

Abstract
Objective
“Ischemic tolerance” can be induced in the retina by “preconditioning” with brief periods of noninjurious retinal ischemia or systemic hypoxia. The present study was undertaken to assess whether tolerance can be induced pharmacologically by deferroxamine (DFX), an iron chelator, which promotes the expression of the transcription factor, hypoxia-inducible factor 1-alpha (HIF-1α), and to identify potential HIF-1α-induced effectors of this endogenous protective response.
Methods
ND4 Swiss-Webster mice were preconditioned with DFX (200 mg/kg, intraperitoneally) as a single dose (SDP) or as repetitive doses (RDP; 6 doses over 2 weeks) and then subjected to 30 min of retinal ischemia (by intraocular pressure elevation) 1 or 4 weeks later. Retinal layer thicknesses and cell counts were quantified 1 week after ischemia. Retinae of additional mice were obtained at various times after SDP or RDP to examine protein-level expression of HIF-1α and adrenomedullin (ADM), a HIF-1α gene target, by immunoblotting and immunohistochemistry.
Results
Ischemia-induced injury was significantly attenuated by SDP 1 week earlier, but not when SDP occurred 4 weeks earlier. However, RDP performed 4 weeks earlier was potently neuroprotective. DFX robustly induced HIF-1α protein expression throughout the inner retina, and levels of HIF-1α protein remained significantly elevated over the 1- and 4-week periods of time between the respective SDP and RDP stimulus and the induction of retinal ischemia. Increases in ADM protein expression were evident throughout the retina following both preconditioning treatments.
Conclusions
DFX preconditions the retina against ischemic injury and multiple doses promote a long-lasting, ischemia-protective phenotype. The widespread and protracted elevations in HIF-1α protein levels and the robust expression of one of its neuroprotective, prosurvival gene targets, ADM, strongly suggest that DFX-induced preconditioning is HIF-1α-dependent. The ability to pharmacologically induce ischemic tolerance in the retina by a clinically well-tolerated drug underscores the potential therapeutic utility of preconditioning for retinal protection in various ischemic retinopathies.
Introduction
Noninjurious brief ischemia or systemic hypoxia are effective preconditioning stimuli known to promote a state of increased ischemic resistance, or “ischemic tolerance,” in the retina.1,2 The endogenous basis for this protection is thought to result from the preconditioning-induced transcriptional activation of a host of survival-promoting genes, as well as post-translational protein processing, that affect metabolic, inflammatory, excitotoxic, apoptotic, and oxidative injury pathways.3 In brain models of ischemic tolerance, cytokines, metabolic inhibitors, anesthetics, and a number of pharmacologic treatments can serve as preconditioning stimuli. However, relatively little is known about pharmacologic approaches to establishing ischemic tolerance in the retina. Ultimately, the clinical applicability of preconditioning to ischemic disease will require pharmacologic therapies that activate endogenous, prosurvival pathways in that tissue.
The present study was undertaken to assess whether deferroxamine (DFX), when administered as a preconditioning stimulus, would afford retinal protection in the setting of ischemia. The efficacy of DFX as a preconditioning agent was shown previously in cerebral ischemia models,4,5 but its utility for retinal preconditioning has yet to be explored. We also aimed to test the hypothesis that the duration of the period of ischemic tolerance so induced could be extended, as we showed previously for hypoxic preconditioning,6 by repetitive DFX treatments. We also sought to explore the mechanistic basis for this protection by focusing on the spatiotemporal expression of the transcription factor, hypoxia-inducible factor 1-alpha (HIF-1α) and one of its gene products, adrenomedullin (ADM). Our examinations of the involvement of this particular transcription factor and one of its cytoprotective, prosurvival gene targets was predicated based on the fact that DFX, like cobalt choride,7 can act as a hypoxia-mimetic and stabilize HIF-1α expression,8,9 thereby driving the expression of hypoxia- or stress-sensitive genes that, we hypothesize, contribute to the ischemia-tolerant phenotype.
Methods
All experiments were performed in accordance with the Association for Research and Vision in Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmology and Vision Research and were approved by the Animal Studies Committee at Washington University School of Medicine (St. Louis, MO). All studies were conducted on 10–12 week-old adult male Swiss-Webster ND4 mice (25–34 g; Harlan Sprague Dawley, Indianapolis, IN).
Single and repetitive DFX preconditioning
Two DFX-based preconditioning protocols were studied for their ability to provide short- and long-term protection of the retina against ischemic injury and for the changes in retinal protein expression these treatments elicited. Mice were randomized to receive either a single DFX preconditioning (SDP), in which one 200 mg/kg intraperitoneal (i.p.) injection of DFX was administered, or repetitive DFX preconditioning (RDP), in which DFX was administered (200 mg/kg, i.p.) 6 separate times over a 2-week period, every other day. Some animals from each of these two groups were sacrificed at various times after SDP or the last RDP stimulus for immunohistochemical or immunoblotting analyses of DFX-induced changes in retinal protein expression. Some animals in the SDP group were subjected to retinal ischemia 1 or 4 weeks later, whereas a 4-week waiting period was followed prior to ischemia in the RDP group. Respective matched sham groups of SDP- and RDP-treated mice not subjected to ischemia were also studied for each of the endpoints examined, as well as nonischemic controls not treated with DFX, but maintained under similar conditions over identical 1- and 4-week time periods.
