

Abstract
To gain insight into the role of the strictly conserved histidine residue, H79, in the reaction mechanism of the methionyl aminopeptidase from Escherichia coli (EcMetAP-I), the H79A mutated enzyme was prepared. Co(II)-loaded H79A exhibits an overall >7000-fold decrease in specific activity. The almost complete loss of activity is primarily due to a >6000-fold decrease in kcat. Interestingly, the Km value obtained for Co(II)-loaded H79A was approximately half the value observed for wild-type (WT) EcMetAP-I. Consequently, kcat/Km values decreased only 3000-fold. On the other hand, the observed specific activity of Mn(II)-loaded H79A EcMetAP-I decreased by ~2.6-fold while kcat decreased by ~3.5-fold. The observed Km value for Mn(II)-loaded H79A EcMetAP-I was ~1.4-fold larger than that observed for WT EcMetAP-I, resulting in a kcat/Km value that is lower by ~3.4-fold. Metal binding, UV–vis, and EPR data indicate that the active site is unperturbed by mutation of H79, as suggested by X-ray crystallographic data. Kinetic isotope data indicate that H79 does not transfer a proton to the newly forming amine since a single proton is transferred in the transition state for both the WT and H79A EcMetAP-I enzymes. Therefore, H79 functions to position the substrate by hydrogen bonding to either the amine group of the peptide linkage or a backbone carbonyl group. Together, these data provide new insight into the catalytic mechanism of EcMetAP-I.
Methionine aminopeptidases (MetAPs) are ubiquitous enzymes responsible for the cleavage of N-terminal methionine residues from polypeptide chains (2–5). Methionine is the universal initiator of protein synthesis, and the removal of methionine is critical in protein maturation processes (6, 7). The physiological importance of MetAP activity is underscored by the cellular lethality upon deletion of MetAP genes in Escherichia coli, Salmonella typhimurium, and Saccharomyces cerevisiae (8–11). Moreover, a MetAP from eukaryotes has been identified as the molecular target for the antiangiogenesis drugs ovalicin and fumagillin, among others (12–16). Therefore, the inhibition of MetAP activity in tumors is an emerging therapy for the prevention of tumor vascularization, which often leads to tumor regression due to oxygen and nutrient deprivation. Used in conjunction with standard cancer therapies, including surgery, radiation, and/or chemotherapy, antiangiogenic drugs are used to shrink localized tumors and protect against metastasis. Targeting angiogenesis for cancer treatment has a number of advantages, including low cellular toxicity and a lack of drug resistance (15), since it is a disease-specific therapy.
MetAPs are organized into two classes (types I and II) on the basis of the absence or presence of an extra 62-amino acid sequence (of unknown function) inserted near the catalytic domain of type II enzymes. The type I MetAPs from E. coli (EcMetAP-I),1 Staphylococcus aureus (SaMetAP-I), Thermotoga maritima (TmMetAP-I), and Homo sapiens (HsMetAP-I) and the type II MetAPs from H. sapiens (HsMetAP-II) and Pyrococcus furiosus (PfMetAP-II) have been crystallographically characterized (15, 17–22). All six display a novel “pita-bread” fold with an internal pseudo-2-fold symmetry that structurally relates the first and second halves of the polypeptide chain to each other. Each half contains an antiparallel β-pleated sheet that is flanked by two α-helical segments. Both domains contribute conserved residues as ligands to the divalent metal ions residing in the active site. In addition, all six structures contain a bis(µ-carboxylato)(µ-aquo/hydroxo)dicobalt(II) core with an additional carboxylate residue at each metal site and a single histidine bound to Co1 (Figure 1) (23, 24). Recently, an X-ray crystal structure of EcMetAP-I was reported with only a single Mn(II) ion bound in the active site (25). This structure was obtained by limiting the amount of metal ion present during crystal growth. The beauty of this structure is that it provides the first structural verification that MetAPs can form mononuclear active sites and are fully active, and the single divalent metal ion resides on the H171 side of the active site as previously predicted by 1H NMR and EXAFS spectroscopy (23, 24).
FIGURE 1.
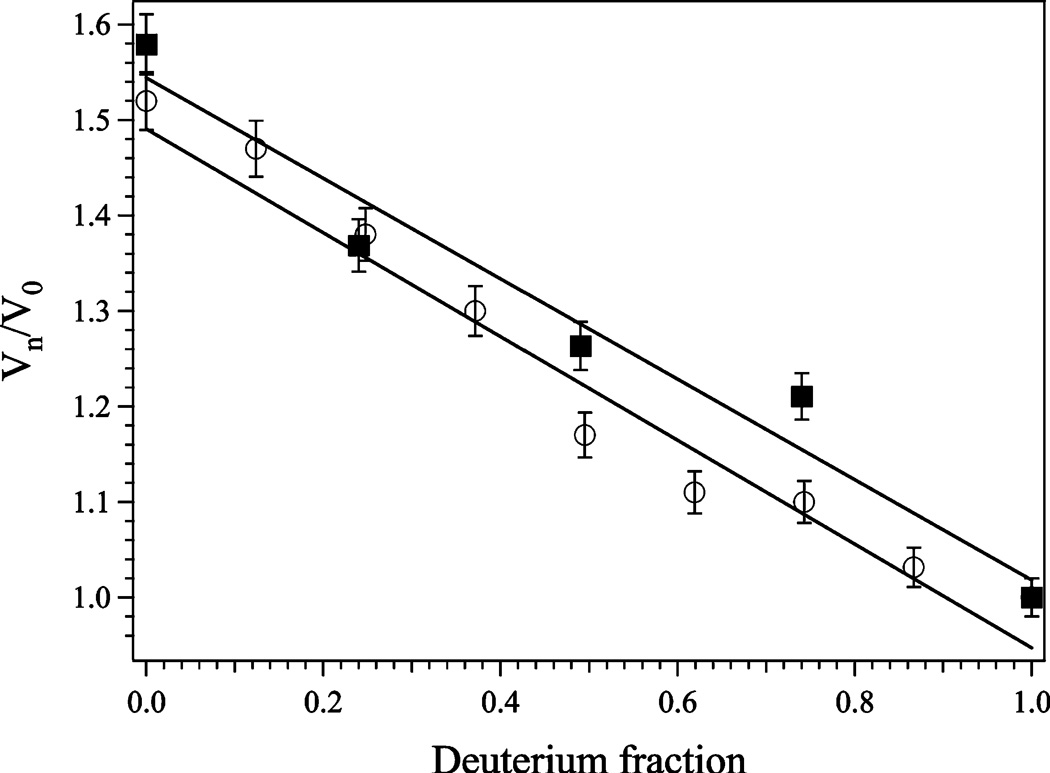
Plot of Vn/V0 vs atom fraction of deuterium for WT (■) and H79A (○) EcMetAP-I at pH 7.5. Vn/V0 is defined as the velocity at n atom fraction of deuterium over the velocity in water. Each point is an average of three replicates.
The catalytic roles of both the active site metal ions as well as active site residues have been proposed (1, 25–27). However, the proposed catalytic mechanism for MetAP assigns no definitive role to the conserved histidine (H79; E. coli numbering), which resides ~8 Å from the first metal binding site (26). On the basis of X-ray crystallographic data for both type I and II MetAPs bound by several substrate, transition state, and product inhibitors as well as fumagillin, H79 has been thought to help position the substrate in the active site and/or to transfer a proton to the newly formed N-terminal amine (15, 28). A significant aspect of designing small molecules that specifically target MetAPs requires a detailed understanding of how potential antiangiogenic inhibitors interact with MetAPs. To determine the catalytic role of H79, we have prepared the H79A mutant EcMetAP-I and characterized the enzyme using kinetic and spectroscopic methods.
MATERIALS AND METHODS
Mutagenesis, Protein Expression, and Purification
All chemicals used in this study were purchased commercially and were of the highest quality available. The H79A-altered EcMetAP-I enzyme was generated as previously reported (16). Recombinant WT and H79A EcMetAP-I were expressed and purified as previously described (29, 30). Purified H79A EcMetAP-I exhibited a single band on SDS–PAGE, indicating an overall purity of >95%. Protein concentrations were estimated from the absorbance at 280 nm using an extinction coefficient of 16450 M−1 cm−1. Apo-EcMetAP-I H79A samples were exchanged into 25 mM HEPES (pH 7.5) containing 150 mM KCl (Centricon-10, Millipore Corp.). Samples were stored frozen at −80 °C until they were needed.
