

Abstract
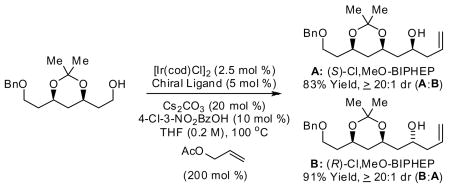
Iterative enantioselective carbonyl allylation from the alcohol oxidation level under the conditions of iridium catalyzed transfer hydrogenation enables chain elongation of 1,3-polyols. High levels of catalyst-directed enantioselectivity and diastereoselectivity are observed.
In the course of exploring C-C bond forming hydrogenations beyond hydroformylation, 1 we recently found that iridium catalyzed transfer hydrogenation of allylic acetates or allenes in the presence of aldehydes results in highly enantioselective carbonyl allylation.2,3,4,5 Whereas reactions from the aldehyde oxidation level employ isopropanol as reductant, alcohols may serve dually as hydrogen donor and aldehyde precursor, enabling asymmetric carbonyl allylation,2a,b,e crotylation2c and reverse prenylation2d directly from the alcohol oxidation level in the absence of any stoichiometric organometallic reagents.
In addition to the step economy associated with bypassing discrete alcohol oxidation and the pre-activation attending stoichiometric use of chiral allylmetal reagents, the ability to perform carbonyl allylation directly from the alcohol oxidation level enables allylations that are not easily achieved using aldehyde electrophiles. For example, whereas double asymmetric allylation of 1,3-diols delivers C2-symmetric adducts with exceptional levels of optical enrichment,2e corresponding allyl additions employing malondialdehydes are unknown. Based on this unique capability, an iterative two-directional elongation of 1,3-diols to furnish 1,3-polyols was achieved. 6, 7 Here, we report related one-directional chain elongations employing mono-protected 1,3-diols as starting materials. In all cases, high levels of catalyst-directed enantioselectivity and diastereoselectivity are observed. 8 This protocol, which involves iterative allylation from the alcohol oxidation level, avoids β-alkoxy aldehyde intermediates, which are often unstable with respect to elimination.
Our initial studies focused on the asymmetric allylation of O-benzyl 1,3-propylene glycol 1a. Under previously reported conditions employing the cyclometallated catalyst generated in situ from [Ir(cod)Cl]2, (S)-Cl,MeO-BIPHEP and 3-nitrobenzoic acid,2b the coupling of allyl acetate (1000 mol%) to 1a at 120 °C delivers the homoallyl alcohol 2a in 82% isolated yield and 94% enantiomeric excess (Table 1, entry 1). Lowering the reaction temperature to 100 °C slightly enhanced the degree of optical enrichment, but decreased the isolated yield of 2a (Table 1, entry 2). At 120 °C, a decrease in the loading of allyl acetate from 10 to 5 equivalents diminished the isolated yield of 2a by only 8% (Table 1, entries 1 and 3). However, upon a further decrease in the loading of allyl acetate from 5 to 2 equivalents, the isolated yield was unchanged (Table 1, entries 3 and 4). Finally, using the iridium C,O-benzoate generated in situ from [Ir(cod)Cl]2, (S)-Cl,MeO-BIPHEP and 4-chloro-3-nitrobenzoic acid, the homoallyl alcohol 2a is obtained in 88% isolated yield and 95% enantiomeric excess (Table 1, entry 5). As decreased reaction temperature (100 °C) or extended reaction time (40 hours) did not improve this result (Table 1, entries 6 and 7), the latter conditions employing the catalyst modified by 4-chloro-3-nitrobenzoic acid at 120 °C were selected for iterative homologation of 2a to form higher 1,3-polyols (Table 1, entry 5).
Table 1.
Asymmetric transfer hydrogenative carbonyl allylation of O-benzyl 1,3-propylene glycol 1a.