Induction of retinal ischemia
Retinal ischemia was performed, as described previously, wherein we provided extensive morphologic and functional endpoints documenting the dependence of retinal injury on ischemic duration in this same strain of mice.2,6 In brief, animals were anesthetized i.p. with chloral hydrate (350 mg/kg; Sigma Chemical Co., St. Louis, MO) and xylazine (4 mg/kg; Burns Veterinary Supply, Inc., Rockville Center, NY), and core body temperature was maintained at 37°C throughout the procedure with a thermoregulated infrared heating lamp, while protecting the surgical eye from this source. Pupils were dilated with 1% tropicamide/2.5% phenylephrine hydrochloride (NutraMax Products, Inc., Gloucester, MA), and after achieving corneal analgesia with 1 drop of 0.5% proparacaine hydrochloride (Bausch & Lomb Pharmaceuticals, Inc., Tampa, FL), a contact lens was placed with saline to protect the cornea from dehydration. A 32-G needle attached to a saline-filled reservoir (0.9% sodium chloride; Baxter, Deerfield, IL) raised above the animal was introduced into the anterior chamber to increase intraocular pressure (IOP) above systolic blood pressure (IOP raised to 90 mmHg) and was used to induce retinal ischemia for 30 min, and a whitening of the anterior segment of the eye and blanching of the retinal arteries, as observed under a surgical microscope, were used as indices of complete retinal ischemia. At the end of the ischemic period, the needle was removed from the anterior chamber, and 1% atropine and 1% Vetropolycin HC ointment (Fougera & Atlanta, Inc., Melville, NY) was applied to the conjunctival sac. The animals were recovered in their home cages for 7 days prior to histologic analyses of injury. The fellow eye of each animal served as a nonischemic control, and results from these controls were pooled with additional data from eyes obtained from anesthetized, but untreated, nonischemic mice. Animals were randomized into nonischemic and ischemic treatment groups, and untreated and DFX-preconditioned treatment groups.
Histology and injury quantification
Thicknesses and cell counts in the different retinal layers were quantified, as described previously.6 In brief, following transcardiac perfusion (5 mL/min) with 50 mL of 10 mM of phosphate-buffered saline (PBS; Sigma ImmunoChemicals, St. Louis, MO) and 100 mL of 4% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA), eyes were enucleated and immersion fixed for 1 h in 4% paraformaldehyde, then transferred to 10% neutral-buffered formalin (Fisher Scientific, Pittsburgh, PA) overnight. Following paraffin embedding, 5-μm-thick serial sections, including the optic nerve, were obtained from appropriately matched samples, using a rotary microtome (Reichert-Jung; Model 2030 Reichert-Jung, Vienna, Austria), dried overnight at 37°C, and stained with hematoxylin and eosin.
Image-analysis software (Image-Pro Plus; Media Cybernetics, Silver Springs, MD) was used to measure the thickness of the different retinal layers, in triplicate, for both superior and inferior quadrants at a fixed distance (750–1000 μm) from the edge of the optic disc; a minimum of three sections were used for each eye. Distances from the outer limiting membrane (OLM) to the inner limiting membrane (ILM) (OLM-ILM) and the thickness of the inner nuclear layer (INL) and the inner plexiform layer (IPL) were quantified. We also counted the total number of morphologically identified living cells in the INL and ganglion cell layer (GCL) in the same topographic region of the retina over lengths of 75 and 250 μm, respectively, to avoid possible regional anatomic variations. All morphometric measures were performed in a masked fashion with regard to prior sham or experimental procedures.
Immunoblotting
Four (4) h, 24 h, 1 week, and 4 weeks after SDP, and 4 h, 24 h, 1 week, 4 weeks, and 8 weeks after RDP, mice were killed by cervical dislocation, and mouse retinae were isolated and frozen as rapidly as possible; whole-cell lysates were prepared from retinae pooled from 2 retinas from 1 mouse for the control and DFX-treated, nonischemic mice. Age- and sex-matched retinae from control mice that did not receive DFX were processed in parallel. Antibodies used included a polyclonal rabbit antimouse HIF-1α antibody (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or monoclonal mouse antimouse β-actin antibody (1:20,000; Sigma-Aldrich, St. Louis, MO), followed by horseradish peroxidase (HRP)-linked antibodies (Cell Signaling Technology, Bevery, MA) to goat antirabbit IgG (1:3000) or horse antimouse IgG (1:3000). Protein in the resultant bands was quantified with Image J software [National Institutes of Health (NIH), Bethesda, MD] and were normalized to protein levels in naïve controls.
Immunohistochemistry
The cellular localization of HIF-1α and ADM expression at various times following SDP and RDP was determined by immunohistochemistry of paraffin-embedded sections of mouse retina close to the central retina. As described earlier,6 following deparaffinization and rehydration, and antigen retrieval by unmasking solution (Vector Laboratories, Burlingame, CA), nonspecific binding was blocked with prediluted normal goat serum (Vecastain Elite ABC kit; Vector Laboratories), and sections were incubated overnight at 4°C with primary antibody [a chicken antimouse HIF-1α antibody (1:50) kindly supplied by Dr. Max Gassmann, Ph.D., Zurich, Switzerland] and a rabbit, antimouse ADM antibody (1:100; Phoenix Pharmaceutics, Inc., Burlingame, CA). For HIF-1α, the secondary antibody was a rabbit antichicken IgY HRP (1:500; Promega, Madison, WI), with a diaminobenzidine-peroxidase conjugate (Vector Laboratories), and hematoxylin counterstaining. For ADM, a goat, antirabbit IgG (1:200; AlexaFluor 568, Invitrogen-Molecular Probes, Inc., Carlsbad, CA) was used as the secondary antibody. Negative control slides were run without primary antibody. Slide sections were examined and photographed by light and fluorescent microscopy and analyzed with Axiovision software (Carl Zeiss, Gottingen, Germany).