Metal Content Measurement
The concentrations of H79A and WT EcMetAP-I samples used for metal analysis were typically 30 µM. Apo-EcMetAP-I samples were incubated under anaerobic conditions with MCl2, where M is Co(II) or Fe(II) (MCl2, 99.999%, Strem Chemicals, Newburyport, MA, or Aldrich, St. Louis, MO), for 30 min prior to exhaustive dialysis under anaerobic conditions against Chelex-100-treated buffer as previously reported (29, 30). Analyses were performed using inductively coupled plasma atomic emission spectrometry (ICP-AES).
Enzymatic Assay of H79A EcMetAP-I
H79A EcMetAP-I was assayed for catalytic activity using the tetrapeptide MGMM as the substrate (8 mM) with a high-performance liquid chromatography (HPLC) assay as previously described with minor modifications (30). The reaction mixture consisted of a 4 µL aliquot of a 100 µM enzyme solution and 3 equiv of a metal solution. Various concentrations of substrate were added to the enzyme solution to a final volume of 20 µL [25 mM HEPES in Chelex-100-treated water (pH 7.5) containing 150 mM KCl]. The reaction time was 24 h. This method is based on the spectrophotometric quantitation of the reaction product tripeptide, GMM, following separation on a C8 HPLC column (Phenomenex, Luna; 5, 4.6 Å ~ 25 cm). The kinetic parameter v (velocity) was determined at pH 7.5 by quantifying the tripeptide GMM at 215 nm in triplicate. One unit of activity is defined as the amount of enzyme that produces 1 µmol of GMM/min. The metal binding titration reaction was carried out using the same conditions with titrated metal concentrations of up to 10 equiv using either Co(II) or Mn(II).
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) measurements were conducted using a MicroCal OMEGA ultrasensitive titration calorimeter. The titrant (CoCl2) and apo-WT and H79A EcMetAP-I solutions were prepared in Chelex-100-treated 25 mM HEPES buffer (pH 7.5) containing 150 mM KCl. Stock buffer solutions were thoroughly degassed before each titration. The enzyme solution (70 µM) was placed in the calorimeter cell and stirred at 200 rpm to ensure rapid mixing. Typically, 3 µL of titrant was delivered over 7.6 s with a 5 min interval between injections to allow for complete equilibration. Each titration was continued until 4.5–6 equiv of Co(II) had been added to ensure that no additional complexes were formed in excess titrant. A background titration, consisting of the identical titrant solution but only the buffer solution in the sample cell, was subtracted from each experimental titration to account for the heat of dilution. These data were analyzed with a two- or three-site binding model using the Windows-based Origin software package supplied by MicroCal.
Spectroscopic Measurements
Electronic absorption spectra were recorded using a Shimadzu UV-3101PC spectrophotometer. All apo-EcMetAP-I samples used in spectroscopic measurements were thoroughly degassed prior to incubation with Co(II) (CoCl2) for ~20 min at 25 °C. Co(II)-containing samples were handled throughout in an anaerobic glovebox (N2/5% H2, 1 ppm O2; Coy Laboratories). Electronic absorption spectra were normalized for protein concentration and the absorption due to uncomplexed Co(II) (512 nm = 6.0 M−1 cm−1). Low-temperature EPR spectroscopy was performed using a Bruker ESP-300E spectrometer equipped with an ER 4116 DM dual-mode X-band cavity and an Oxford Instruments ESR-900 helium flow cryostat.
Solvent Isotope Effect
All buffers were prepared from a freshly opened bottle of 99.9% [2H]H2O (Aldrich). The buffers and Co(II) salts used in the preparation of all deuterated buffers were in the anhydrous form. The pHs of all the buffers used were adjusted via addition of NaOD or DCl [both at 99% deuterium content (Acros Organics, Geel, Belgium)] and corrected for deuteration by adding 0.4 to the reading of the pH electrode (p2H = p1H meter reading + 0.4) (31). The kinetic assay was performed as previously described with the exception of the replacement of H2O with D2O.
RESULTS
Metal Content and Specific Activity of H79A EcMetAP-I
The number of tightly bound divalent metal ions was determined for H79A EcMetAP-I by ICP-AES. Apoenzyme samples (30 µM), to which 2–30 equiv of Co(II) had been added under anaerobic conditions, were dialyzed for 3 h at 4 °C with Chelex-100-treated, metal-free HEPES buffer [25 mM HEPES and 150 mM KCl (pH 7.5)]. ICP-AES revealed 1.0 ± 0.1 equiv of cobalt-associated H79A-altered EcMetAP-I, similar to the amount of WT EcMetAP-I (23).
The specific activity of H79A EcMetAP-I was determined with the tetrapeptide substrate MGMM. Since the H79A enzyme was stored as the apo form, it was preincubated with 3 equiv of Co(II) for ~30 min prior to all kinetic experiments. The apo form of H79A EcMetAP-I was catalytically inactive. Kinetic parameters were determined for Co(II)- and Mn(II)-loaded H79A and WT EcMetAP-I (Table 1). Co(II)-loaded H79A exhibits an overall >7000-fold decrease in specific activity. The almost complete loss of activity is primarily due to a >6000-fold decrease in kcat. Interestingly, the Km value obtained for Co(II)-loaded H79A was approximately half the value observed for WT EcMetAP-I. Consequently, kcat/Km values decreased only 3000-fold. Kinetic parameters were also determined for Mn(II)-loaded H79A and WT EcMetAP-I (Table 1). For Mn(II)-loaded H79A EcMetAP-I, the specific activity decreased by ~2.6-fold while kcat decreased by ~3.5-fold. The observed Km value for Mn(II)-loaded H79A EcMetAP-I was only ~1.4 times larger than that observed for WT EcMetAP-I, resulting in a kcat/Km value that is lower by only ~3.4-fold.
Table 1.
Kinetic Parameters for H79A EcMetAP-I toward MGMM in the Presence of 3 equiv of Mn(II) or Co(II) at pH 7.5
| metal ion | kinetic parameter | wild type | H79A |
|---|---|---|---|
| Co(II) | Km (mM) | 3.2 ± 0.1 | 1.5 ± 0.1 |
| kcat (s−1) | 18.3 ± 0.1 | 0.003 ± 0.001 | |
| kcat/Km (M−1 s−1) | 5720 | 2 | |
| Mn(II) | Km (mM) | 1.3 ± 0.2 | 1.8 ± 0.1 |
| kcat (s−1) | 4.6 ± 0.1 | 1.8 ± 0.1 | |
| kcat/Km (M−1 s−1) | 3500 | 1000 |
Solvent Isotope Effect
The solvent isotope effect was determined for both WT and H79A EcMetAP-I using MGMM as the substrate at pH 7.6. kcat values for MGMM were measured at several ratios of D2O and H2O, and the results were plotted as the atom fraction of deuterium versus Vn/V0, where Vn is the observed velocity at n fraction of deuterium and V0 is the observed velocity in water (Figure 1). Proton inventories were obtained by fitting the experimental data to equations derived from the Gross-Butler equation (eq 1):
| (1) |
where n is the atom fraction of deuterium, Vn and V0 are the same as described above, vT is the number of protons transferred in the transition state, vR is the number of protons transferred in the reactant state, and ϕ is the fractionation factor defined as
| (2) |
where are the mole fractions of deuterons and protons in the ith transition or reactant state, respectively, while the subscripts T and R refer to the transition and reactant state, respectively (32, 33). At pH 7.6, the best fit for both WT and H79A EcMetAP-I exhibited linearity, suggesting that one proton is transferred in the transition state of the catalytic reaction (Figure 1). The fit yielded fractionation factors of 0.80 and 0.79 for WT and H79A EcMetAP-I, respectively.
Since the largest deviations for theoretical proton inventory curves occur at atom fractions of 0.5 (34, 35), calculation of the midpoint partial solvent isotope effect can provide insight into the number of protons involved in the catalytic reaction. The following equations, derived by Elrod (34), allowed for the calculation of midpoint partial solvent isotope effects when the experimental data were obtained at different atom fractions:
| (3) |
| (4) |
| (5) |
where nm = 0.49 (the H2O/D2O ratio at the midpoint), Vm/V1 equals the midpoint partial solvent isotope effect, and V0/V1 represents the total isotope effect (velocity in 100% H2O/velocity in 100% D2O). The experimental and calculated midpoint partial isotope effects are presented in Table 2. At pH 7.6, comparison of the theoretical and experimental midpoint partial isotope effects for both WT and H79 EcMetAP-I suggests that one proton is transferred at this pH. However, the midpoint partial solvent isotope effect calculations could not strongly distinguish between one proton transfer and general solvation effects (Table 2).
