

Direct oxidation of C–H bonds has the potential to emerge as a powerful approach for introducing oxygen and nitrogen functionality in the synthesis of complex molecules.[1] Functional-group manipulations (FGMs) that are required for carrying oxygenated functionalities throughout a synthetic route may be avoided by installing them directly into the hydrocarbon framework.[2] For a hydrocarbon oxidation strategy to reach its full potential, C–H-oxidation reactions with high levels of chemo-, regio- and stereoselectivity must be identified and developed. We recently described a DMSO-promoted, Pd(OAc)2-catalyzed allylic oxidation reaction that furnishes E allylic acetates from α-olefins with high regio- and stereoselectivity, and with outstanding tolerance of functional groups.[3] Herein, we describe advances in this methodology for the oxidation of C–H bonds which enabled the development of a new hydrocarbon oxidation strategy for the rapid assembly of polyol frameworks. This strategy has been validated in an enantioselective, de novo synthesis of differentially protected L-galactose from a commercial, achiral starting material in which all new oxygen functionality has been installed through oxidation reactions of C–H and C=C bonds.
A short hydrocarbon oxidation strategy for the synthesis of chiral polyols is presented in Scheme 1. Key to the efficiency of this strategy is the rapid access to chiral (E)-2-butene-1,4-diols such as 2, which may be directly elaborated to polyol structures through asymmetric dihydroxylation (AD). Compounds like 2 are particularly attractive building blocks because of their dense functionalization, dissonant relationship between the oxygen atoms,[4] and the ease with which they can be further elaborated through the established oxidation chemistry of olefins. Compounds similar to 2 have been routinely employed as intermediates to install a diverse range of structures in the synthesis of natural products; for example, five- and six-membered mono- and polycyclic ethers,[5] epoxy alcohols,[6] and, most extensively, contiguous polyol structures.[7] Notably, compounds analogous to 2 have been used as intermediates in several stereodivergent syntheses of the hexoses, an important class of polyols (Scheme 2).[8a,9] State-of-the-art syntheses of 2 based on Wittig-type olefinations[10] or cross-metathesis reactions[11] suffer from lengthy sequences, in part because of the difficulty in accessing enantioenriched α-hydroxyaldehyde and α-hydroxy olefin starting materials. Alternatively, 2 may be synthesized directly from protected chiral homoallylic alcohols such as 3 through the DMSO/PdII-promoted allylic oxidation by using the strategy presented in Scheme 1. [2,3a] Homoallylic alcohols 3 are readily accessible from the asymmetric allylation of aldehydes[12,13] or the regioselective vinylation of chiral epoxides (Scheme 1).[14]
Scheme 1.
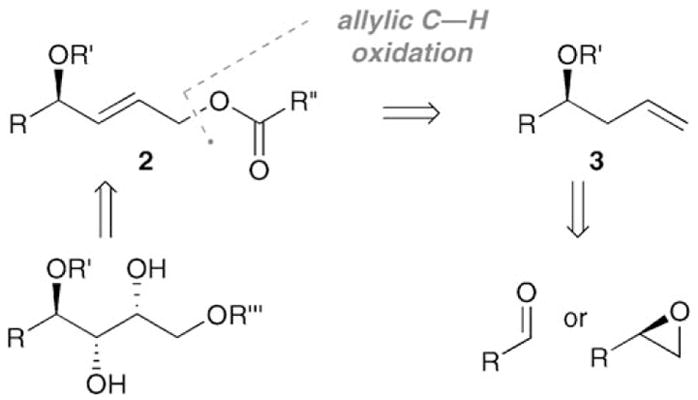
A hydrocarbon oxidation strategy for the streamlined construction of polyols.
Scheme 2.
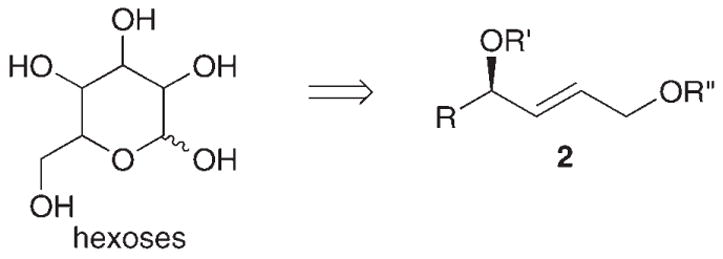
Chiral (E)-2-butene-1,4-diols as key intermediates in stereodivergent hexose syntheses.