Statistical analyses
Differences in the morphologic endpoints and protein expression by immunoblotting were compared by the nonparametric Kruskas-Wallis analysis of variance rank-sum test. A value of P < 0.05 was accepted as significant.
Results
Ischemic injury and DFX-mediated protection
Histologic analyses shown in Figure 1 reveal that both SDP and RDP regimens we used were profoundly neuroprotective in the ischemic retina. Relative to controls (n = 15), in eyes without any prior DFX preconditioning treatment (n = 9), 30 min of retinal ischemia reduced OLM-ILM thickness by 24% and the thickness of the INL and IPL by 16 and 36%, respectively (all P < 0.02). Loss of cells in the INL and GCL was 21 and 32%, respectively (both P < 0.001).
FIG. 1.
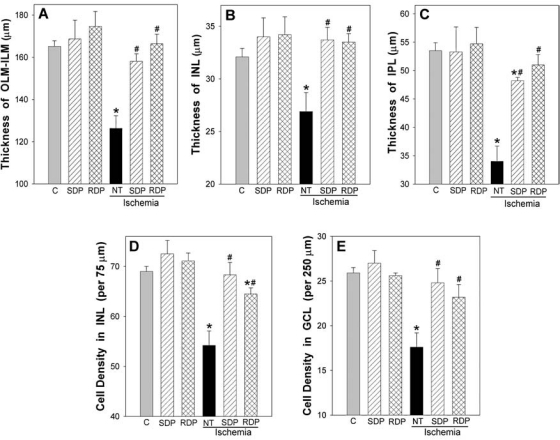
Histologic analyses of retinal ischemic injury and protection by single deferroxamine (DFX) preconditioning (SDP, hatched bars) and repetitive DFX preconditioning (RDP, cross-hatched bars). Retinal layer thicknesses are shown in the top row (A–C), and retinal cell counts in the inner nuclear layer (INL) and ganglion cell layer (GCL) are shown in the bottom row (D and E, respectively), as measured in retinal thin sections obtained 1 week after ischemia or at matched time points in nonischemic mice. Note that the significant thinning of all retinal layers and the cell loss in the INL and GCL in response to 30-min ischemia in untreated animals (NT, solid black bars; n = 9) was nearly completely abrogated by prior preconditioning with SDP 1 week earlier (n = 5) or RDP four weeks earlier (n = 7). Layer thicknesses and cell counts in the nonischemic fellow eyes of ischemic mice with SDP and RDP are also shown (with no significant changes). *P < 0.05 versus controls (C, gray bars; n = 15); #P < 0.05 versus untreated ischemia.
When SDP was performed 1 week before retinal ischemia (n = 5), all morphologic indices of retinal ischemic injury were significantly reduced. Relative to ischemia, the extent of protection realized by SDP was a 73–131% recovery for the different retinal thicknesses (P < 0.02) and a 87–95% recovery for cell counts (P < 0.05). In all instances, except IPL thickness, these metrics were not significantly different in magnitude from those measured in respective nonischemic control mice. The ability of SDP to protect the retina against ischemic injury was lost if the time between the SDP stimulus and the ischemic event was extended to 4 weeks. In these 9 mice (data not shown), trends toward recoveries in retinal layer thicknesses (INL and IPL) were observed, but only the overall thickness metric (OLM-ILM) was significantly improved (by 53%, relative to the 82% recovery in OLM-ILM thickness measured when ischemia was induced 1 week after SDP). No significant protection was observed with respect to INL and GCL cell counts at this time.
However, when RDP was used as the preconditioning stimulus, retinal neuroprotection was evidenced 4 weeks later. A 87–127% recovery of ischemia-induced retinal thinning was measured in these mice (P < 0.004), along with a 68–70% recovery in INL and GCL cell counts (P < 0.05), when ischemia was induced 4 weeks after the last of the six RDP stimuli (n = 7). These measures did not differ significantly from those in nonischemic controls.
Examination of nonischemic fellow eyes revealed that neither the SDP or RDP regimens significantly affected any of the five morphometric endpoints relative to nonischemic controls (Fig. 1).
HIF-1α expression following DFX preconditioning
Figure 2 provides immunohistochemical evidence for the widespread induction of HIF-1α throughout the retina in response to a single systemic injection of DFX. During baseline, normoxic, untreated conditions, some HIF-1α protein appeared to be expressed very faintly in some ganglion cell bodies and axons, in scattered cells in the inner nuclear layer, and in the inner segments of the photoreceptors (Fig. 2B). In response to DFX, HIF-1α protein levels were rapidly (within 1 h) upregulated in the GCL, INL, and ONL layers, the photoreceptor inner segments, and the retinal pigment epithelial cells, with a less robust expression in the nerve fiber layer and outer plexiform layer, (OPL), and no staining in the IPL (Fig. 2C). In the GCL and INL, both the cytoplasm and nucleus are positive for HIF-1α, whereas in the ONL, it is the cytoplasm that appears positive, surrounding unstained nuclei.
