

syn-1,2-Amino alcohols are prevalent motifs in a diverse range of important small molecules such as natural products, pharmaceuticals, and asymmetric catalysts. The majority of methods for selectively constructing 1,2-amino alcohols rely on functional group interconversions or C–C bond-forming reactions using preoxidized materials.1 Selective methods for directly installing nitrogen functionality into inert C–H bonds have the potential to streamline the synthesis of these important compounds by avoiding the functional group manipulations (FGMs) required for working with oxidized materials.2,3 Herein we present a novel route for accessing chiral syn-1,2-amino alcohols enabled by the discovery of a Pd-(II)/sulfoxide-catalyzed, diastereoselective allylic C–H amination reaction of chiral homoallylic N-tosyl carbamates (eq 1). To the best of our knowledge, this represents the first example of a general and stereoselective, catalytic allylic C–H amination reaction.4 We demonstrate that this reaction enables the streamlined synthesis of a diverse collection of syn-1,2-amino alcohols because of its broad scope and the versatility of its vinyl anti-oxazolidinone products.
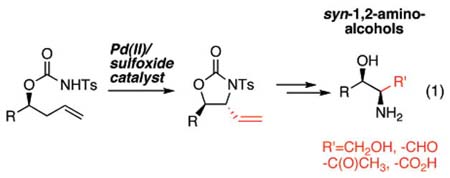
Palladium(II)-promoted addition of nitrogen nucleophiles to olefins (aminopalladation) is a well-established process that has led to many interesting catalytic reactions such as oxidative amination,5 aminoacetoxylation,6 aminohalogenation,7 and diamination.8 Alternatively, Pd(II)-promoted allylic C–H amination processes are rare.4b To avoid aminopalladation, the C–H cleavage step is generally done in the absence of a nitrogen nucleophile resulting in a two-step sequence that is stoichiometric in palladium.9
We previously discovered that the addition of bis-sulfoxide ligands to Pd(OAc)2 promotes allylic C–H cleavage of α-olefins versus oxypalladation in the presence of weak oxygen nucleophiles (i.e., carboxylic acids). This finding has resulted in several general allylic C–H esterification reactions of this important olefin class.10 We hypothesized that eq 1 could effect a similar catalytic allylic C–H amination reaction of α-olefins using weak nitrogen nucleophiles such as N-tosyl carbamates. We reasoned that the weak Lewis basicity of this nucleophile would prevent it from interfering with the electrophilic C–H cleavage step. Moreover, because the N–H proton of N-tosyl carbamate is relatively acidic,11 we anticipated that it may become activated toward functionalization by deprotonation with the acetate counterion of the Pd(II) catalyst. Importantly, acetate may be regenerated throughout the catalytic cycle during benzoquinone (BQ) mediated reoxidation of palladium.12 Herein we report the successful development of this allylic C–H amination reaction and the exploration of its scope and synthetic utility. Furthermore, we provide evidence in support of a mechanism that proceeds via a Pd(II)/sulfoxide-promoted allylic C–H cleavage to furnish a π-allylPd intermediate followed by acetate counterion-promoted functionalization with the tethered N-tosyl carbamate nucleophile.
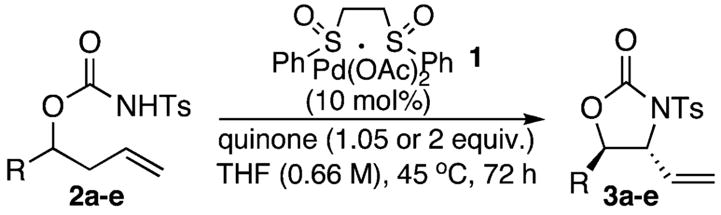
Our study began by examining the allylic C–H amination of 2-methylhex-5-en-3-yl N-tosylcarbamate 2a under standard allylic C–H oxidation conditions (Table 1).13 We found that 10 mol % phenyl bis-sulfoxide/Pd(OAc)2 1/BQ (2 equiv) formed the desired oxazolidinone product 3a in a modest overall yield (37%) with a good level of diastereoselectivity (7:1 anti/syn) (Table 1, entry 1). Significantly, in the absence of bis-sulfoxide ligand, only 3% of the allylic amination product was formed (entry 2). Increasing the substrate concentration (0.33 M → 0.66 M) improved the yield to 50% (entry 3). A screen of commercially available quinones revealed phenyl-benzoquinone (PhBQ) was superior to BQ (entry 4), and when added in slight excess (1.05 equiv) provides a 72% overall yield with only a modest decrease in diastereoselectivity (6:1 diastereomeric ratio (dr), entry 5). The addition of 5 mol % extra ligand provided a slightly higher yield (entry 6).