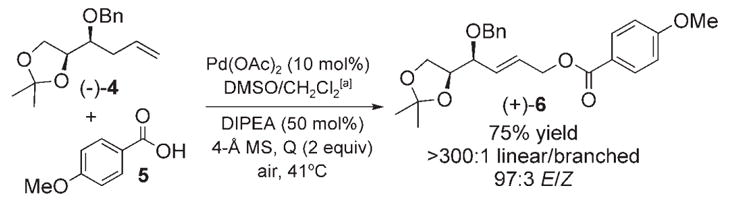
4-Methoxybenzoate derivatives of chiral (E)-2-butene-1,4-diols 2 are unique among allylic alcohol derivatives in their ability to undergo AD with excellent reagent-controlled diastereoselectivity and minimal acyl transfer.[8] We recently demonstrated that the DMSO/PdII-promoted linear allylic oxidation of protected chiral homoallylic alcohols with acetic acid furnishes the acetate derivatives of 2 with excellent regio-and stereoselectivities and with no erosion of optical purity.[2] To avoid FGMs, we set out to identify conditions for allylic oxidation of α-olefins using p-anisic acid (5) to directly generate 4-methoxybenzoate derivatives of 2 from α-olefins (Scheme 1). As shown in Table 1, preliminary studies with α-olefin 4 suggested that acid 5 may be a competent nucleophile in the DMSO/PdII-promoted linear allylic oxidation reaction to form the hexose precursor 6, if the challenges associated with high acid loadings and low yields were resolved (15 equiv of 5, 23% yield, Table 1, entry 1). We were encouraged by the observation that significant amounts of the α-olefin starting material remained at the end of the reaction, thus suggesting that the acid-labile acetonide functionality was tolerant of these conditions. The addition of DIPEA, a noncoordinating base additive, effected a significant increase in yield (45%, Table 1, entry 2). Although the exact role of the base is currently unclear, we hypothesize that it increases concentrations of the benzoate anion. The yield was also increased on switching oxidants from benzoquinone (BQ) to phenyl-benzoquinone (PhBQ, 55%, Table 1, entry 3). Finally, on increasing the reaction concentration to 2.0M, we achieved further increases in yields, and were able to use fewer equivalents of carboxylic acid (2.0M, 3 equiv 5, 75% yield, Table 1, entry 6).
Table 1.
Investigation into the linear allylic oxidation reaction to form the hexose precursor 6.
 | ||||
|---|---|---|---|---|
| Entry | DMSO/CH2Cl2 Molarity [M] | Quinone (Q) | Acid 5 (equiv) | Yield [%][c] |
| 1 | 0.33 | BQ | 15 | 23[d] |
| 2 | 0.33 | BQ | 15 | 45 |
| 3 | 0.33 | PhBQ | 15 | 55 |
| 4 | 0.6 | PhBQ | 10 | 66 |
| 5 | 1.0 | PhBQ | 5 | 67 |
| 6[b] | 2.0 | PhBQ | 3 | 75 |
| 7[b] | 2.0 | PhBQ | 3 | 71[e] |
| 8 | 2.0 | PhBQ | 3 | 63[f] |
| 9 | 3.0 | PhBQ | 1.5 | 50 |
DMSO/CH2Cl2 (3.2:1).
Linear to branched allylic ester and E/Z ratios were determined by HPLC for the material obtained from entries 6 and 7 on comparison with branched or acetonide-free E and Z standard compounds: linear/branched >300:1, E/Z =30:1 and 36:1 (for entries 6 and 7, respectively).
Yield of isolated product from the reactions carried out on a 1 mmol scale (4, 262 mg). Yields and selectivities represent an average of at least 2 runs.
With no DIPEA added.
[Pd(CH3CN)4](BF4)2 (10 mol%), 13% of 4 was recovered.
Pd(OAc)2 (5 mol%). Bn =benzyl, DIPEA=N,N-diisopropylethylamine, MS =molecular sieves.
The linear allylic oxidation reaction is exceptionally stereo- and regioselective, with the major product formed being the linear E isomer (linear/branched >300:1, E/Z = 30→36:1, entries 6 and 7, Table 1). The reaction is also preparatively convenient, with all reactions carried out in an air atmosphere with no precautions taken to exclude moisture or O2. Significantly, with these newly developed conditions, the catalyst loading may be decreased to 5 mol% with only a minor decrease in yield (63%, Table 1, entry 8). Moreover, useful yields are obtained from fragment couplings with 1.5 equivalents of carboxylic acid at 3.0M concentrations of α-olefin (50%, Table 1, entry 9). The only by-product we observed in the reaction using Pd(OAc)2 as the PdII source was allylic acetate, which can be eliminated by using [Pd(CH3CN)4](BF4)2 (Table 1, entry 7).