FIG. 2.
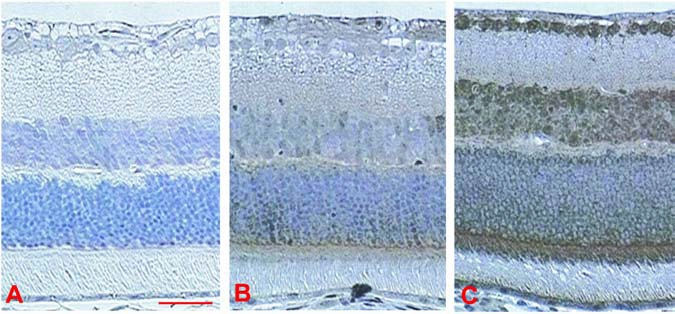
Representative immunohistochemistry for retinal hypoxia-inducible factor 1-alpha (HIF-1α) protein expression in control mice and 1 hour after a single preconditioning dose of deferroxamine (SDP). (A) Negative control without primary antibody. (B) Normal control with primary antibody, showing only faint and scattered expression of HIF-1α. (C) At 1 h after SDP, with widespread expression of HIF-1α evident throughout the retina. Scale bar = 50 μm.
Figure 3 summarizes the changes in HIF-1α protein expression we observed following SDP and RDP, as determined by Western blotting. Note that SDP resulted in a robust, significant increase in retinal HIF-1α protein expression within 4 h, which remained elevated for 24 h. In fact, HIF-1α protein levels were still significantly elevated above baseline 1 week after the single DFX injection, at a time when the retina was still protected from retinal ischemic injury; however, 4 weeks following SDP, when ischemic tolerance was no longer evident, HIF-1α levels had returned to baseline. From 4 h to 1 week following the last of the 6 DFX injections that comprise the RDP stimulus, retinal HIF-1α expression was also significantly elevated to a similar magnitude as that induced by a single DFX injection. Moreover, retinal HIF-1α levels were still significantly increased above baseline 4 weeks after RDP, a time when the retina was still ischemia tolerant. However, 8 weeks after RDP, HIF-1α levels had returned to baseline, and the retina was no longer protected by an RDP stimulus administered 8 weeks earlier.
FIG. 3.
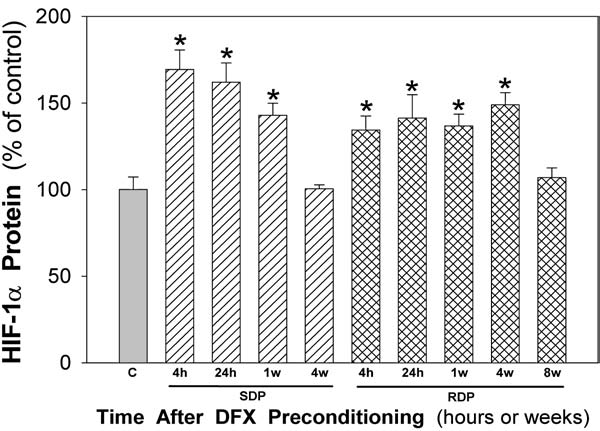
Immunoblot analyses of retinal hypoxia-inducible factor 1-alpha (HIF-1α) protein expression at various times (4 or 24 h and 1, 4, or 8 weeks) after single deferroxamine (DFX) preconditioning (SDP, hatched bars) or repetitive DFX preconditioning (RDP, cross-hatched bars), relative to controls (C, gray bar). Responses were normalized to HIF-1α protein levels in nonpreconditioned controls collected concurrently and run simultaneously. *P < 0.05 versus control. n = 6–9 mice for experimental samples, n = 11 for controls.
ADM expression following DFX preconditioning
The widespread retinal distribution of ADM protein expression at various times after SDP and RDP is shown by the immunohistochemistry results in Figure 4. In general, these stimuli led to a widespread expression of ADM throughout the retina, and in both instances, this expression was long lasting. Under baseline conditions (normoxia) without DFX, expression of ADM protein was noted at mild levels in the cytoplasm of scattered cells in parts of the GCL, INL, ONL, and retinal pigment epithelium (Fig. 4A). At 4 h after a single dose of DFX (Fig. 4B), ADM expression was elevated in all retinal regions that expressed it basally. By 24 h after DFX, ADM expression was similar in the GCL and somewhat diminished in the INL and ONL relative to 4 h; by 24 h, ADM expression was then noted in the IPL and more robustly in the photoreceptor outer segments (Fig. 4C). One (1) week after SDP, an elevated ADM expression was still evident in the GCL, as well as in the INL, ONL, and IPL, and in the retinal pigment epithelium, at levels similar to those observed at 4 and 24 h; but the expression in the photoreceptor outer segments was attenuated relative to 24 h (Fig. 4D). Four (4) weeks following the last of the RDP treatments, ADM expression was still robustly elevated in the GCL, INL, and ONL, as well as in the IPL, photoreceptor outer segments, and RPE, at levels similar to that observed at 1 week following SDP (Fig. 4E).
FIG. 4.