Table 2.
Midpoint Solvent Isotope Effect Data for WT and H79A EcMetAP-I Using MGMM as the Substratea
| calculated midpoint solvent isotope effect |
|||||
|---|---|---|---|---|---|
| enzyme | V0/V1 | midpoint solvent isotope effect (Vm/V1) | one proton | two protons | general solvation |
| WT | 1.6 | 1.26 | 1.27 | 1.28 | 1.25 |
| H79A | 1.5 | 1.17 | 1.21 | 1.24 | 1.22 |
Experimental and theoretical midpoint isotope effects were calculated for a 0.49 atom fraction of deuterium.
Metal Binding Properties of H79A EcMetAP-I
To examine the metal binding properties of the H79A EcMetAP-I enzyme, we titrated Mn(II) and Co(II) into apo-H79A EcMetAP-I (Figure 2). The dissociation constant (Kd) for the first divalent metal binding event for both Mn(II) and Co(II) was obtained by fitting these titration data to eq 6 (36):
| (6) |
where p is the number of sites for which interaction with M(II) is governed by the intrinsic dissociation constant Kd and r is the binding function calculated by conversion of the fractional saturation (fa) using eq 7 as previously described (23).
| (7) |
Cs, the free metal concentration, was calculated using eq 8
| (8) |
where CTS and CA are the total molar concentrations of metal and enzyme, respectively. Kd values were obtained for both Mn(II) and Co(II) by fitting these data via an iterative process that allowed both Kd and p to vary (Figure 2). The best fits obtained provided p values of ~1 and Kd values of 0.4 ± 0.1 and 0.5 ± 0.1 µM for Mn(II) and Co(II) binding, respectively.
FIGURE 2.
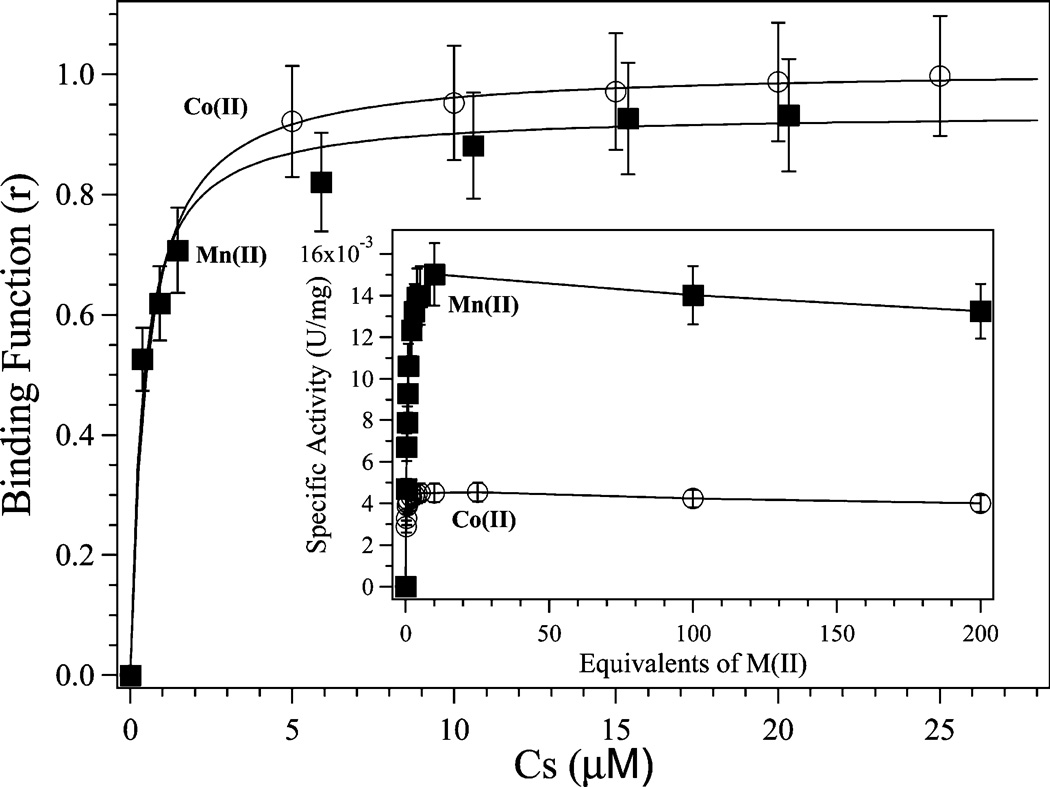
Plot of binding function r vs Cs (the free metal ion concentration) for Co(II) and Mn(II) binding to H79A EcMetAP-I. The solid lines represent the theoretical fit to the data using eq 1. These fits provide a p value of ~1 and Kd values of 0.5 ± 0.1 and 0.4 ± 0.1 µM for Co(II) and Mn(II), respectively. The inset shows a plot of specific activity (units per milligram) vs equivalents of Co(II) and Mn(II). All activity assays were conducted using 8 mM MGMM in 25 mM HEPES (pH 7.5) and 150 mM KCl.
In addition, isothermal titration calorimetry (ITC) was employed to determine the association constants (Ka) for both Co(II) and Mn(II) binding to H79A EcMetAP-I at 25.0 ± 0.2 °C (Figure 3). Ka values were obtained by fitting the ITC data, after subtraction of the background heat of dilution, via an iterative process using Origin. This software package uses a nonlinear least-squares algorithm that allows the concentrations of the titrant and the sample to be fit to the heat flow per injection to an equilibrium binding equation for two sets of noninteracting sites. The Ka value, the metal/enzyme stoichiometry (n), and the change in enthalpy (ΔH°) were allowed to vary during the fitting process (Table 3 and Figure 3). The relationship between Ka and the dissociation constant is defined as
| (9) |
The best fits obtained for H79A EcMetAP-I provided an overall n value of 3, indicating three noninteracting sites. Attempts to fit these data with n values of 1 or 2 provided poor fits. For Co(II) and Mn(II) binding to H79A EcMetAP-I, the observed Kd values for the first binding event are 1.4 ± 1.5 and 5.3 ± 1.4 µM, respectively, which are within error of those observed for WT EcMetAP-I. The second and third metal binding events are equivalent for H79A EcMetAP-I and provided Kd values of 1.6 ± 0.8 and 0.08 ± 0.02 mM for Co(II) and Mn(II), respectively.
FIGURE 3.
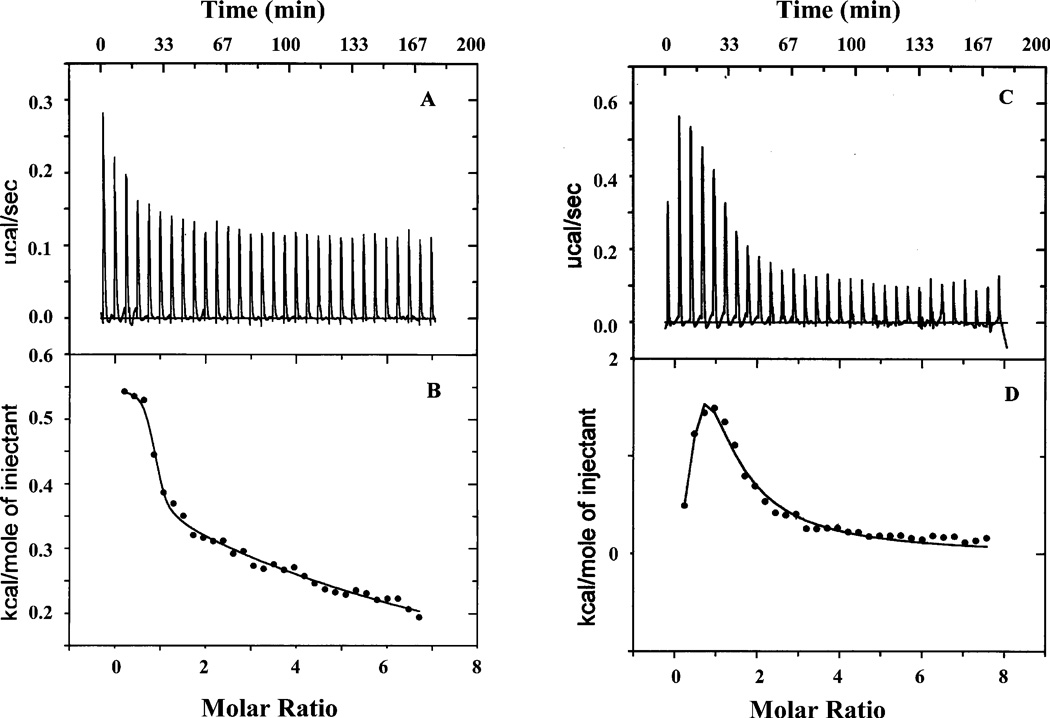
Isothermal titration calorimetry (ITC) titrations of 70 µM solutions of H79A EcMetAP-I with 5 mM Co(II) (A) or 5 mM Mn(II) (C). The best fits of the data in panels A and C are shown in panels B and D, respectively, after subtraction of the heat of dilution.