We set out to test the efficiency of this overall strategy for polyol construction in the context of a short, de novo synthesis of the differentially protected L-galactose (−)-11 (Scheme 3). Bulk chemical (Z)-2-butene-1,4-diol (7) was epoxidized with m-chloroperbenzoic acid (mCPBA) to give the meso-epoxide 8 in 74% yield. The meso-epoxide 8 was then desymmetrized by using the enantioselective Payne rearrangement with oligomeric (R,R)-[(salen)CoIII]OTf catalyst (salen =N,N′-bis(salicylidene)ethylenediamine, Tf =trifluoromethanesulfonyl) and subsequently ketalized in situ to give the chiral epoxyketal (S,S)-9.[15,16] Regioselective opening at the terminal position of the epoxyketal with vinylcuprate and subsequent protection of the intermediate alcohol gave the benzyl ether protected homoallylic alcohol (−)-4 in 54% overall yield (3 steps, 99% ee).[14,15b] Linear allylic C–H oxidation of (−)-4 using 10 mol% [Pd(CH3CN)4](BF4)2 in DMSO under the optimized conditions (2M, PhBQ, 50 mol% DIPEA) with 3 equivalents of p-anisic acid 5 furnished the 4-methoxybenzoate-derived (E)-2-butene-1,4-diol (+)-6 in 71% yield (along with 13% recovered (−)-4) as essentially one isomer (linear/branched =>300:1; E/Z =36:1) and with no erosion of enantiopurity.[17] Alternatively, on using 10 mol% Pd(OAc)2 in DMSO under the same conditions, (+)-6 was obtained in 75% yield with around 10% of the allylic acetate product that was arduous to separate by silica gel chromatography. Asymmetric dihydroxylation of (+)-6 proceeded smoothly to give the polyoxygenated (−)-10 in 96% yield and greater than 20:1 d.r. (1H NMR).[18] Protection of the diol (−)-10 as the silyl ether, followed by removal of the p-methoxybenzoate ester with DIBAL, Swern oxidation of the resulting primary alcohol, and removal of the isopropyl-idene ketal group with Zn(NO3)·6H2O[19] gave the differentially protected L-galactose (−)-11 in 74% yield (over 4 steps).[20] The enantioselective, de novo synthesis of (−)-11 thus proceeded in a total of 10 linear steps with 20% overall yield from the commercially available 7.
Scheme 3.
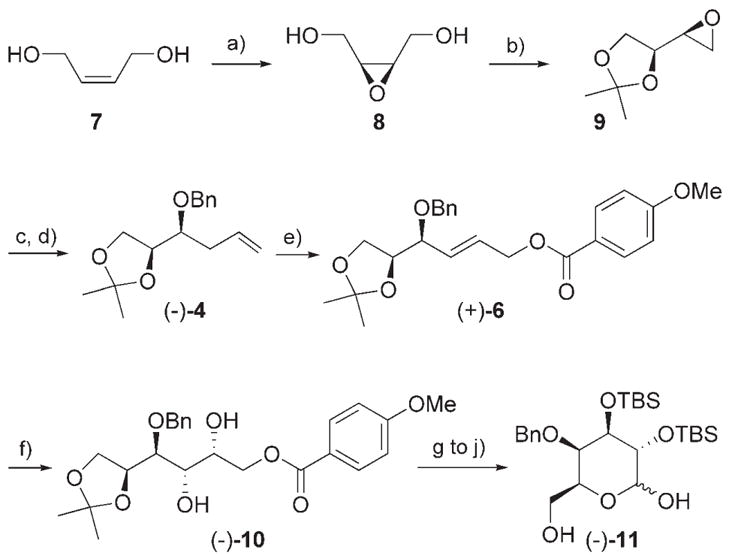
Total synthesis of differentially protected L-galactose (−)-11 (see the Supporting Information for full experimental details). a) mCPBA, CH2Cl2, 0 °C→RT (74%); b) oligomeric (R,R)-[(salen)CoIII]-OTf (0.05 mol%), CH3CN; then 2-methoxypropene, p-TsOH·H2O (1 mol%), 0°C; c) CuBr (10 mol%), CH2=CHMgBr, THF, −40 °C; d) NaH, TBAI (10 mol%), DMF, 0°C, BnBr, 0°C→RT, (54%, 3 steps from 8, 99% ee); e) [Pd(CH3CN)4](BF4)2 (10 mol%), PhBQ (2 equiv), 5 (3 equiv), DIPEA (0.5 equiv), 4-& MS, DMSO/CH2Cl2 (3:1), 41 °C, 72h (71%, linear/branched >300:1, E/Z =97:3); f) K2OsO4·2H2O (1 mol%), (DHQD)2PHAL (5 mol%), K3Fe(CN)6, K2CO3, MeSO2NH2, tBuOH/H2O, 0 °C (96%, >20:1 d.r.); g) TBSOTf, 2,6-lutidine, CH2Cl2, 0°C→RT, (90%); h) DIBAL, CH2Cl2, −78 °C, (98%); i) (COCl)2, DMSO, NEt3, −65 °C, (90%), j) Zn(NO3)2·6H2O, CH3CN, 50 °C, (93%). DHQD =dihydroquinidine, DIBAL = diisobutylaluminum hydride, DMF =N,N-dimethylformamide, PHAL =phthalazine, TBAI =tetra-n-butylammonium iodide, TBS =tert-butyldimethylsilyl, Ts =toluene-4-sulfonyl.