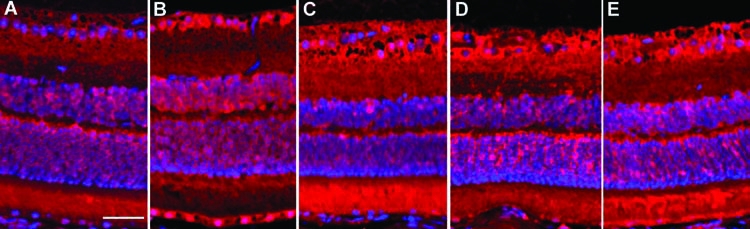
Fluorescent immunohistochemistry for adrenomedullin (ADM) expression in representative retinae of mice at various times following single deferroxamine (DFX) preconditioning (SDP) or repetitive DFX preconditioning (RDP). (A) Non-preconditioned control. (B–D) At 4 h, 24 h, and 1 week after SDP. (E) At 4 weeks after RDP. ADM immunoreactivity shown in red against a blue DAPI nuclear counterstain. Scale bar = 50 μm.
Discussion
Results of the present study are the first to document DFX preconditioning as a therapeutic approach for establishing retinal ischemic tolerance. A single dose of this agent robustly protected the retina from ischemic damage for at least 1 week, but protection was no longer evident 4 weeks later. However, multiple doses of DFX over a 2-week period provided equally robust ischemic protection lasting at least 1 month after the last DFX treatment. The temporal profile over which these two ischemia-tolerant states were established by DFX paralleled that of retinal HIF-1α protein expression, suggesting that the continued expression of one or more of its downstream targets contributed to the neuro-protective phenotype. Indeed, we documented an elevation of ADM protein expression throughout the retina following both of our DFX preconditioning regimens, occurring along a time course similar to that of HIF-1α. These findings identify DFX, already approved for human use, as a potential pharmacologic trigger for preconditioning the retina of patients at risk for various ischemic retinopathies.
In the central nervous system (CNS), and all other tissues examined, a transient period of increased resistance to ischemic injury, known as “ischemic tolerance,” can be induced by an assortment of “preconditioning” stimuli.3,10 The ability of brief ischemia1,2,11,12 to precondition the rat retina against ischemic injury is now well established; systemic hypoxia is also effective,2,6 at least in mice. However, these stimuli are not ideal options for promoting ischemic tolerance in humans. Relative to investigations using brief ischemia or hypoxia, studies documenting pharmacologic preconditioning of the retina are relatively sparse. Mitochondrial KATP channel openers13,14 effectively precondition rat retina. Cobalt chloride, which activates the transcription factor, HIF-1α,15 increases the level of retinal vascular glucose transporters16 and upregulates heat-shock protein-27,7 which promotes retinal ischemic tolerance in rats.7 Our present study, identifying two different DFX preconditioning protocols that promote short- and long-lasting ischemic tolerance in mouse retina, provides the first documentation of DFX-mediated preconditioning in the eye.
DFX was originally identified decades ago for iron-chelation therapy in patients with transfusion-dependent anemias (see below), but is increasingly becoming recognized for its “hypoxia-mimetic” qualities as a result of its ability to stabilize the hypoxia-driven transcription factor, HIF-1α, under normoxic conditions. This results from its ability to disrupt the association between the iron-dependent prolyl hydroxylase enzyme and HIF-1α, so that ongoing ubiquitation-mediated degradation of the latter is prevented8,9; HIF-1α then translocates to the nucleus and drives the expression of hypoxia-sensitive genes, whose protein products promote an assortment of prosurvival actions.9,17 Although the DFX-mediated chelation of iron may also attenuate Fenton reaction chemistry, thereby reducing superoxide radical generation,18 it is difficult to postulate how an attenuation in reactive oxygen species (ROS) would drive gene expression related to preconditioning, particularly since ROS promote HIF-1α expression19 and participate in triggering ischemic tolerance.20 It is also unclear how, if DFX has a significant effect with respect to interrupting the cell cycle,21 such a response would contribute to DFX-mediated preconditioning. Rather, given the widespread evidence for a role for HIF-1α as a proximal mediator of preconditioning in the retina6,7,22 and brain,4,23,24 we contend that it is the ability of DFX to stabilize HIF-1α in a dose-dependent, temporally unique manner that is responsible for its protective effects in our mouse model of retinal ischemia.