Table 1.
Allvlic C–H Amination Reaction Optimization and Scope
 | ||||
|---|---|---|---|---|
| entry | R | quinone (equiv.) | isolated yielda | drb(anti:syn) |
| 1 | i-Pr (2a) | BQ (2)c | 37% | 7:1 |
| 2 | i-Pr | BQ (2)c,d | 3%e | -- |
| 3 | i-Pr | BQ(2) | 50% | 7:1 |
| 4 | i-Pr | PhBQ (2) | 66% | 6:1 |
| 5 | i-Pr | PhBQ(1.05) | 72% | 6:1 |
| 6 |
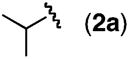
|
PhBQ (1.05)f | 76% | 6:1 |
| 7 |
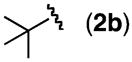
|
PhBQ(1.05)f | 8% | 18:1 |
| 8 |
|
PhBQ (1.05)f | 86% | 1.6:1 |
| 9 |
|
PhBQ(1.05)f | 84% | 7:1 |
| 10 |
|
PhBQ (1.05)f | 84% | 1.8:1e |
Average of two runs at 0.3 mmol.
Determined by GC analysis of the crude reaction mixture.
Reaction run at 0.33 M.
Reaction run using 10 mol % Pd(OAc)2 (no sulfoxide ligand).
Determined by NMR analysis of the crude reaction mixture.
Reaction run using 5 mol % additional bis-sulfoxide ligand.
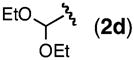
A variety of homoallylic N-tosyl carbamate substrates containing one stereogenic center were cyclized to test the generality of this method (Table 1, entries 6–10).14 One branching element adjacent to the N-tosyl carbamate provides the best balance of reactivity and diastereoselectivity (entries 6 and 9). Unbranched substrates provided good yields but poor levels of diastereoselectivity (R = n-Pr and TBDPSOCH2, entries 8 and 10), while a bulky quaternary alkyl substituent provided a poor yield but excellent diastereoselectivity (R = t-Bu, entry 7). The mildness of this method and its potential utility for generating densely functionalized compounds is illustrated by the allylic C–H amination of a substrate containing a proximal diethyl acetal moiety (entry 9). Significantly, diastereomerically pure oxazolidinone products can be obtained using standard column chromatography.
An important feature of this chemistry is that it enables the rapid synthesis of stereochemically defined oxazolidinones. Not only does this product comprise the skeleton of a very important class of antibiotics,15 but oxazolidinones can be further elaborated to a wide range of medicinally and biologically relevant 1,2-amino alcohols. For example, (+)-(2R,3S)-3-hydroxyleucine, an unnatural β-hydroxy-α-amino acid present in the antitumor agent lactacystin,16 is readily obtained from anti-oxazolidinone (−)-3a. Enantioenriched homoallylic N-tosyl carbamate (−)-2a, easily accessed via asymmetric allylboration, undergoes allylic C–H amination with good diastereoselectivity (6:1) to give a 61% isolated yield of the diastereomerically pure anti-(−)-3a with no erosion in enantiomeric purity (Scheme 1). The vinyl moiety of (−)-3a was further elaborated to carboxylic acid (+)-4 via ruthenium tetraoxide-catalyzed oxidative cleavage. Mild, reductive detosylation and oxazolidinone hydrolysis under acidic conditions afforded hydroxyleucine (+)-5 (Scheme 1).17
Scheme 1a.