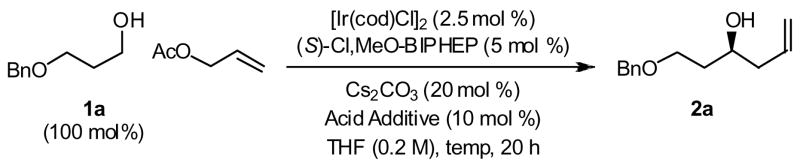 | |||||
|---|---|---|---|---|---|
| entry | acid additive | allyl acetate | temp °C | yield (%) | ee (%) |
| 1 | 3-N2-BzOH | 1000 (mol %) | 120 | 82 | 94 (S) |
| 2 | 3-NO2-BzOH | 1000 (mol %) | 100 | 76 | 96 (S) |
| 3 | 3-NO2-BzOH | 500 (mol %) | 120 | 74 | 94 (S) |
| 4 | 3-NO2-BzOH | 200 (mol %) | 120 | 74 | 95 (S) |
| ⇨5 | 4-Cl-3-NO2-BzOH | 200 (mol %) | 120 | 88 | 95 (S) |
| 6 | 4-Cl-3-NO2-BzOH | 200 (mol %) | 100 | 45 | 95 (S) |
| 7b | 4-Cl-3-NO2-BzOH | 200 (mol %) | 100 | 79 | 95 (S) |
All reactions were performed in 13 × 100 mm pressure tubes. The cited yields are of material isolated by silica gel chromatography and represent the average of two runs. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. See Supporting Information for experimental details.
The reaction was allowed to proceed for 40 hours.
The iterative synthesis of higher 1,3-polyols requires exceptional levels of catalyst-directed diastereoselectivity. To explore this prospect, homoallyl alcohol 2a was converted to the corresponding tert-butyldimethylsilyl ether 2b and subjected to ozonolysis in methanol solvent employing a small quantity of Sudan III as indicator (3–5 drops of a 1.5 mM solution in methanol).9 Upon complete consumption of 2b, as revealed through the change from a pink to a colorless solution, the reaction mixture was treated with sodium borohydride to deliver the alcohol 2c in 92% isolated yield. Upon exposure of 2c to the allylation conditions optimized for compound 1a (Table 1) employing the catalyst modified by (S)-Cl,MeO-BIPHEP at 120 °C, the product of carbonyl allylation (R,S)-3a was obtained in 55% isolated yield as 15:1 ratio of diastereomers. By lowering the reaction temperature to 100 °C and extending the reaction time to 40 hours under otherwise identical conditions, (R,S)-3a was obtained in 79% isolated yield as a 17:1 ratio of diastereomers. Under these same conditions, but using the catalyst modified by (R)-Cl,MeO-BIPHEP, the diastereomeric adduct (R,R)-3a was obtained in 71% isolated yield as a 15:1 ratio of diastereomers. Upon use of the achiral iridium catalyst ligated BIPHEP, (R,S)-3a and (R,R)-3a are produced in an equimolar ratio (Scheme 1).
Scheme 1.

Catalyst-directed diastereoselectivity in the transfer hydrogenative carbonyl allylation of 2c and synthesis of higher homologues 4a.a
aSee Supporting Information for experimental details.
With compounds (R,S)-3a and (R,R)-3a in hand, the stereoselective synthesis of higher homologues was undertaken. Exposure of (R,S)-3a or (R,R)-3a to methanol in the presence of p-toluenesulfonic acid (10 mol%) with subsequent introduction of 2,2-dimethoxypropane delivers the diastereomeric acetonides (R,S)-3b and (R,R)-3b, respectively, which are isolated as single diastereomers. Ozonolysis of (R,S)-3b and (R,R)-3b followed by NaBH4 reduction in accordance with the aforementioned procedure9 provides (R,S)-3c and (R,R)-3c. Transfer hydrogenative carbonyl allylation of (R,S)-3c or (R,R)-3c employing the catalyst modified by (S)-Cl,MeO-BIPHEP at 100 °C delivers the products of carbonyl allylation (R,S,S)-4a and (R,R,S)-4a, respectively. Using the enantiomeric catalyst modified by (R)-Cl,MeO-BIPHEP, (R,S)-3c or (R,R)-3c are transformed to homoallylic alcohols (R,S,R)-4a and (R,R,R)-4a, respectively. In each case, the minor diastereomer could not be detected by 1H NMR analysis. Upon use of the achiral iridium catalyst ligated BIPHEP, (R,S)-3c and (R,R)-3c are converted to equimolar quantities of (R,S,S)-4a, (R,S,R)-4a and (R,R,S)-4a, (R,R,R)-4a, respectively (Scheme 1).