Indeed, our immunoblot measures of HIF-1α in the present study confirm that systemically administered DFX promotes significant, and relatively long-lasting, increases in the expression of this transcription factor in the retina, similar to that shown elsewhere in the CNS4,5 and other tissues25 at comparable doses. Moreover, in those in vivo studies, as well as in ours, it was conclusively demonstrated that DFX preconditioning promoted ischemic tolerance. A similarly protracted HIF-1α response was documented previously in the rat brain in response to global ischemia, wherein the expression of HIF-1α protein was elevated for at least 1 week.26 In the retina, we measured a prolonged period of HIF-1α expression following hypoxic preconditioning, the magnitude and duration of which was related to whether the hypoxic stimulus was presented singly or repetitively.6 The present results with DFX largely parallel these latter findings, but with potentially important differences. The duration of the resultant HIF-1α upregulation was greater with both SDP and RDP relative to single and repetitive hypoxic preconditioning; with RDP, but not SDP, this increase in HIF-1α expression lasted at least 4 weeks. And, although the extent to which HIF-1α expression was elevated in the retina was similar with SDP and RDP, and was of relatively moderate magnitude, a significantly greater increase in HIF-1α expression followed repetitive, relative to single, hypoxic preconditioning.6 While considerable evidence supports a rapid “on response” for HIF-1α expression that, in most tissues, is proportionally dependent on the extent of oxygen-tension reduction,9,17,27 our present results with DFX are similar to our previous study with hypoxia, in that the “off response” does not appear to be controlled in the same way, with HIF-1α expression continuing well into normoxic recovery in a fashion dependent on the characteristics of the preceding hypoxic stimulus.28,29 Recent studies of HIF-1α regulation following intermittent hypoxia also point to responses that are uniquely related to the nature of the triggering stimulus.30 These magnitude- and duration-dependent differences in HIF-1α expression may be just one more reflection of the many different ways in which this transcription factor is translationally regulated by hypoxia, DFX, and other stimuli.31,32
Our immunohistochemical studies to localize HIF-1α following DFX preconditioning also revealed interesting, stimulus-dependent differences in expression patterns relative to hypoxic preconditioning. In the present study, DFX treatment led to HIF-1α expression in the cells of the GCL, INL, and photoreceptor inner segments, whereas a more widespread induction of HIF-1α, including ONL cells and some plexiform layer expression, resulted following hypoxic preconditioning.6 However, only limited time points were studied in both studies, so we cannot rule out that expression patterns may have been similar at other times, or that intraretinal pharmacokinetics influenced the response to DFX. In any event, the increases in retinal HIF-1α protein expression following DFX preconditioning, that we documented, implicate the downstream gene targets of HIF-1α as likely effectors of the ischemia-tolerant state so induced.
The administration or pharmacologic upregulation of several HIF-1α gene targets, including erythropoeitin,33 heme oxygenase-1,6,34 and vascular endothelial growth factor,35 protects the ischemic retina. In the present study, we confirmed that a robust, widespread increase in the expression of the peptide ADM, a HIF-1α target gene and member of the calcitonin gene-related peptide family, persists over protracted periods of time following SDP and RDP, temporally coincident with the pattern of HIF-1α expression that was triggered by each preconditioning stimulus. Many of the ADMs cytoprotective and hemodynamic effects in the CNS36,37 are consistent with a role for this peptide as a key mediator of ischemic tolerance in the brain and retina. For example, the ADM upregulates nitric oxide production by endothelial cells, resulting in vasodilation in many tissues, including the retina.38,39 Proangiogenic effects of the ADM are realized secondary to its ability to stimulate, in an autocrine and paracrine manner, endothelial proliferation, and migration and survival, via receptors located on endothelial and vascular smooth muscle cells linked to changes in intracellular calcium mobilization, and activation of cAMP and the opening of potassium channels; its effects on cell migration and survival in retinal pigment epithelial cells, however, remains controversial.40,41 Cell protection has been documented in neuronal, astrocytic, and vascular CNS models of in vitro ischemia,42,43 as well as in in vivo stroke models.44,45 Although not causally demonstrated, to date, the fact that hypoxia,46,47 DFX (present study), cytokines, lipopolysaccharide, and other preconditioning stimuli increases ADM mRNA and/or ADM protein in the CNS is consistent with the hypothesis that ADM is an effector molecule in ischemic tolerance.
Other effector molecules involved in promoting the two distinct temporal windows of retinal ischemic tolerance following our respective DFX-based preconditioning protocols remain to be clarified. We emphasize that this protection is different from that afforded by postischemic treatment of the retina with DFX and related DFX conjugates, as demonstrated previously by others,48–50 and propose that the respective mechanisms underlying the protection in the preconditioning and postischemic treatment models are, in fact, largely distinct. We also provided evidence herein that, at least based on careful morphologic assessments, the single and repetitive DFX dosing regimens we used to trigger ischemic tolerance did not cause retinal injury in the absence of ischemia. Although brief ischemia or hypoxia, when titrated appropriately, also can precondition without apparent harm to the retina,1,2,6 DFX is an attractive preconditioning agent with respect to considering the translational aspects of preconditioning because of its longstanding history of use in humans with various hemoglobinopathies and other pathologic iron disorders, as well as in patients with transfusion-dependent anemias.51 On the other hand, the dose we used for preconditioning in the present study was about 10 times that used clinically in such populations; so, given the concerns about DFX toxicity in patients treated chronically with DFX,52 and more specifically the retina-related side effects,53 it will ultimately be necessary to determine dose-response profiles for DFX's preconditioning effects in different experimental models and to more extensively monitor potentially injurious ocular outcomes resulting from such treatment. The obvious aim of these studies would be to eventually identify acute DFX-dosing regimens that can effectively precondition the human retina without the side effects associated with chronic DFX-treatment paradigms.
Conclusions
Single or multiple doses of DFX can serve as preconditioning stimuli to promote short- or long-lasting periods of ischemic tolerance in retina. In this study, we provided evidence consistent with the notion that DFX stabilizes the transcription factor HIF-1α in a dose-dependent manner, and thereby drives temporally unique expression profiles for cytoprotective molecules, such as ADM, to promote this protective response. The capacity to pharmacologically induce endogenous adaptive mechanisms and promote a sustained change in retinal phenotype with clinically well-tolerated agents, such as DFX, should underscore its potential therapeutic utility in preventing or reducing injury in glaucoma and other ischemic retinopathies.
Acknowledgments
This study was supported by NIH RO3 EY 014938 (to J.M.G.), EY02687 (Department of Ophthalmology and Visual Sciences, Washington University), and a Grant-In-Aid from The Glaucoma Foundation (New York, NY) (to J.M.G.).