Several stereodivergent, de novo syntheses of the hexoses from 7 have employed chiral (E)-2-butene-1,4-diols analogous to 6 as intermediates. The C–H-oxidation route to 6 (5 steps, 28% overall yield) compares favorably, in number of steps and overall yield, with the Wittig olefination routes previously reported (for example: 11 steps, 18% overall yield,[9] and 9 steps, 16% overall yield[8a]). The strategy developed herein, along with these previous syntheses, provides access to hexose stereoisomers that are complementary to those obtained through aldol-based approaches.[21]
In summary, we have reported a mild and efficient hydrocarbon oxidation strategy for the preparation of chiral polyols, a process validated by an efficient, enantioselective synthesis of the differentially protected L-galactose (−)-11. The synthesis used a highly regio- and stereoselective linear allylic C–H-oxidation reaction that generated 4-methoxybenzoate derivatives of chiral (E)-2-butene-1,4-diols directly from readily available chiral homoallylic alcohols and carboxylic acids. We anticipate that the simple and mild nature of this transformation (that is 2→3, Scheme 1) will render it useful for the synthesis of polyoxygenated motifs in complex molecules.
Experimental Section
Representative procedure for the Pd(OAc)2-catalyzed linear allylic C–H oxidation of (−)-4 to (+)-6: Catalyst Pd(OAc)2 (22.4 mg, 0.1 mmol, 10 mol%), PhBQ (368 mg, 2.0 mmol, 2 equiv), p-anisic acid (456 mg, 3.0 mmol, 3 equiv), 4-& MS (200 mg), (−)-4 (262 mg, 1 mmol, 1 equiv), DMSO (0.380 mL), CH2Cl2 (0.120 mL), DIPEA (0.087 mL, 0.5 mmol, 0.5 equiv), and a teflon stir bar were added sequentially to a borosilicate vial (40 mL). The vial was then capped and the reaction mixture stirred at 41°C for 72 h. Care was taken to keep all reagents off of the walls of the vial and in maintaining the temperature between 40°C and 43°C. After 72 h, the reaction was quenched with saturated aqueous NH4Cl solution (1 mL), stirred for 30 minutes, and then transferred to a separating funnel, and the vial rinsed with ethyl acetate (10 mL). The mixture was diluted with hexanes (40 mL; causing significant precipitation of phenyldihydroquinone as a black solid that was later removed by filtration). The organic phase was washed with H2O (50 mL) and aq Na2CO3 solution (5%, 2 × 50 mL). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Subsequent transfers were performed using Et2O to minimize transfer of phenyldihydroquinone. Purification by column chromatography (silica gel, 30% Et2O in hexanes) gave (+)-6 as an amber oil (309 mg, 75% average yield of 2 runs). The ratios of the linear/branched products (>300:1) were determined by HPLC on comparison with the branched-product standard[22] (Agilent Zobrax Eclipse XDB-C8 column, 35% iPrOH in H2O, 1 mLmin−1, tR(linear) = 15.7 min, tR(branched, two diastereomers) = 18.0 and 19.0 min). E/Z ratios (30:1) were determined by HPLC using the E and Z diols without the acetonide group (Symmetry C-18 column, 40% CH3CN in H2O, 1.0 mLmin−1, tR(E) = 10.1 min, tR(Z) = 11.3 min). The linear acetate product (32 mg, 10%) was also formed in this procedure and was difficult to separate from (+)-6.