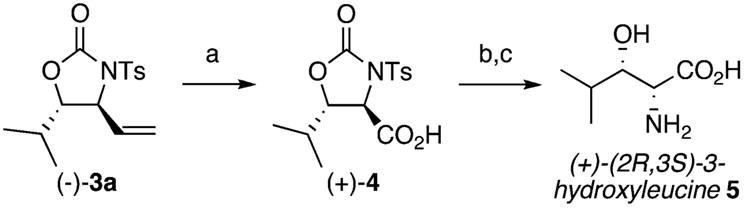
a Conditions: (a) RuCl3 (5 mol %), NaIO4 (5 equiv), CH3CN/CCl4/H2O (2:2:3), room temp (82%); (b) sodium naphthalene (8 equiv), DME, −78 °C; (c) 6 M HCl, 100 °C (63%, two steps).
In substrates containing multiple stereogenic centers, the diastereomeric outcome is controlled by the stereocenter that bears the N-tosyl carbamate. Both syn- and anti-diols (−)-6 and (+)-7 gave anti-oxazolidinone products [(−)-8 and (−)-9, respectively] with good to modest diastereoselectivities and in excellent yields (Scheme 2). Debenzylation of stereochemically and functionally dense oxazolidinone (−)-8 with BCl3 afforded a known diol intermediate (−)-10 used in the synthesis of 1,4-dideoxy-1,4-imino-L-xylitol, a pyrrolidine with promising activity as a glycosidase inhibitor (see Supporting Information).18
Scheme 2a.
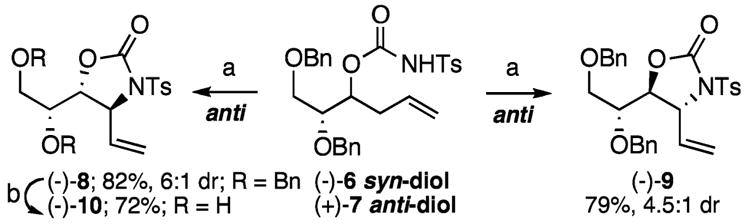
a Conditions: (a) 1 (10 mol %), 5 mol % additional bis-sulfoxide, PhBQ (1.05 equiv), THF (0.66 M), 45 °C, 72 h; (b) BCl3 (5 equiv), CH2Cl2, room temp, 72%.
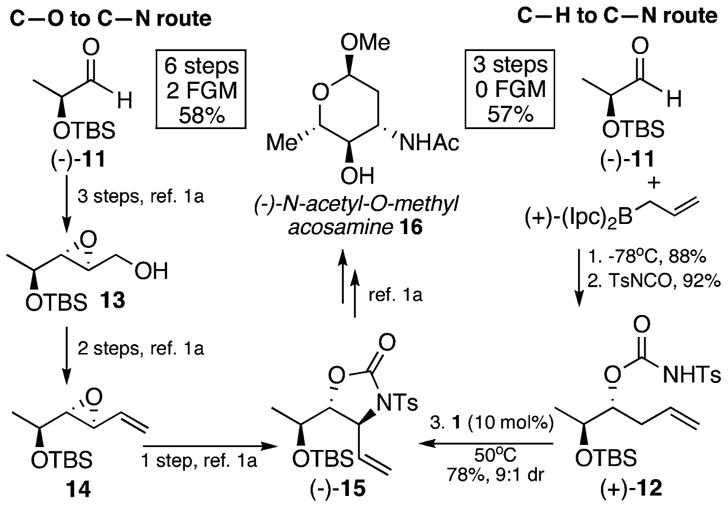
We hypothesized that routes based on selective C–H to C–N bond-forming reactions would streamline the syntheses of nitrogen containing compounds. To evaluate this, we compared the route enabled by allylic C–H amination relative to a previous state-of-the-art route based on allylic C–O substitution for the synthesis of oxazolidinone (−)-15 (Scheme 3). This heterocycle is a known intermediate toward L-acosamine derivative (−)-16, an aminosugar that is key to the medicinal properties of clinically used chemotherapeutic agent epirubicin.19 Starting from aldehyde (−)-11, enantiopure allylic C–H amination substrate (+)-12 was synthesized in only two steps with no FGMs. By way of contrast, beginning from the same starting material enantiopure allylic C–O substitution substrate 14 had required five steps with two FGMs.1a Hydrocarbon (+)-12 readily cyclized via allylic C–H amination with very good diastereoselectivity (9:1 dr) to give a 70% isolated yield of desired anti-(−)-15. Significantly, the C–H to C–N route to enantio- and diastereomerically pure (−)-15 proceeded in half the total number of steps, no FGMs, and comparable overall yield to the alternative C–O to C–N bond-forming route.1a
Scheme 3.