The authors thank Beryl Ojwang and Belinda McMahon for their expert technical assistance.
References
- 1.Roth S. Li B. Rosenbaum P.S., et al. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest. Ophthalmol. Vis. Sci. 1998;39:777–785. [PubMed] [Google Scholar]
- 2.Zhu Y. Ohlemiller K.K. McMahan B.K., et al. Mouse models of retinal ischemic tolerance. Invest. Ophthalmol. Vis. Sci. 2002;43:1903–1911. [PubMed] [Google Scholar]
- 3.Gidday J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 4.Bergeron M. Gidday J.M. Yu A.Y., et al. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann. Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- 5.Prass K. Ruscher K. Karsch M., et al. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J. Cereb. Blood Flow Metab. 2002;22:520–525. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Y. Zhang Y. Ojwang B.A., et al. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: Role of HIF-1a and heme oxygenase-1. Invest. Ophthalmol. Vis. Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
- 7.Whitlock N.A. Agarwal N. Ma J.X. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest. Ophthalmol. Vis. Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- 8.Jiang B.H. Zheng J.Z. Leung S.W., et al. Transactivation and inhibitory domains of hypoxia-inducible factor 1α. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- 9.Schofield C.J. Ratcliffe P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005;338:617–626. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 10.Roth S. Endogenous neuroprotection in the retina. Brain Res. Bull. 2004;62:461–466. doi: 10.1016/j.brainresbull.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Nonaka A. Kiryu J. Tsujikawa A., et al. Inhibitory effect of ischemic preconditioning on leukocyte participation in retinal ischemia-reperfusion injury. Invest. Ophthalmol. Vis. Sci. 2001;42:2380–2385. [PubMed] [Google Scholar]
- 12.Zhu Y. Ohlemiller K.K. McMahan B.K., et al. Constitutive nitric oxide synthase activity is required to trigger ischemic tolerance in mouse retina. Exp. Eye Res. 2005;82:153–163. doi: 10.1016/j.exer.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Ettaiche M. Heurteaux C. Blondeau N., et al. ATP-sensitive potassium channels (K(ATP)) in retina: A key role for delayed ischemic tolerance. Brain Res. 2001;890:118–129. doi: 10.1016/s0006-8993(00)03152-8. [DOI] [PubMed] [Google Scholar]
- 14.Roth S. Dreixler J.C. Shaikh A.R., et al. Mitochondrial potassium ATP channels and retinal ischemic preconditioning. Invest. Ophthalmol. Vis. Sci. 2006;47:2114–2124. doi: 10.1167/iovs.05-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan Y. Hilliard G. Ferguson T., et al. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 16.Badr G.A. Zhang J.Z. Tang J., et al. Glut1 and glut3 expression, but not capillary density, is increased by cobalt chloride in rat cerebrum and retina. Brain Res. Mol. Brain Res. 1999;64:24–33. doi: 10.1016/s0169-328x(98)00301-5. [DOI] [PubMed] [Google Scholar]
- 17.Ke Q. Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol. Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 18.Halliwell B. Protection against tissue damage in vivo by desferrioxamine: What is its mechanism of action? Free Radic. Biol. Med. 1989;7:645–651. doi: 10.1016/0891-5849(89)90145-7. [DOI] [PubMed] [Google Scholar]
- 19.Bell E.L. Klimova T.A. Eisenbart J., et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Duffy A.E. Bordelon Y.M. McLaughlin B. Killer proteases and little strokes—how the things that do not kill you make you stronger. J. Cereb. Blood Flow Metab. 2007;27:655–668. doi: 10.1038/sj.jcbfm.9600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farinelli S.E. Greene L.A. Cell-cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J. Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimm C. Wenzel A. Groszer M., et al. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat. Med. 2002;8:718–724. doi: 10.1038/nm723. [DOI] [PubMed] [Google Scholar]
- 23.Bernaudin M. Nedelec A.S. Divoux D., et al. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Sharp F.R. Ran R. Lu A., et al. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1:26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dendorfer A. Heidbreder M. Hellwig-Burgel T., et al. Deferoxamine induces prolonged cardiac preconditioning via accumulation of oxygen radicals. Free Radic. Biol. Med. 2005;38:117–124. doi: 10.1016/j.freeradbiomed.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 26.Chavez J.C. LaManna J.C. Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: Potential role of insulin-like growth factor-1. J. Neurosci. 2002;22:8922–8931. doi: 10.1523/JNEUROSCI.22-20-08922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stroka D.M. Burkhardt T. Desbaillets I., et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001;15:2445–2453. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 28.Jewell U.R. Kvietikova I. Scheid A., et al. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001;15:1312–1314. [PubMed] [Google Scholar]