To illustrate the utility of this methodology in a related context, O-p-methoxybenzyl neopentyl glycol 5a was subjected to transfer hydrogenative carbonyl allylation employing the catalyst modified by (R)-Cl,MeO-BIPHEP at 120 °C. The homoallylic alcohol 6a was produced in 80% isolated yield and 95% enantiomeric excess. Conversion of 6a to the tert-butyldimethylsilyl ether 6b, followed by removal of the p-methoxybenzyl ether delivers the primary neopentyl alcohol 6c. Transfer hydrogenative carbonyl allylation of 6c employing the catalyst modified by (R)-Cl,MeO-BIPHEP at 120 °C provides the homoallyl alcohol (R,R)-7a in 78% isolated yield in a 12:1 diastereomeric ratio. Using the catalyst modified by (S)-Cl,MeO-BIPHEP at 120 °C, 6c is converted to the homoallyl alcohol (S,R)-7a in 82% isolated yield in an 11:1 diastereomeric ratio. Using the achiral iridium catalyst modified by BIPHEP, (R,R)-7a and (S,R)-7a are formed in roughly equal proportion. Thus, catalyst-directed chain elongation may be conducted from either terminus of the 1,3-diol precursor with equal efficiency (Scheme 2).
Scheme 2.

Transfer hydrogenative chain elongation from both termini of neopentyl glycol 5a.
aSee supporting information for experimental details.
In summary, under the conditions of transfer hydrogenative carbonyl allylation, the stereochemical bias of enantiomeric iridium catalysts modified by (R)-or (S)-Cl,MeO-BIPHEP is found to override the intrinsic diastereofacial bias of transient β-chiral aldehydes. Based on this finding, a concise enantio- and diastereoselective synthesis of 1,3-polyols was achieved via iterative chain elongation. The utility of this approach stems from the ability to circumvent use of chirally modified allylmetal reagents, which require multi-step preparation, and the ability to perform chain elongation directly from the alcohol oxidation level, which avoids discrete generation of β-alkoxy aldehydes that are often unstable. Future studies will focus on the development of related C-C bond forming transfer hydrogenations, including imine additions from the amine oxidation level.10
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation, the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable, and the NIH-NIGMS (RO1-GM069445) for partial support of this research. The Higher Education Commission, Pakistan is acknowledged for generous fellowship support (AH). Dr. Oliver Briel of Umicore is thanked for the generous donation of [Ir(cod)Cl]2.
Footnotes
Supporting Information Available. Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- 2.For enantioselective carbonyl allylation, crotylation and reverse prenylation via iridium catalyzed transfer hydrogenation, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48 doi: 10.1002/anie.200901648. In Press.
- 3.For reviews on enantioselective carbonyl allylation, see: Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Ramachandran PV. Aldrichim Acta. 2002;35:23.Kennedy JWJ, Hall DG. Angew Chem Int Ed. 2003;42:4732. doi: 10.1002/anie.200301632.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.