Representative procedure for the [Pd(CH3CN)4](BF4)2-catalyzed linear allylic C–H oxidation of (−)-4 to (+)-6: Catalyst [Pd(CH3CN)4](BF4)2 (44.4 mg, 0.1 mmol, 10 mol%), PhBQ (368 mg, 2.0 mmol, 2 equiv), p-anisic acid (456 mg, 3.0 mmol, 3 equiv), 4-& MS (200 mg), DMSO (0.380 mL), CH2Cl2 (0.120 mL), DIPEA (0.122 mL, 0.7 mmol, 0.7 equiv), and a teflon stir bar were added sequentially to a borosilicate vial (40 mL). The vial was then capped and the reaction mixture stirred at 41°C for 1 h. The reaction was cooled to room temperature and (−)-4 (262 mg, 1 mmol, 1 equiv) was added. The vial was capped and stirred at 41°C for a further 72 h. Care was taken to keep all reagents off of the walls of the vial and in maintaining the temperature between 40°C and 43°C. The workup and isolation procedures were identical to those described on using Pd(OAc)2, to give (+)-6 (71% average yield of 2 runs). Approximately 13% of (−)-4 was also recovered. Ratios of linear/branched and E/Z products were determined as described above and found to be similar to those determined for Pd(OAc)2 (linear/branched > 300:1, E/Z = 36:1).
Footnotes
Financial support was provided by the Henry Dreyfus Foundation, the University of Illinois (UIUC), the Merck Research Laboratories, and the NIH/NIGMS (GM076153). D.J.C. gratefully acknowledges the UIUC for Roger Adams and C. S. Marvel fellowships. We thank Prof. E. N. Jacobsen (Harvard University) and Dr. C. P. Stevenson (Merck) for a gift of oligomeric (R,R)-[(salen)CoIII]OTf catalyst and helpful advice on the Payne rearrangement, respectively. We thank Johnson Matthey for a generous gift of Pd(OAc)2. We are also grateful to M. S. Chen for checking our experimental procedure for the allylic oxidation reaction (Table 1, entry 7).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dustin J. Covell, Department of Chemistry, Roger Adams Laboratory, University of Illinois, Urbana, IL 61801 (USA)
Nicolaas A. Vermeulen, Department of Chemistry, Roger Adams Laboratory, University of Illinois, Urbana, IL 61801 (USA)
Nathan A. Labenz, Harvard College, Cambridge, MA 02138 (USA)
M. Christina White, Department of Chemistry, Roger Adams Laboratory, University of Illinois, Urbana, IL 61801 (USA), Fax: (+1)217-244-8024, E-mail: white@scs.uiuc.edu.
References
- 1.a) Wender PA, Hilinski MK, Mayweg AVW. Org Lett. 2005;7:79. doi: 10.1021/ol047859w. [DOI] [PubMed] [Google Scholar]; b) Zhang Q, Rich JO, Cotterill IC, Pantaleone DP, Michels PC. J Am Chem Soc. 2005;127:7286. doi: 10.1021/ja051682z. [DOI] [PubMed] [Google Scholar]; c) Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]; d) Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J Am Chem Soc. 2002;124:11856. doi: 10.1021/ja027311p. [DOI] [PubMed] [Google Scholar]; e) Breslow R, Baldwin S, Flechter T, Kalicky P, Liu S, Washburn W. J Am Chem Soc. 1973;95:3251. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 2.Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223. doi: 10.1021/ol047800p. [DOI] [PubMed] [Google Scholar]
- 3.Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n. for a related N, N-dimethylacetamide system, see: Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Angew Chem. 2006;118:495. doi: 10.1002/anie.200502886.Angew Chem Int Ed. 2006;45:481.
- 4.For an explanation of the term dissonant relationship, see: Evans DA, Andrews GC. Acc Chem Res. 1974;7:147.
- 5.For example, see: Jiang L, Burke SD. Org Lett. 2002;4:3411. doi: 10.1021/ol026504e.Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem. 2001;113:202.Angew Chem Int Ed. 2001;40:196.