Table 3.
ITC Data for the Mn(II)- and Co(II)-Loaded H79A EcMetAP-I Enzymes
| na | Ka1, Ka2 (M−1) | Kd1, Kd2 (µM) | ΔH°1, ΔH°2 (kcal/mol) | TΔS°1, TΔS°2 (kcal/mol) | ΔG°1, ΔG°2 (kcal/mol) | |
|---|---|---|---|---|---|---|
| [CoCo(EcMetAP-I)] | 1 | 6.36 × 105 | 1.6 | 15700 | 15708 | −7.8 |
| 2 | 7.05 × 101 | 14200 | 1.1 × 106 | 1.1 × 106 | −2.5 | |
| [MnMn(EcMetAP-I)] | 2 | 3.31 × 105 | 3.0 | 2600 | 2607 | −7.4 |
| 1 | 2.27 × 102 | 4400 | 33800 | 33803 | −3.2 | |
| [CoCo(H79A)] | 1 | 7.35 × 105 | 1.4 | 549 | 557 | −7.9 |
| 2 | 6.41 × 102 | 1600 | 9770 | 9774 | −3.8 | |
| [MnMn(H79A)] | 1 | 1.90 × 105 | 5.3 | −461 | −454 | −7.1 |
| 2 | 1.34 × 104 | 80 | 20900 | 20905 | −5.5 |
All fits were performed using an n value of 3, providing two distinct binding events with individual n values of 1 and 2.
The heat of reaction, measured during the ITC experiment, was converted into other thermodynamic parameters using the Gibbs free energy relationship:
| (10) |
The thermodynamic parameters obtained for titrations of Co(II) and Mn(II) into WT and H79A EcMetAP-I reveal changes that affect both of the metal binding sites (Table 3). Substitution of H79 with A makes the process of binding of the metal ions, particularly for the second metal ion, more spontaneous based on the more negative Gibbs free energies (ΔG).
UV–Vis Spectra of Co(II)-Loaded H79A
The electron absorption spectrum of H79A EcMetAP-I was recorded as a function of Co(II) concentration and compared to that of WT EcMetAP-I (Figure 4). For H79A EcMetAP-I, the addition of 1 equiv of Co(II) produced maxima at 520 nm (ε = 58 M−1 cm−1), 560 nm (ε = 72 M−1 cm−1), and 620 nm (ε = 34 M−1 cm−1). These absorption maxima are blue-shifted by ~75 nm relative to those observed for WT EcMetAP-I. Upon addition of a second equivalent of Co(II) to H79A EcMetAP-I, the relative energies of all three observed maxima remained relatively constant; however, the molar absorptivities increased for each absorption band [520 nm (ε = 109 M−1 cm−1), 560 nm (ε = 124 M−1 cm−1), and 620 nm (ε = 66 M−1 cm−1)]. The observed increase is consistent with both metal ions residing in five-coordinate environments (24).
FIGURE 4.
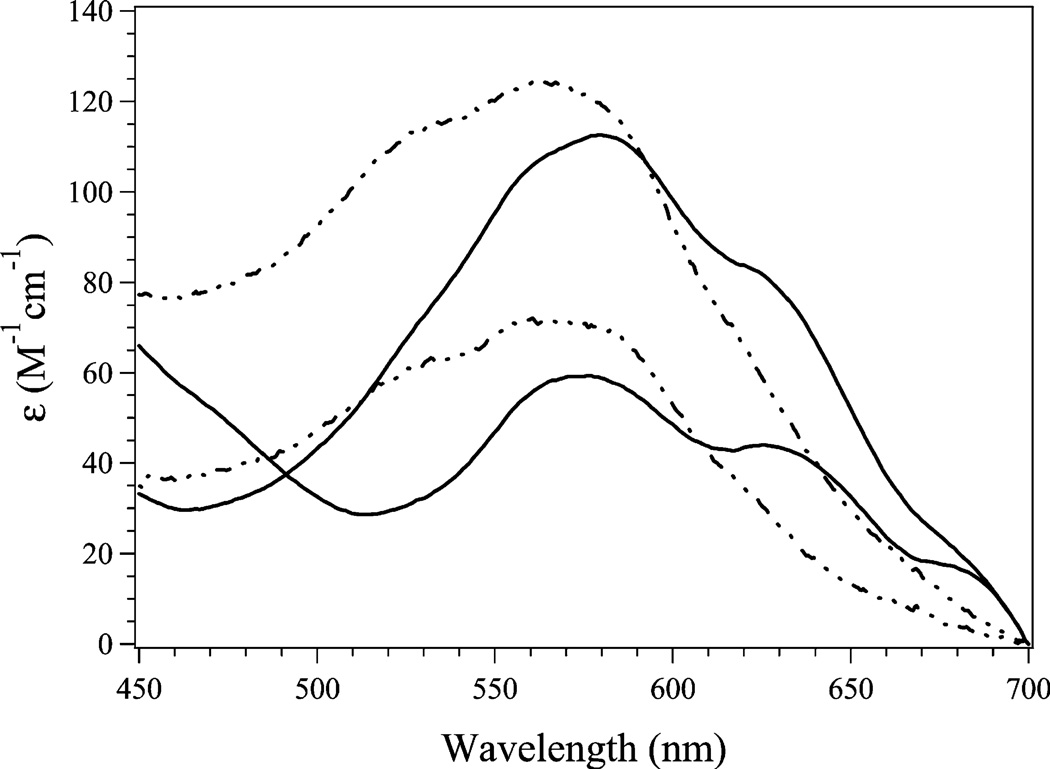
UV–vis spectra of H79A (–--–) and WT (—) EcMetAP-I in the presence of 1 and 2 equiv of Co(II). All spectra were recorded in 25 mM HEPES (pH 7.5) and 150 mM KCl.
EPR Spectra of Co(II)- and Mn(II)-Loaded H79A
The EPR spectrum of H79A EcMetAP-I to which 1 equiv of Co(II) was added {“[Co_(H79A EcMetAP-I)]”} provided an S = 3/2 species, with only the MS = 1±1/2〉 doublet populated from 6 to 60 K, and an essentially axial zero-field splitting (Figure 5A). The signal was very similar to, though distinguishable from, that of [Co_(WT EcMetAP-I)] (Figure 5C). Both EcMetAP-I signals displayed Curie law temperature dependence from 10 to 60 K. The observed EPR spectrum for [CoCo(H79A EcMetAP-I)] (Figure 5B) was indistinguishable from that of [Co_(H79A EcMetAP-I)] and also exhibited Curie law temperature dependence. This behavior mimicked that of the wild-type enzyme; the spectrum of [CoCo(WT EcMetAP-I)] and its temperature dependence were indistinguishable from those of [Co_(WT EcMetAP-I)]. Each of the spectra of Co(II) in the various forms of EcMetAP-I exhibited microwave power-dependent rapid passage effects below 10 K; comparison of the extent to which the finishing baseline of the first integral of spectra recorded at 8 K overshot the starting baseline indicated that the susceptibility to rapid passage effects was very similar for each of the species and that their relaxation properties were all, therefore, similar.
FIGURE 5.