- 29.Berra E. Richard D.E. Gothie E., et al. HIF-1-dependent transcriptional activity is required for oxygen-mediated HIF-1alpha degradation. FEBS Lett. 2001;491:85–90. doi: 10.1016/s0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 30.Semenza G.L. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp. Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- 31.Sandau K.B. Zhou J. Kietzmann T., et al. Regulation of the hypoxia-inducible factor 1alpha by the inflammatory mediators nitric oxide and tumor necrosis factor-alpha in contrast to desferroxamine and phenylarsine oxide. J. Biol. Chem. 2001;276:39805–39811. doi: 10.1074/jbc.M107689200. [DOI] [PubMed] [Google Scholar]
- 32.Brahimi-Horn C. Mazure N. Pouyssegur J. Signalling via the hypoxia-inducible factor-1alpha requires multiple post-translational modifications. Cell. Signal. 2005;17:1–9. doi: 10.1016/j.cellsig.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Junk A.K. Mammis A. Savitz S.I., et al. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegazy K.A. Dunn M.W. Sharma S.C. Functional human heme oxygenase has a neuroprotective effect on adult rat ganglion cells after pressure-induced ischemia. NeuroReport. 2000;11:1185–1189. doi: 10.1097/00001756-200004270-00008. [DOI] [PubMed] [Google Scholar]
- 35.Nishijima K. Ng Y.S. Zhong L., et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kis B. Abraham C.S. Deli M.A., et al. Adrenomedullin in the cerebral circulation. Peptides. 2001;22:1825–1834. doi: 10.1016/s0196-9781(01)00533-2. [DOI] [PubMed] [Google Scholar]
- 37.Serrano J. Alonso D. Fernandez A.P., et al. Adrenomedullin in the central nervous system. Microsc. Res. Tech. 2002;57:76–90. doi: 10.1002/jemt.10053. [DOI] [PubMed] [Google Scholar]
- 38.Dorner G.T. Garhofer G. Huemer K.H., et al. Effects of adrenomedullin on ocular hemodynamic parameters in the choroid and the ophthalmic artery. Invest. Ophthalmol. Vis. Sci. 2003;44:3947–3951. doi: 10.1167/iovs.02-0855. [DOI] [PubMed] [Google Scholar]
- 39.Kaneko Y. Saito M. Mori A., et al. Vasodilator effects of adrenomedullin on retinal arterioles in streptozotocin-induced diabetic rats. J. Ocul. Pharmacol. Ther. 2006;22:317–322. doi: 10.1089/jop.2006.22.317. [DOI] [PubMed] [Google Scholar]
- 40.Udono-Fujimori R. Udono T. Totsune K., et al. Adrenomedullin in the eye. Regul. Pept. 2003;112:95–101. doi: 10.1016/s0167-0115(03)00027-2. [DOI] [PubMed] [Google Scholar]
- 41.Huang W. Wang L. Yuan M., et al. Adrenomedullin affects two signal transduction pathways and the migration in retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2004;45:1507–1513. doi: 10.1167/iovs.03-0731. [DOI] [PubMed] [Google Scholar]
- 42.Xia C.F. Yin H. Borlongan C.V., et al. Postischemic infusion of adrenomedullin protects against ischemic stroke by inhibiting apoptosis and promoting angiogenesis. Exp. Neurol. 2006;197:521–530. doi: 10.1016/j.expneurol.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 43.Chen L. Kis B. Busija D.W., et al. Adrenomedullin protects rat cerebral endothelial cells from oxidant damage in vitro. Regul. Pept. 2005;130:27–34. doi: 10.1016/j.regpep.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe K. Takayasu M. Noda A., et al. Adrenomedullin reduces ischemic brain injury after transient middle cerebral artery occlusion in rats. Acta. Neurochir. 2001;143:1157–1161. doi: 10.1007/s007010100007. [DOI] [PubMed] [Google Scholar]
- 45.Miyashita K. Itoh H. Arai H., et al. The neuroprotective and vasculo-neuro-regenerative roles of adrenomedullin in ischemic brain and its therapeutic potential. Endocrinology. 2006;147:1642–1653. doi: 10.1210/en.2005-1038. [DOI] [PubMed] [Google Scholar]
- 46.Cormier-Regard S. Nguyen S.V. Claycomb W.C. Adrenomedullin gene expression is developmentally regulated and induced by hypoxia in rat ventricular cardiac myocytes. J. Biol. Chem. 1998;273:17787–17792. doi: 10.1074/jbc.273.28.17787. [DOI] [PubMed] [Google Scholar]
- 47.Bernaudin M. Tang Y. Reilly M., et al. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J. Biol. Chem. 2002;277:39728–39738. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- 48.Gehlbach P. Purple R.L. Enhancement of retinal recovery by conjugated deferoxamine after ischemia-reperfusion. Invest. Ophthalmol. Vis. Sci. 1994;35:669–676. [PubMed] [Google Scholar]
- 49.Ophir A. Berenshtein E. Kitrossky N., et al. Protection of the transiently ischemic cat retina by zinc-desferrioxamine. Invest. Ophthalmol. Vis. Sci. 1994;35:1212–1222. [PubMed] [Google Scholar]
- 50.Banin E. Berenshtein E. Kitrossky N., et al. Gallium-desferrioxamine protects the cat retina against injury after ischemia and reperfusion. Free Radic. Biol. Med. 2000;28:315–323. doi: 10.1016/s0891-5849(99)00227-0. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg E.D. Therapeutic potential of iron chelators in diseases associated with iron mismanagement. J. Pharm. Pharmacol. 2006;58:575–584. doi: 10.1211/jpp.58.5.0001. [DOI] [PubMed] [Google Scholar]
- 52.Porter J.B. Monitoring and treatment of iron overload: State of the art and new approaches. Semin. Hematol. 2005;42:S14–S18. doi: 10.1053/j.seminhematol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Haimovici R. D'Amico D.J. Gragoudas E.S., et al. The expanded clinical spectrum of deferoxamine retinopathy. Ophthalmology. 2002;109:164–171. doi: 10.1016/s0161-6420(01)00947-2. [DOI] [PubMed] [Google Scholar]