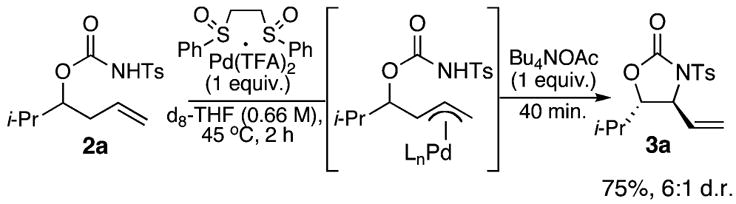
Stoichiometric studies were performed to test the hypothesis that allylic C–H amination proceeds via a C–H cleavage mechanism involving a π-allylPd intermediate. When a stoichiometric mixture of N-tosyl carbamate 2a and Pd(OAc)2/bis-sulfoxide 1 were heated for 2 h, 1H NMR analysis indicated approximately 24% conversion of starting material 2a to oxazolidinone product 3a.20 Significantly, a π-allylPd species was not observed. We next evaluated Pd(TFA)2/bis-sulfoxide in an analogous experiment (Scheme 4). Using this alternative palladium(II) source having a counterion that is a much weaker base, 1H NMR analysis at the same 2 h time point revealed predominant formation of a π-allylPd complex (ca. 61%) and no significant formation of product 3a (<5%).21 Importantly, the addition of an external acetate source to this π-allylPd intermediate led to the formation of 3a with a similar yield and identical diastereoselectivity to that observed under standard catalytic conditions (Scheme 4 and Table 1, entry 5).
Scheme 4.
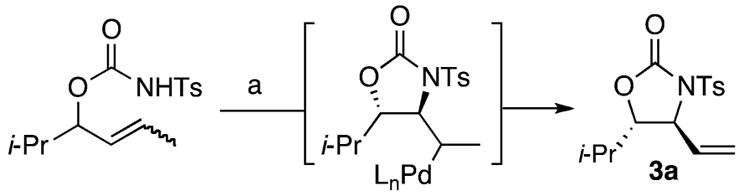
An alternative mechanism involving olefin isomerization followed by aminopalladation and β-hydride elimination was also considered. In that vein, internal (E)- and (Z)- olefins 17 and 18 were independently synthesized and subjected to the reaction conditions (Table 2). In contrast to α-olefin substrate 2a, both internal olefins afford oxazolidinone product 3a with poorer yields and higher diastereoselectivities that decrease over time (Table 2, entry 3 vs entries 1 and 2). The latter is most likely due to formation of Pd–H in the oxidative aminopalladation pathway that mediates olefin isomerization to 2a. Significantly, with α-olefin substrate 2a, olefin isomers 17 or 18 were not detected under standard reaction conditions,22 and no change in the diastereoselectivity of oxazolidone product 3a is observed during the course of the reaction (Table 2, entry 3).
Table 2.
Testing for a Possible Aminopalladation Mechanism
 | ||||
|---|---|---|---|---|
| entry | olefin isomer | isolated yield 3a | 5 h drb (anti/syn) | 72 h drb (anti/syn) |
| 1 | E isomer (17) | 20% | 9:1 | 8:1 |
| 2 | Z isomer (18) | 9% | 13:1 | 11:1 |
| 3 | α-olefin (2a) | 72% | 6:1 | 6:1 |
Reaction run using 1 (10 mol %), PhBQ (1.05 equiv), THF (0.66 M), 45°C, 72 h.
GC.