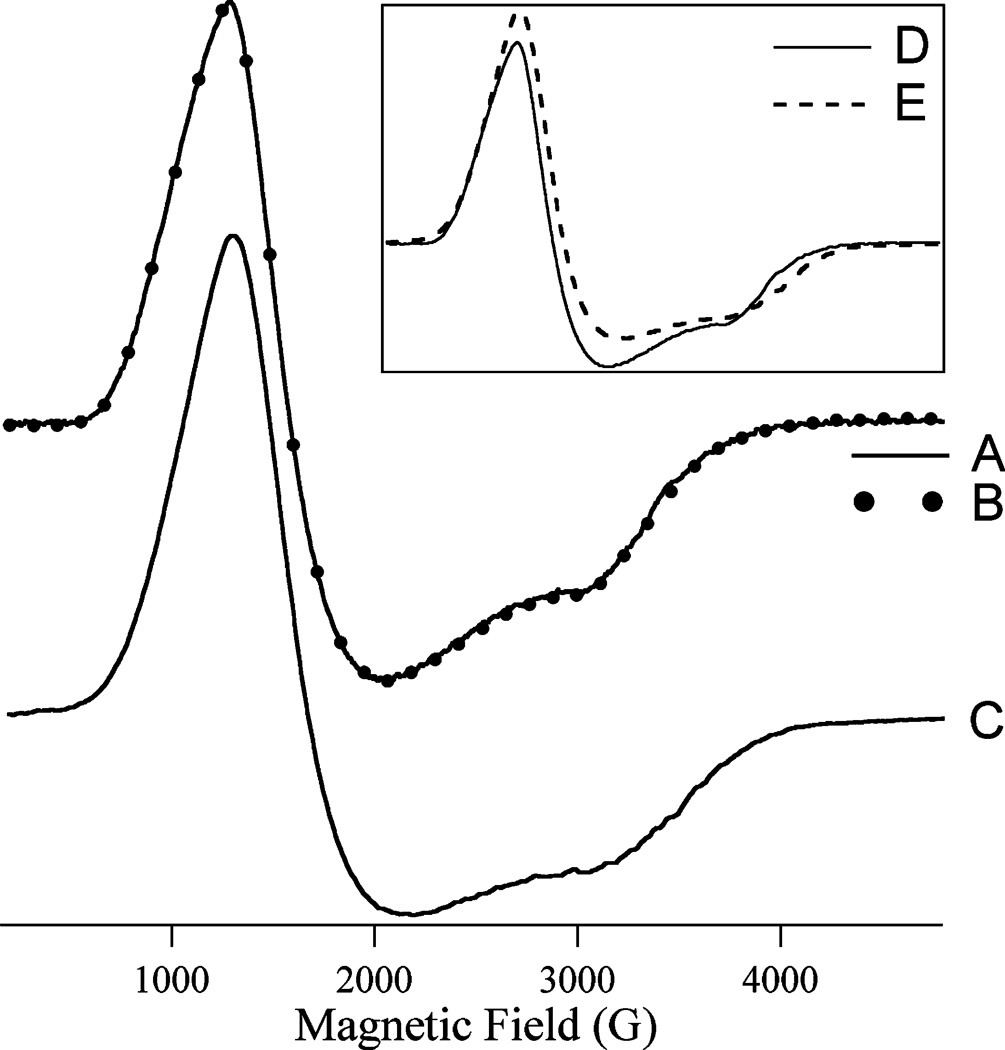
X-Band EPR spectra of Co(II)-substituted forms of EcMetAP-I: (A) [Co_(H79A EcMetAPI)], (B) [CoCo(H79A EcMetAP-I)], and (C) [CoCo(WT EcMetAP-I)]. Spectra A and B were recorded at 8 K with a 0.2 mW microwave power; spectrum C was recorded at 9 K with a 0.2 mW microwave power. The amplitude of trace A is shown multiplied by 1.88 compared to spectrum B. The inset shows the spectra of [CoCo(H79A EcMetAP-I)] (D) and [CoCo(WT EcMetAP-I)] (E) overlaid. All the spectra were recorded at a field modulation amplitude of 1.2 mT, a modulation frequency of 100 kHz, and a sweep rate of 10.2 mT s−1, and the samples were in 25 mM HEPES buffer and 150 mM KCl (pH 7.5).
The EPR spectra of [Mn_(H79A EcMetAP-I)] (Figure 6A,E) and [MnMn(H79A EcMetAP-I)] (Figure 6B,F) were indistinguishable from each other but were subtly different from the mutually indistinguishable spectra of [Mn_(WT EcMetAP-I)] (Figure 6C,G) and [MnMn(WT EcMetAP-I)] (Figure 6D,H). Close examination of the spectra (e.g., the expanded regions of Figure 6A) revealed no evidence of an ~45 G (4.5 mT) pattern due to inter-Mn(II) spin coupling. In addition, spectra from both WT and H79A EcMetAP-I exhibited Curie law temperature dependence over a wide temperature range, and in contrast to WT EcMetAP-I, no additional transitions were observed when the temperature was increased to 60 K. The Mn(II) signals from WT EcMetAP-I were noticeably susceptible to rapid passage effects at low temperatures (6–8 K), while those of H79A EcMetAP-I were not, indicating somewhat different relaxation properties.
FIGURE 6.
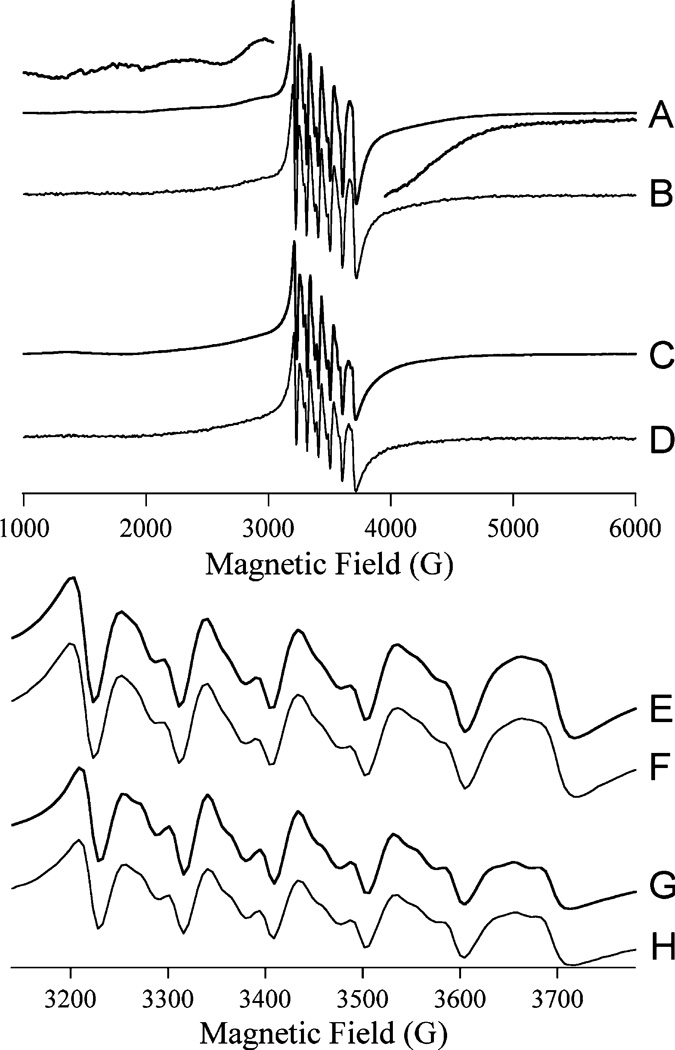
X-Band EPR spectra of Mn(II)-substituted forms of EcMetAP-I: (A) [Mn_(H79A EcMetAP-I)], (B) [MnMn(H79A EcMetAP-I)], (C) [Mn_(WT EcMetAP-I)], and (D) [MnMn(WT EcMetAP-I)]. The high- and low-field wings of the spectrum of [Mn_(H79A EcMetAP-I)] are shown with a larger (5 times) amplitude, above and below, respectively, trace A. Traces E–H are of the geff ~ 2 regions of spectra A–D, respectively. Spectra were recorded at 8 K with a microwave power of 0.2 mW, a field modulation amplitude of 1.2 mT, a modulation frequency of 100 kHz, and a sweep rate of 10.2 mT s−1, and the samples were in 25 mM HEPES buffer and 150 mM KCl (pH 7).
DISCUSSION
The active site histidine residues, H178 and H79, are strictly conserved in all MetAPs sequenced to date (6, 28). It has been suggested that both of these active site His residues play important roles in the catalytic removal of N-terminal methionine residues from polypeptide chains by MetAPs. While the catalytic role of H178 has been examined in some detail (1), the contributions of H79 to the hydrolysis reaction catalyzed by EcMetAP-I remain unclear. Several catalytic functions have been prescribed to H79, some of which are (1, 6, 19, 28, 37) (i) proper positioning of the substrate, (ii) stabilizing the transition state through hydrogen bond formation, (iii) hydrogen bonding to the backbone amide of the scissile peptide bond, and (iv) acting as a general acid by providing a proton to the backbone amide of the scissile peptide bond forming the new N-terminal amine (1, 6). To investigate the role of H79 in catalysis, we have generated the H79A EcMetAP-I enzyme and characterized this enzyme by kinetic and spectroscopic methods.