- 6.For example, see: Crimmins MT, Caussanel F. J Am Chem Soc. 2006;128:3128. doi: 10.1021/ja060018v.
- 7.For example, see: Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589. doi: 10.1021/ja0533646.Ikemoto N, Schreiber SL. J Am Chem Soc. 1992;114:2524.
- 8.a) Guzman-Perez A, Corey EJ. Tetrahedron Lett. 1997;38:5941. [Google Scholar]; b) Corey EJ, Guzman-Perez A, Noe MC. J Am Chem Soc. 1995;117:10805. [Google Scholar]
- 9.a) Ko SY, Lee AWM, Masamune S, Reed LA, III, Sharpless KB, Walker FJ. Science. 1983;220:949. doi: 10.1126/science.220.4600.949. [DOI] [PubMed] [Google Scholar]; b) Ko SK, Lee AWM, Masamune S, Reed LA, III, Sharpless KB, Walker FJ. Tetrahedron. 1990;46:245. [Google Scholar]
- 10.For example, see: Tsuboi K, Ichikawa Y, Naganawa A, Isobe M, Ubukata M, Isono K. Tetrahedron. 1997;53:5083.
- 11.a) Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]; b) Keller VA, Kim I, Burke SD. Org Lett. 2005;7:737. doi: 10.1021/ol0473400. [DOI] [PubMed] [Google Scholar]
- 12.For syntheses of chiral (E)-2-buten-1,4-diols with good to moderate enantioselectivities from allylation with chiral (E)-γ-(dimethylphenylsilyl)allylboronate reagents followed by epoxidation/Petersen elimination, see: Roush WR, Grover PT. Tetrahedron Lett. 1990;31:7567.Roush WR, Pinchuk AN, Micalizio GC. Tetrahedron Lett. 2000;41:9413.
- 13.For representative asymmetric allylation reactions, see: Xia G, Yamamoto H. J Am Chem Soc. 2006;128:2554. doi: 10.1021/ja058454p.Denmark SE, Fu J, Lawler MJ. J Org Chem. 2006;71:1523. doi: 10.1021/jo052203h.Kubota K, Leighton JL. Angew Chem. 2003;115:976. doi: 10.1002/anie.200390252.Angew Chem Int Ed. 2003;42:946.Racherla US, Brown HC. J Org Chem. 1991;56:401.Roush WR, Walts AE, Hoong LK. J Am Chem Soc. 1985;107:8186.
- 14.Mukai C, Kim JS, Uchiyama M, Sakamoto S, Hanaoka M. J Chem Soc Perkin Trans. 1998;1:2903. [Google Scholar]
- 15.a) White DE, Jacobsen EN. Tetrahedron: Asymmetry. 2003;14:3633. [Google Scholar]; b) Stevenson CP. PhD thesis. Harvard University (USA); 2005. [Google Scholar]; d) Ready JM, Jacobsen EN. Angew Chem. 2002;114:1432. doi: 10.1002/1521-3773(20020415)41:8<1374::aid-anie1374>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:1374. [Google Scholar]; d) Wu MH, Hansen KB, Jacobsen EN. Angew Chem. 1999;111:2167. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2012::AID-ANIE2012>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1999;38:2012. [Google Scholar]
- 16.This reaction was optimized using commercially available (R, R)-[(salen)CoIII]OAc (2 mol%) to give the desired chiral epoxyketal in 50% overall yield (95% ee). Lower yields may be attributed to epoxide opening by MeOH during the ketalization step with higher catalyst loadings of the monomeric catalyst. See the Supporting Information for details.
- 17.The diastereomer which corresponds to racemization of the allylic center was independently synthesized and was not detected in the HPLC analysis of the products (see the Supporting Information for details).
- 18.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]
- 19.Vijayasaradhi S, Singh J, Aidhen I. Synlett. 2000:110. [Google Scholar]
- 20.The silyl groups were removed and (−)-11 was peracetylated to give 1,2,3,6-O-tetraacetyl-4-O-benzyl-L-galactopyranose, the spectroscopic data of which was found to correspond to those reported for the known compound (see the Supporting Information for details): Ermolenko L, Sasaki NA. J Org Chem. 2006;71:693. doi: 10.1021/jo0521192.
- 21.Northrup AB, MacMillan DWC. Science. 2004;305:1752. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 22.a) Chen MS, Prabagaran N, Labenz N, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; b) Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r. [DOI] [PMC free article] [PubMed] [Google Scholar]