Collectively, this data strongly supports a mechanism for allylic C–H amination involving Pd(II)/bis-sulfoxide promoted allylic C–H cleavage to form a π-allylPd intermediate followed by acetate-mediated functionalization. The acetate most likely acts as a base to deprotonate the N-tosyl carbamate nucleophile.23 A key to this catalytic amination reactivity is the ability to use catalytic quantities of acetate base that can be regenerated via quinone-mediated Pd-(0) oxidation. The use of stoichiometric base significantly attenuates this reactivity, most likely by interfering with the electrophilic C–H cleavage step of the catalytic cycle.24
In summary, this method represents the first general and stereoselective Pd(II)-catalyzed allylic C–H amination reaction. The good levels of diastereoselectivity and functional group tolerance demonstrated for this method enable the synthesis of densely functionalized anti-oxazolidinone products that can be rapidly transformed into useful syn-1,2-amino alcohols. Mechanistic studies support a Pd(II)/bis-sulfoxide mediated C–H cleavage to form a π-allylPd intermediate followed by a Pd(II) counterion-mediated deprotonation of the nitrogen nucleophile to achieve functionalization. The latter aspect of this mechanism may provide a general approach for achieving activation of weak nucleophiles without attenuating the reactivity of an electrophilic metal catalyst.25 Current studies are focused on further exploration of the scope and mechanism of this reaction.
Acknowledgments
M.C.W. thanks the Henry Dreyfus Foundation, the A.P. Sloan Foundation, the University of Illinois (UIUC), Merck Research Laboratories, and the NIH/NIGMS (Grant GM076153) for financial support. We thank Johnson Matthey for a generous gift of Pd(OAc)2. We thank Mr. G. Rice for checking the experimental procedure in Table 1, entry 6.
Footnotes
Supporting Information Available: Detailed experimental procedures and full characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.C–O to C–N catalyzed by Pd(0): Trost BM, Sudhakar AR. J Am Chem Soc. 1987;109:3792.Trost BM, Patterson DE. J Org Chem. 1998;63:1339.Hayashi T, Yamamoto A, Ito Y. Tetrahedron Lett. 1988;29:99.Bystrom SE, Aslanian R, Backvall JE. Tetrahedron Lett. 1985;26:1749.Hayashi T, Kishi K, Yamamoto A, Ito Y. Tetrahedron Lett. 1990;31:1743.Evans PA, Robinson JE, Nelson JD. J Am Chem Soc. 1999;121:6761.Ohmura T, Hartwig JF. J Am Chem Soc. 2002;124:15164. doi: 10.1021/ja028614m.Olefin aminohydroxylation: Kolb HC, Sharpless KB. In: Transition Metals for Organic Synthesis. 2. Beller M, Bolm C, editors. Wiley-VCH Verlag; Weinheim, Germany: 2004. p. 309.Mannich: Trost BM, Terrell LR. J Am Chem Soc. 2003;125:338. doi: 10.1021/ja028782e.Matsunaga S, Yoshida T, Morimoto H, Kumagai N, Shibasaki M. J Am Chem Soc. 2004;126:8777. doi: 10.1021/ja0482435.Ramasastry SSV, Zhang H, Tanaka F, Barbas CF. J Am Chem Soc. 2007;129:288. doi: 10.1021/ja0677012.
- 2.Streamlining syntheses via late stage C–H oxidation see: Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223. doi: 10.1021/ol047800p.Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew Chem, Int Ed. 2006;45:8217. doi: 10.1002/anie.200603321.Hoffman RW. Synthesis. 2006;21:3531.Elegant examples of late stage C–H hydroxylation and amination see: Wender PA, Hilinski MK, Mayweg AVW. Org Lett. 2005;7:79. doi: 10.1021/ol047859w.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510. doi: 10.1021/ja0368305.
- 3.C-H amination via metal nitrenes: Espino CG, Du Bois J. Angew Chem, Int Ed. 2001;40:598. doi: 10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9.Kim M, Mulcahy JV, Espino CG, Du Bois J. Org Lett. 2006;8:1073. doi: 10.1021/ol052920y.Lebel H, Huard K, Lectard S. J Am Chem Soc. 2005;127:14198. doi: 10.1021/ja0552850.
- 4.For single examples of catalytic allylic C–H amination: (a) ref 3b. Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584. doi: 10.1021/jo952088i.