The specific activity of the H79A-altered EcMetAP-I enzyme was determined in the presence of the tetrapeptide MGMM. Kinetic parameters for both Co(II)- and Mn(II)-loaded WT and H79A enzymes indicate that a significant amount of the observed effect on activity is due to a decrease in kcat. The kcat values for Co(II)- and Mn(II)-loaded H79A EcMetAP-I decreased by ~6100- and ~2.6-fold, respectively. Interestingly, the observed Km value for Co(II)-loaded H79A EcMetAP-I decreased ~2-fold compared to that of WT EcMetAP-I; however, the observed Km value for Mn(II)-loaded H79A EcMetAP-I remained nearly identical to that of WT EcMetAP-I. Consequently, the resulting catalytic efficiency (kcat/Km) for Co(II)- and Mn(II)-loaded H79A EcMetAP-I decreased by ~2900- and ~3.5-fold, respectively. The more drastic loss of catalytic activity observed for the Co(II)-loaded H79A enzyme compared to the Mn(II)-loaded H79A enzyme highlights the differences in the preferred coordination geometry (i.e., five- vs six-coordinate) for Co(II)- versus Mn(II)-loaded EcMetAP-I enzymes. These data are also in line with previous studies on the Co(II)-loaded H79A-altered EcMetAP-I enzyme (19, 28). They are also consistent with those reported for the Co(II)-loaded H231N HsMetAP-II enzyme (H231 is equivalent to H79) in which a complete loss of activity was observed (37).
To determine the effects of altering H79 on the metal binding properties of EcMetAP-I, the number of tightly bound metal ions and the Kd values for both metal binding sites were determined and compared to those of WT EcMetAP-I. On the basis of ICP-AES analysis, ~1.0 metal ion is tightly associated with the H79A EcMetAP-I enzyme, as in WT EcMetAP-I (23). Similarly, both the Co(II)- and Mn(II)-loaded H79A EcMetAP-I enzymes exhibit Kd values identical, within error, to that of WT EcMetAP-I. These data confirm that H79 is not directly involved in binding of the first divalent metal ion. For H79A and WT EcMetAP-I, two additional weak metal binding events are also observed, the last of which is likely a remote Co(II) binding site identified in the X-ray crystal structure of EcMetAP-I (16, 20). This remote metal binding site, or third metal binding site, was also observed in the structure of HsMetAP-I (22). In both enzymes, this remote site is on the outer edge of the enzyme and becomes at least partially occupied at Co(II) concentrations near 1 mM. Interestingly, ITC data for the second Co(II) and Mn(II) binding events indicate that altering H79 to an Ala significantly increases the affinity of the second divalent metal ion binding site by factors of ~10 and ~25, respectively. The second β-sheet backbone of MetAP-I, which contains H79, also contains the active site ligands D108 and D97 which may perturb the metal ligating ability of those residues which are involved in binding the second divalent metal ion.
The observed electronic absorption and EPR spectra of H79A with 1 and 2 equiv of Co(II) are similar to those of WT EcMetAP-I and are consistent with the X-ray crystal structure of H79A EcMetAP-I, which suggests that both Co(II) ions are pentacoordinate (19). The slight blue shift observed in the electronic absorption spectrum of Co(II)-loaded H79A EcMetAP-I versus WT EcMetAP-I is possibly due to the perturbation of the β-sheet peptide backbone that contains the metal ligands D108 and D97. Although the differences between the electronic absorption and EPR spectra of WT and H79A EcMetAP-I are slight, the very large difference in kcat between these species suggests that the structural origins of these spectroscopic differences have catalytic relevance. The Co(II) concentration-normalized intensities of EPR spectra obtained for [Co_(H79A EcMetAP-I)] and [CoCo(H79A EcMetAP-I)] were indistinguishable. As with the EPR spectra, the d–d region of the electronic absorption spectrum of H79A EcMetAP-I changed only in Co(II) concentration-dependent intensity upon addition of additional Co(II) to [Co_(H79A EcMetAP-I)]. Entirely analogous behavior has been reported for WT EcMetAP-I (23), and these data suggest that for both WT and H79A EcMetAP, the natures and relative proportions of the Co(II) environments do not change upon addition of up to 2 equiv of Co(II).
Rationalization of these data with those obtained from ITC, kinetic, and X-ray crystallographic studies, at first, appears to be difficult. However, the most logical explanation leads to the conclusion that metal binding to MetAPs is cooperative and that discrepancies have arisen due to the concentrations of the enzyme samples used in the various experiments. For example, ITC data do not reveal cooperative binding of divalent metal ions to EcMetAP-I or PfMetAP-II but, instead, indicate that one metal ion binds with a much higher affinity than subsequent metal ions. It should be noted that ITC titrations are typically run with enzyme concentrations of ~70 µM, and most often reveal two sets of binding sites (23). Likewise, initial activity assays carried out on Ec-MetAP-I and PfMetAP-II used enzyme concentrations of ~20 µM, a value 2 orders of magnitude larger than the Kd value determined for the first metal binding site of 0.2 or 0.4 µM assuming a Hill coefficient of 1.3 or 2.1, respectively (38, 39). However, a Kd value between 2.5 and 4.0 µM was reported if it was assumed that only a single Co(II) binding site exists in the low-concentration regime, which is within error of ITC and kinetic Kd values. Spectroscopic and most X-ray crystallographic measurements were carried out at much higher enzyme (~1 mM) and metal concentrations, where a significant concentration of dinuclear sites will undoubtedly be present. Under the conditions utilized in ITC experiments, any cooperativity in divalent metal binding will not be detectable but may appear in EPR and electronic absorption data. Since activity titrations and ITC data are not particularly sensitive to the type of binding (i.e., cooperativity vs two independent binding sites), the weak cooperativity observed by Larrabee et al. (38) will not be observed in these experiments but is entirely consistent with the EPR and electronic absorption data and, indeed, with recent X-ray crystallographic data. Most X-ray structures of MetAPs were determined with a large excess of divalent metal ions, so only dinuclear sites are observed. However, crystallographic data obtained for EcMetAP-I using metal ion:enzyme ratios of 0.5:1 reveal metal ion occupancies of 71% bound to the M1 site and 28% bound to the M2 site, consistent with cooperative binding (25).
While studies on the Co(II) form of MetAP may provide some mechanistic insight, Mn(II) or Fe(II) is more likely the physiological metal ion (29, 40, 41). EPR spectra obtained with 1 and 2 equiv of Mn(II) were indistinguishable (apart from intensity) and, similar to Co(II), suggest cooperativity in binding under the conditions that were employed. EPR spectra recorded on the Mn(II)-loaded H79A Ec-MetAP-I enzyme suggested that the Mn(II) ions reside in a mixed oxygen/nitrogen coordination environment, as in the WT enzyme (42). However, in contrast to WT EcMetAP-I and the type II MetAP from P. furiosus, no evidence of inter-Mn(II) spin coupling was observed for the Mn(II)-loaded H79A EcMetAP-I enzyme from either parallel mode EPR, temperature dependence, or close examination of the spectra for a 45 G hyperfine pattern. In addition, the relaxation behaviors of Mn(II) in the WT and H79A enzymes differed. These data suggest subtle differences in the architecture of the dinuclear site in the WT versus H79A enzyme, possibly pertaining to a bridging ligand that, in WT EcMetAP-I, mediates exchange coupling between the Mn(II) ions. That H79A EcMetAP-I is only 2.5-fold less active than the WT enzyme suggests, in turn, that the putative bridging ligand is unimportant for activity and is, therefore, unlikely to be the source of the nucleophile.
