochondrial DNA content increase in response to cigarette smoking. Cancer Epidemiol Biomarkers Prev. 2006;15(1):19–24. doi: 10.1158/1055-9965.EPI-05-0210. [DOI] [PubMed] [Google Scholar]