- 5.Hegedus LS, Allen GF, Waterman EL. J Am Chem Soc. 1976;98:2674.Ronn M, Backvall JE, Andersson PG. Tetrahedron Lett. 1995;36:7749. (c) ref. 4b. Overman LE, Remarchuk TP. J Am Chem Soc. 2002;124:12. doi: 10.1021/ja017198n.Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J Am Chem Soc. 2005;127:2868. doi: 10.1021/ja0433020.
- 6.(a) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (b) Lui G, Stahl SS. J Am Chem Soc. 2006;128:7179. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]
- 7.Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618. [Google Scholar]
- 8.(a) Backvall JE. Tetrahedron Lett. 1978;19:163. [Google Scholar]; (b) Streuff J, Hovelmann CH, Nieger M, Muniz K. J Am Chem Soc. 2005;127:14586. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (c) Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 9.(a) Akermark B, Akermark G, Hegedus LS, Zetterberg K. J Am Chem Soc. 1981;103:3037. [Google Scholar]; (b) Trost BM. Tetrahedron. 1977;33:2615. and references therein. [Google Scholar]
- 10.(a) Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; (b) Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Delcamp JH, White MC. J Am Chem Soc. 2006;128:15076. doi: 10.1021/ja066563d. [DOI] [PubMed] [Google Scholar]; (d) Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]
- 11.pKa (H2O) of EtOC(O)NHTs = 3.7. Taylor LD, Pluhar M, Rubin LE. J Polym Sci, Part B: Polym Phys. 1967;5:77.
- 12.2 equiv AcOH + BQ + Pd(0) → DHQ + Pd(OAc)2. Only ca. 1% of the allylic acetate product observed (Supporting Information, SI-23).
- 13.Other homoallylic carbamates –OC(O)NH2, OC(O)NHC(O)CCl3, and –OC(O)NH(p-OMe)Ph with less acidic N–H bonds showed poor reactivities. Addition of 10 mol% Hunig’s base to p-anisyl carbamate substrate gave no significant improvement (SI-9 and ref 24).
- 14.(a) 1,1-disubstituted and 1,2-disubstituted olefin substrates proceeded with poor conversions (30%) and yields (ca. 8–12% as mixtures of oxidative amination products). (b) Methyl 2-(tosylcarbamoyloxy)pent-4-enoate gave a complex mixture of products (ca. 30% anti-oxazolidinone identified) (SI-8,9).
- 15.Ross JE, Fritsche TR, Sader HS, Jones RN. Int J Antimicrob Agents. 2007;29:295. doi: 10.1016/j.ijantimicag.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Nagamitsu T, Sunazuka T, Tanaka H, Omura S, Sprengeler PA, Smith AB. J Am Chem Soc. 1996;118:3584. [Google Scholar]
- 17.For alternative syntheses see: Bonini C, Righi G. Tetrahedron. 2002;58:4981.
- 18.Lei A, Liu G, Lu X. J Org Chem. 2002;67:974. doi: 10.1021/jo0161429. [DOI] [PubMed] [Google Scholar]
- 19.Weymouth-Wilson AC. Nat Prod Rep. 1997;14:99. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 20.Unlike allylic C–H oxidation, Pd/sulfoxide-catalyzed allylic C–H amination does not require quinone for functionalization and therefore does not proceed via a serial ligand catalysis mechanism. See ref 10a.
- 21.1-Phenyl-3-buten-1-N-tosyl carbamate substrate gives 4-phenyl-1,3-butadiene. This result is consistent with β-N-tosyl carbamate elimination from a π-allylPd intermediate (SI-22).
- 22.1H NMR monitoring at 5, 24, 48, and 72 h (signal/noise > 500:1) (SI-24). The possibility that trace levels of 17 and/or 18 form under standard reaction conditions and contribute to formation of 3a cannot be rigorously excluded.
- 23.Previous work (ref 1a and ref 1b) in Pd(0)-mediated allylic substitution reactions has shown that the poor nucleophilic properties of carbamates require the use of their respective anions to achieve reactivity with π-allylpalladium intermediates.
- 24.Addition of 1 equiv of Bu4NOAc to the catalytic reaction conditions gave 3a in 20% yield.
- 25.For an alternative approach to this see: Lafrance M, Fagnou K. J Am Chem Soc. 2006;128:16496. doi: 10.1021/ja067144j.