A fundamental aspect of the catalytic mechanism of MetAPs that has not been addressed to date is the chemical identity of the rate-limiting step. Kinetic isotope effect studies are an excellent way of gaining an understanding of the nature of the rate-limiting step as well as probing the transition state of catalytic reactions (43). Primary isotope effects are observed if a bond to the labeled atom is made or broken during the reaction, whereas secondary isotope effects describe processes at other positions. Therefore, we examined the solvent isotope effect of WT and H79A EcMetAP-I using MGMM as the substrate at pH 7.6 (31) by substituting hydrogen (1H) with deuterium (2H). The presence of D2O lowers the catalytic activity of both WT and H79A EcMetAP-I, resulting in solvent isotope effects of 1.6 and 1.5, respectively. This is similar to the solvent isotope effect observed for carboxypeptidase A (1.3) (44), the aminopeptidase from Aeromonas proteolytica (2.8) (45), and the β-lactamase from Bacteriodes fragilis (2.6) (46). These results are consistent with a proton being transferred in the rate-limiting step of the reaction. Since the fractionation factor (ϕ) is characteristic of a proton–oxygen bond (neutral oxygen, 0.8–1.2) with a conventional isotope effect equal to 1 (33), the ϕ value of 0.80 suggests that an O–H bond is broken in the rate-limiting step. Since the rate-limiting step in peptide hydrolysis has previously been shown to be product formation (47), this may represent the protonation of the leaving group by a proton obtained from the hydroxide that reorganizes to a carbonyl group in the transition state after attacking the scissile peptide bond leading to the collapse of the transition state (Figure 7). Both WT and H79A EcMetAP-I exhibit similar solvent isotope effects, suggesting that H79 is not involved in the transfer of a proton during catalytic turnover. Therefore, its function is likely limited to stabilization of the transition state by hydrogen bonding which facilitates the formation of the leaving group. These data are consistent with the proposed catalytic mechanism for MetAP and indicate that proton transfer processes limit the catalytic reaction (Figure 7).
FIGURE 7.
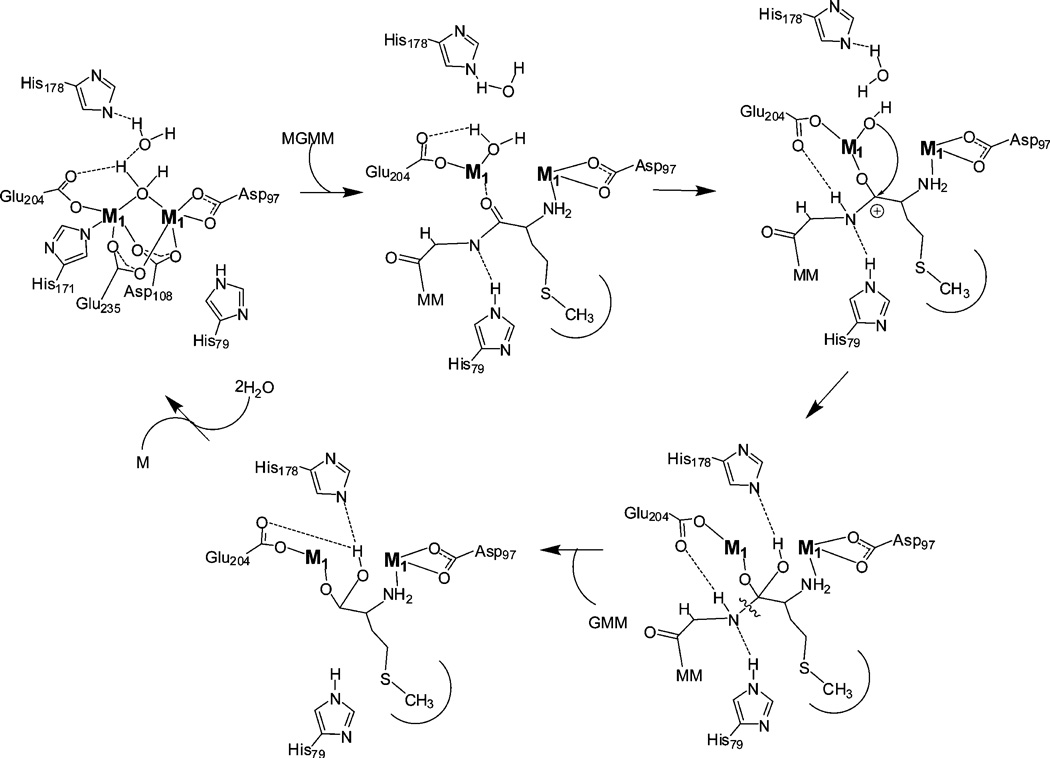
Proposed catalytic mechanism for MetAPs assuming cooperative metal binding.
In summary, several catalytic mechanisms for EcMetAP-I have been proposed (19, 26) on the basis of kinetic, spectroscopic, X-ray crystallographic, and molecular modeling studies. In all of these mechanisms, donation of a proton by E204 or H79 to the leaving group amino nitrogen was proposed to cause the collapse of the transition state into the corresponding products. However, on the basis of data presented herein, H79 does not transfer a proton to the newly forming amine since a single proton is transferred in the transition state for both WT and H79A EcMetAP-I (Figure 7). Therefore, H79 is not essential as a general acid during catalysis, which suggests that the single observed proton transfer is likely due to E204 transferring a proton to the peptide backbone amide. The starkly different effects of H79A substitution on the catalytic activities of Mn(II)- and Co(II)-containing EcMetAP-I suggest that the role of H79 is dependent on some property of the metal ion that is not shared by Co(II) and Mn(II). Where Co(II) is five-coordinate in EcMetAP-I, Mn(II) prefers six-coordinate geometries. A role for H79 in positioning the substrate, by hydrogen bonding to either the amine group of the peptide linkage or a backbone carbonyl group, would indeed cause the catalytic efficiency to be very sensitive to the geometry of the metal ion. Analogues of both of these putative interactions between H79 and substrate have been observed in X-ray crystallographic studies with inhibitors (6). This role is also consistent with the observed differences in activity for different length peptides as well as the observed difference in kcat values for Co(II)- versus Mn(II)-loaded H79A enzymes.
Footnotes
This work was supported by the National Science Foundation (CHE-0652981, R.C.H.) and the National Institutes of Health (AI056231, B.B.). The Bruker ESP-300E EPR spectrometer was purchased with funds provided by the National Science Foundation (BIR-9413530), and XSophe was purchased with funds from the National Institutes of Health (NIH RR01008, B.B.).
Abbreviations: EcMetAP-I, methionine aminopeptidase from E. coli; PfMetAP-II, methionine aminopeptidase from P. furiosus; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; ICP-AES, inductively coupled plasma atomic emission spectroscopy.
REFERENCES
- 1.Copik AJ, Swierczek SI, Lowther WT, D’souza V, Matthews BW, Holz RC. Kinetic and Spectroscopic Characterization of the H178A Mutant of the Methionyl Aminopeptidase from Escherichia coli. Biochemistry. 2003;42:6283–6292. doi: 10.1021/bi027327s. [DOI] [PubMed] [Google Scholar]
- 2.Bradshaw RA. Protein translocation and turnover in eukaryotic cells. Trends Biochem. Sci. 1989;14:276–279. doi: 10.1016/0968-0004(89)90063-7. [DOI] [PubMed] [Google Scholar]
- 3.Meinnel T, Mechulam Y, Blanquet S. Methionine as Translation Start Signal: A Review of the Enzymes of the Pathway in Escherichia coli. Biochimie. 1993;75:1061–1075. doi: 10.1016/0300-9084(93)90005-d. [DOI] [PubMed] [Google Scholar]
- 4.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: The methionine aminopeptidase and Nα-acetyl transferase families. Trends Biochem. Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 5.Arfin SM, Bradshaw RA. Cotranslational processing and protein turnover in eukaryotic cells. Biochemistry. 1988;27(21):7979–7984. doi: 10.1021/bi00421a001. [DOI] [PubMed] [Google Scholar]
- 6.Lowther WT, Matthews BW. Metalloaminopeptidases: Common Functional Themes in Disparate Structural Suroundings. Chem. Rev. 2002;102:4581–4607. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 7.Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 8.Chang S-YP, McGary EC, Chang S. Methionine Aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989;171(7):4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Y-H, Teichert U, Smith JA. Molecular cloning, sequencing, deletion, and overexpression of a methionine aminopeptidase gene from Saccharomyces cerevisiae. J. Biol. Chem. 1992;267:8007–8011. [PubMed] [Google Scholar]
- 10.Li X, Chang Y-H. Amino terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc. Natl. Acad. Sci. U.S.A. 1995;92:12357–12361. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller CG, Kukral AM, Miller JL, Movva NR. pepM is an essential gene in Salmonella typhimurium. J. Bacteriol. 1989;171:5215–5217. doi: 10.1128/jb.171.9.5215-5217.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taunton J. How to starve a tumor. Chem. Biol. 1997;4:493–496. doi: 10.1016/s1074-5521(97)90320-3. [DOI] [PubMed] [Google Scholar]