- 4.For selected reviews of carbonyl allylation based on the reductive coupling of metallo-π-allyls derived from allylic alcohols, ethers or carboxylates, see: Masuyama Y. Palladium-Catalyzed Carbonyl Allylation via p-Allylpalladium Complexes. In: Lanny S, editor. Advances in Metal-Organic Chemistry; Liebeskind. Vol. 3. JAI Press; Greenwich: 1994. pp. 255–303.Tamaru Y. Palladium-Catalyzed Reactions of Allyl and Related Derivatives with Organoelectrophiles. In: Negishi Eii, Meijere Armin de., editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 2. Wiley; New York: 2002. pp. 1917–1943.Tamaru Y. Novel Catalytic Reactions involving p-Allylpalladium and -Nickel as the Key Intermediates: Umpolung and ß-Decarbopalladation of p-Allylpalladium and Nickel-Catalyzed Homoallylation of Carbonyl Compounds with 1,3-Dienes. In: Tsuji J, editor. Perspectives in Organopalladium Chemistry for the XXI Century. Elsevier; Amsterdam: 1999. pp. 215–231.Kondo T, Mitsudo T-a. Curr Org Chem. 2002;6:1163.Tamaru Y. Eur J Org Chem. 2005;13:2647.Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur J Org Chem. 2007;22:3599.
- 5.For selected examples of carbonyl allylation via catalytic Nozaki-Hiyama-Kishi coupling of allylic halides, see: Fürstner A, Shi N. J Am Chem Soc. 1996;118:2533.Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537.McManus HA, Cozzi PG, Guiry PJ. Adv Synth Catal. 2006;348:551.Hargaden GC, Müller-Bunz H, Guiry PJ. Eur J Org Chem. 2007:4235.Hargaden GC, O’Sullivan TP, Guiry PJ. Org Biomol Chem. 2008;6:562. doi: 10.1039/b715834c.
- 6.For a review on two-directional chain elongation in target-oriented synthesis, see: Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9.
- 7.For selected reviews of the synthesis of 1,3-diol substructures, see: Masamune S, Choy W. Aldrichim Acta. 1982;15:47.Oishi T, Nakata T. Synthesis. 1990:635.Schneider C. Angew Chem Int Ed. 1998;37:1375. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1375::AID-ANIE1375>3.0.CO;2-H.Bode SE, Wohlberg M, Müller M. Synthesis. 2006:557.
- 8.For selected examples of catalyst directed diastereoselectivity, see: Minami N, Ko SS, Kishi Y. J Am Chem Soc. 1982;104:1109.Ko SY, Lee AWM, Masamune S, Reed LA, III, Sharpless KB, Walker FJ. Science. 1983:949. doi: 10.1126/science.220.4600.949.Kobayashi S, Ohtsubo A, Mukaiyama T. Chem Lett. 1991:831.Hammadi A, Nuzillard JM, Poulin JC, Kagan HB. Tetrahedron: Asymmetry. 1992;3:1247.Doyle MP, Kalinin AV, Ene DG. J Am Chem Soc. 1996;118:8837.Trost BM, Calkins TL, Oertelt C, Zambrano J. Tetrahedron Lett. 1998;39:1713.Balskus EP, Jacobsen EN. Science. 2007;317:1736. doi: 10.1126/science.1146939.Han SB, Kong JR, Krische MJ. Org Lett. 2008;10:4133. doi: 10.1021/ol8018874.
- 9.Veysoglu T, Mitscher LA, Swayze JK. Synthesis. 1980:807. [Google Scholar]
- 10.The collective alcohol-unsaturate C-C coupling we describe (ref. 1c,d) may be viewed as “hydrohydroxyalkylations.” For related amine-unsaturate C-C coupling, or “hydroaminoalkylation,” see: Maspero F, Clerici MG. Synthesis. 1980:305.Nugent WA, Ovenall DW, Homes SJ. Organometallics. 1983;2:161.Herzon SB, Hartwig JF. J Am Chem Soc. 2007;129:6690. doi: 10.1021/ja0718366.Herzon SB, Hartwig JF. J Am Chem Soc. 2008;130:14940. doi: 10.1021/ja806367e.Kubiak R, Prochnow I, Doye S. Angew Chem Int Ed. 2009;48:1153. doi: 10.1002/anie.200805169.Bexrud JA, Eisenberger P, Leitch DC, Payne PR, Schafer LL. J Am Chem Soc. 2009;131:2116. doi: 10.1021/ja808862w.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.