- 13.Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 14.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the mathionine aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of the human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 16.Lowther WT, McMillen DA, Orville AM, Matthews BW. The anti-angiogenic agent fumagillin covalently modifies a conserved active site histidine in the Escherichia coli methionine aminopeptidase. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12153–12157. doi: 10.1073/pnas.95.21.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douangamath A, Dale GE, D’Arcy A, Almstetter M, Eckl R, Frutos-Hoener A, Henkel B, Illgen K, Nerdinger S, Schulz H, MacSweeney A, Thormann M, Treml A, Pierau S, Wadman S, Oefner C. J. Med. Chem. 2004;47:1325–1328. doi: 10.1021/jm034188j. [DOI] [PubMed] [Google Scholar]
- 18.Tahirov TH, Oki H, Tsukihara T, Ogasahara K, Yutani K, Ogata K, Izu Y, Tsunasawa S, Kato I. Crystal structure of the methionine aminopeptidase from the hyperthermophile Pyrococcus furiosus. J. Mol. Biol. 1998;284:101–124. doi: 10.1006/jmbi.1998.2146. [DOI] [PubMed] [Google Scholar]
- 19.Lowther WT, Orville AM, Madden DT, Lim S, Rich DH, Matthews BW. Escherichia coli Methionine Aminopeptidase: Implications of crystallographic analyses of the native, mutant and inhibited enzymes for the mechanism of catalysis. Biochemistry. 1999;38:7678–7688. doi: 10.1021/bi990684r. [DOI] [PubMed] [Google Scholar]
- 20.Roderick LS, Matthews BW. Structure of the Cobalt-Dependent Methionine Aminopeptidase from Escherichia coli: A new type of proteolytic enzyme. Biochemistry. 1993;32:3907–3912. doi: 10.1021/bi00066a009. [DOI] [PubMed] [Google Scholar]
- 21.Spraggon G, Schwarzenbacher R, Kreusch A, McMullan D, Brinen LS, Canaves JM, Dai X, Deacon AM, Elsliger MA, Eshagi S, Floyd R, Godzik A, Grittini C, Grzechnik SK, Jaroszewski L, Karlak C, Klock HE, Koesema E, Kovarik JS, Kuhn P, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Page R, Quijano K, Rezezadeh F, Robb A, Sims E, Stevens RC, van den Bedem H, Velasquez J, Vincent J, von Delft F, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Lesley SA, Wilson IA. Proteins. 2004;56:396–400. doi: 10.1002/prot.20084. [DOI] [PubMed] [Google Scholar]
- 22.Addlagatta A, Hu X, Liu JO, Matthews BW. Structural basis for the finctonal differences between type I and type II human methionine aminopeptidases. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 23.D’souza VM, Bennett B, Copik AJ, Holz RC. Characterization of the Divalent Metal Binding Properties of the Methionyl Aminopeptidase from Escherichia coli. Biochemistry. 2000;39:3817–3826. doi: 10.1021/bi9925827. [DOI] [PubMed] [Google Scholar]
- 24.Cosper NJ, D’souza V, Scott R, Holz RC. Structural Evidence that the Methionyl Aminopeptidase from Escherichia coli is a Mononuclear Metalloprotease. Biochemistry. 2001;40:13302–13309. doi: 10.1021/bi010837m. [DOI] [PubMed] [Google Scholar]
- 25.Ye QZ, Xie SX, Ma ZQ, Huang M, Hanzlik RP. Structural Basis of Catalysis by Monometalated Methionine Aminopeptidase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9470–9475. doi: 10.1073/pnas.0602433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowther TW, Zhang Y, Sampson PB, Honek JF, Matthews BW. Insights into the mechanism of E. coli methionine aminopeptidase from the structural analysis of reaction products and phosphorous-based transition state analogs. Biochemistry. 1999;38:14810–14819. doi: 10.1021/bi991711g. [DOI] [PubMed] [Google Scholar]
- 27.Huang M, Xie SX, Ma ZQ, Hanzlik RP, Ye QZ. Metal mediated inhibition of methionine aminopeptidase by quinolinyl sulfonamides. Biochem. Biophys. Res. Commun. 2006;339:506–513. doi: 10.1016/j.bbrc.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 28.Li J-Y, Cui Y-M, Chen L-L, Gu M, Li J, Nan FJ, Ye QZ. Mutations at the S1 Sites of Methionine Aminopeptidases from Escherichia coli and Homo sapiens Reveal the Residues Critical for Substrate Specificity. J. Biol. Chem. 2004;279:21128–21134. doi: 10.1074/jbc.M401679200. [DOI] [PubMed] [Google Scholar]
- 29.D’souza VM, Holz RC. The Methionyl Aminopeptidase from Escherichia coli is an Iron(II) Containing Enzyme. Biochemistry. 1999;38:11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- 30.D’souza VM, Bennett B, Holz RC. Characterization of the Divalent Metal Binding Properties of the Methionyl Aminopeptidase from Escherichia coli. Biochemistry. 2000;39:3817–3826. doi: 10.1021/bi9925827. [DOI] [PubMed] [Google Scholar]
- 31.Salomaa P, Schaleger LL, Long FA. Solvent Deuterium Isotope Effects on Acid-Base Equilibria. J. Am. Chem. Soc. 1964;86:1–7. [Google Scholar]
- 32.Albery WJ. Proton Transfer Reactions. London: Chapman & Hall; 1975. [Google Scholar]
- 33.Venkatasubban KS, Schowen RL. The proton inventory technique. CRC Crit. Rev. Biochem. 1984;17:1–44. doi: 10.3109/10409238409110268. [DOI] [PubMed] [Google Scholar]
- 34.Elrod JP, Hogg JL, Quinn DM, Venkatasubban KS, Schowen RL. Protionic reorganization and substrate structure in catalysis by serine proteases. J. Am. Chem. Soc. 1980;102:3917–3922. [Google Scholar]
- 35.Izatt RM, Christensen JJ. Heats of proton ionization, pK, and related thermodynamic quantities. 3rd ed. Vol. 1. Cleveland, OH: CRC Press; 1976. [Google Scholar]
- 36.Winzor DJ, Sawyer WH. Quantitative Characterization of Ligand Binding. New York: Wiley-Liss; 1995. [Google Scholar]
- 37.Griffith EC, Su Z, Niwayama S, Ramsay CA, Chang Y-H, Liu JO. Molecular recognition of angiogenesis inhibitors fumagillin and ovalicin by methionine aminopeptidase 2. Proc. Natl. Acad. Sci. U.S.A. 1998;95:15183–15188. doi: 10.1073/pnas.95.26.15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larrabee JA, Leung CH, Moore R, Thamrong.nawasawat T, Wessler BH. Magnetic Circular Dichroism and Cobalt(II) Binding Equilibrium Studies of Escherichia coli Methionyl Aminopeptidase. J. Am. Chem. Soc. 2004;126:12316–12324. doi: 10.1021/ja0485006. [DOI] [PubMed] [Google Scholar]
- 39.Hu XV, Chen X, Han KC, Mildvan AS, Liu JO. Kinetic and Mutational Studies of the Number of Interacting Divalent Cations Required by Bacterial and Human Methionine Aminopeptidases. Biochemistry. 2007;46:12833–12843. doi: 10.1021/bi701127x. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- 41.Chai SC, Wang W-L, Ye Q-Z. Fe(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J. Biol. Chem. 2008;283:26879–26885. doi: 10.1074/jbc.M804345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whiting AK, Boldt YR, Hendrich MP, Wackett LP, Que L. Manganese(II)-dependent extradiol-cleaving chatechol dioxygenase from Arthrobacter globiformis CM-2. Biochemistry. 1996;35:160–170. doi: 10.1021/bi951979h. [DOI] [PubMed] [Google Scholar]
- 43.Cleland WW. Use of isotope effects to elucidate enzyme mechanisms. Crit. Rev. Biochem. 1982;13:385–427. doi: 10.3109/10409238209108715. [DOI] [PubMed] [Google Scholar]
- 44.Kaiser ET, Kaiser BL. Carboxypeptidase A. Mechanistic analysis. Acc. Chem. Res. 1972;5:219–224. [Google Scholar]
- 45.Bzymek KP, Holz RC. The Catalytic Role of Glutamate-151 in the Leucine Aminopeptidase from Aeromonas proteolytica. J. Biol. Chem. 2004;279:31018–31025. doi: 10.1074/jbc.M404035200. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Fast W, Valentine AM, Benkovic SJ. Metallo-β-lactamase: Structure and mechanism. Curr. Opin. Chem. Biol. 1999;3:614–622. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 47.Chen G, Edwards T, D’souza VM, Holz RC. Mechanistic studies on the aminopeptidase from Aeromonas proteolytica: A two-metal ion mechanism for peptide hydrolysis. Biochemistry. 1997;36:4278–4286. doi: 10.1021/bi9618676. [DOI] [PubMed] [Google Scholar]